

Abstract
Chronic wasting disease (CWD), a transmissible prion disease that affects elk and deer, poses new challenges to animal and human health. Although the transmission of CWD to humans has not been proven, it remains a possibility. If this were to occur, it is important to know whether the “acquired” human prion disease would show a phenotype including the scrapie prion protein (PrPSc) features that differ from those associated with human sporadic prion disease. In this study, we have compared the pathological profiles and PrPSc characteristics in brains of CWD-affected elk and deer with those in subjects with sporadic Creutzfeldt-Jakob disease (CJD), as well as CJD-affected subjects who might have been exposed to CWD, using histopathology, immunohistochemistry, immunoblotting, conformation stability assay, and N-terminal protein sequencing. Spongiform changes and intense PrPSc staining were present in several brain regions of CWD-affected animals. Immunoblotting revealed three proteinase K (PK)-resistant bands in CWD, representing different glycoforms of PrPSc. The unglycosylated PK-resistant PrPSc of CWD migrated at 21 kDa with an electrophoretic mobility similar to that of type 1 human PrPSc present in sporadic CJD affecting subjects homozygous for methionine at codon 129 (sCJDMM1). N-terminal sequencing showed that the PK cleavage site of PrPSc in CWD occurred at residues 82 and 78, similar to that of PrPSc in sCJDMM1. Conformation stability assay also showed no significant difference between elk CWD PrPSc and the PrPSc species associated with sCJDMM1. However, there was a major difference in glycoform ratio of PrPSc between CWD and sCJDMM1 affecting both subjects potentially exposed to CWD and non-exposed subjects. Moreover, PrPSc of CWD exhibited a distinct constellation of glycoforms distinguishable from that of sCJDMM1 in two-dimensional immunoblots. These findings underline the importance of detailed PrPSc characterization in trying to detect novel forms of acquired prion disease.
Chronic wasting disease (CWD),2 first described in 1967, was identified in 1977 as a transmissible spongiform encephalopathy or prion disease that affects both captive and free-ranging cervids (1, 2). Three species are known to be affected, mule deer (Odocoileus hemionus), white-tailed deer (O. virginianus), and Rocky Mountain elk (Cervus elaphus nelsoni) (3).
In addition to progressive loss of body weight, CWD is clinically characterized by abnormal behavior, polydipsia, hypersalivation, and occasionally ataxia and tremor (2, 4). The major histopathological features include spongiform degeneration, astrogliosis, and neuronal loss, as in most other forms of prion disease (2, 4). However, in CWD these lesions are reported to have a distinct distribution (5, 6). In the central nervous system, the spongiform degeneration is apparently most prominent in subcortical regions, including the thalamus and hypothalamus, lower brain stem, cerebellum, and spinal cord, whereas the cerebral cortex is less affected or unaffected (5, 6). Kuru plaques, often surrounded by vacuoles as in the “florid” plaques of variant Creutzfeldt-Jakob disease (vCJD), the human prion disease acquired from the consumption of prion-contaminated beef, have been convincingly shown only in captive mule deer (7, 8). The disease-associated prion protein isoform (PrPSc) is detected by immunohistochemistry with a topography similar to, but wider than, that of the histological lesions (5–7). The immunostained PrPSc deposits have been reported to be dispersed in the parenchyma with a punctate or “synaptic” pattern, in addition to the loose clusters forming the so-called plaque-like pattern as well as packed clusters in the kuru plaques (7–11). The PrPSc deposits may line along the surface of neuronal cell bodies and processes as well as in the parenchyma immediately surrounding small vessels (7). Consistent PrPSc immunostaining has also been reported in the peripheral nervous system, especially the vagus and sympathetic nerves, lymphoreticular system, and visceral organs (5, 10).
CWD appears to be freely transmitted among the three susceptible species of cervids by direct or indirect horizontal contact (2, 4, 11). The oral port of infection through forage contaminated by feces or saliva from infected animals is considered the most likely mode of transmission (2–4, 11). Intra-species transmissibility and increased surveillance have contributed to the discovery that geographic distribution and prevalence of CWD are greater than previously suspected (2–4). In free-ranging and captive animals, CWD has now been detected in fourteen states in the United States and in two Canadian provinces, with a prevalence of up to 20% (2, 4, 12). Furthermore, CWD has been transmitted to a number of experimental animals, including laboratory mice, ferrets, mink, goats, and cattle (13, 14). The expanded geographic distribution and increased prevalence as well as the infectivity of CWD have raised questions concerning CWD transmissibility to humans. These questions are particularly alarming in North America where almost all the CWD-affected animals have been observed and a large number of hunters and their families regularly consume cervid meat. Although prion disease has been demonstrated in at least 27 hunters or their family members, as a group these cases were heterogeneous in terms of the clinical and pathological phenotypes, the PrP genotype, and the PrPSc subtype (15).3 Furthermore, the “hunter” cases appeared to fit rather well in the known subtypes of human prion disease (16). However, the phenotype of the human prion disease that might be acquired from CWD is not known; it might mimic any of the subtypes of sporadic CJD, making the detection of this hypothetical “CWD-acquired CJD” difficult. Thus far, the findings with the CJD in the hunter cases (15) contrast with the reports on vCJD, where affected cases have highly homogeneous features (17, 18). The homogeneity of vCJD has been attributed to the exposure of the affected individuals to the same type of PrPSc or prion strain associated with bovine spongiform encephalopathy (BSE) (19). Indeed, features of the BSE-associated PrPSc are reproduced in vCJD (20). By analogy, it is likely that the possible CWD-acquired CJD may reproduce the features of the PrPSc associated with CWD. To gain insights into important pathological features and molecular characteristics of PrPSc in CWD-affected elk and deer, we have carried out a comparative study of the histopathological, immunohistochemical, and PrPSc characteristics of CWD and sporadic CJD subjects as well as CJD-affected subjects who might have been exposed to CWD.
MATERIALS AND METHODS
Reagents and Antibodies
All chemicals were purchased from Sigma unless specified otherwise. Immobilized pH gradient strips (pH 3–10, 11 cm long), ampholine pH 3–10, and reagents for enhanced chemiluminescence were from Amersham Biosciences. Mouse monoclonal antibodies (mAb) against PrP 8H4 (21) and 3F4 (Signet Laboratories, Dedham, MA) were used in this study.
Brain Tissues
Tissue samples from free-ranging animals were collected from the wild, including seven mule deer (O. hemionus), one white-tailed deer (O. virginianus), and six Rocky Mountain elk (C. elaphus nelsoni). Brain tissues were stored at −80 °C until use. Brains from animals affected by CWD were diagnosed and characterized by histopathology, immunohistochemistry, and immunoblotting. Human brain tissue was processed and characterized as previously reported (16, 22). Brain tissue was obtained from five cases of elk and deer hunters who also had a history of regularly consuming elk and deer meat; one of these cases was also known to hunt in an area where CWD has been proven to be endemic. They were all homozygous methionine at codon 129, had PrPSc type 1 and a phenotype consistent with CJD, with an average age of onset of 71 years (range 61–84 years) and an average duration of 5 months (range 2–5 months). Sporadic cases of human CJD having either PrPSc type 1 or 2 were also used.
Molecular Genetics
Genomic DNA was extracted from frozen brain tissues. The open reading frame of cervid PrP gene was sequenced from both alleles as described previously (16).
Histology and Immunohistochemistry
Cervid brains removed at autopsy were fixed in formalin and sampled. Tissue blocks were kept in 98% formic acid for 1 h, followed by a second formalin fixation and paraffin embedding. They were then sectioned and prepared for histological examination according to standard techniques. For PrPSc immunohistochemistry, sections were immersed in 1.5 mM hydrochloric acid and microwaved for 15 min at 20% power. Sections were then rinsed and incubated for 2 h in a 4 M guanidine thiocyanate solution at 4 °C. The endogenous peroxidase was blocked by incubation with 8% hydrogen peroxide in distilled water for 10 min. After rinsing, sections were incubated with mAb 8H4 at 1:600, washed, and incubated with Dako Cytomation Envision plus Dual Link System Peroxidase for 30 min. Diaminobenzidine tetrahydrochloride was used to visualize the immunoreactivity.
One- and Two-dimensional Immunoblot
Cervid and human brain homogenates (10%, w/v) were made in ice-cold lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% Nonidet P 40, 0.5% sodium deoxycholate, 10 mM Tris-HCl, pH 7.5), followed by centrifugation at 1,000 × g for 10 min at 4 °C to remove debris. An aliquot of the brain homogenate was digested with proteinase K (PK) at 50 μg/ml for 1 h at 37 °C. The digestion was terminated by the addition of 3 mM phenylmethylsulfonyl fluoride and an equal volume of 2×gel loading buffer (6% sodium dodecyl sulfate (SDS), 5% β-mercaptoethanol, 4 mM EDTA, 20% glycerol, 125 mM Tris-HCl, pH 6.8), followed by boiling for 10 min. In some cases, deglycosylation of proteins was performed following precipitation with 5 volumes of methanol. The methanol-precipitated proteins were denatured and incubated with recombinant peptide N-glycosidase F according to the manufacturer’s protocol (Roche Applied Science). Deglycosylated samples were then mixed with an equal volume of 2×gel loading buffer and boiled for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using 12% Tris-glycine mini-gels (Invitrogen). PrPSc was also characterized by two-dimensional gel electrophoresis as described previously (23). Briefly, PK-treated samples were boiled in gel loading buffer, followed by precipitation by 9 volumes of chilled acetone at −20 °C for 2 h, followed by centrifugation at 16,000 × g for 15 min at 4 °C. The pellets were resuspended in 10% trichloroacetic acid for 2 h at room temperature and were centrifuged again. The pellets were resuspended in 200 μl of rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, 0.5% ampholine pH 3–10, and trace amount of bromphenol blue) and were loaded onto the immobilized pH gradient strips for rehydration at 50 V for 14 h. The first-dimensional isoelectric focusing was performed on the rehydrated immobilized pH gradient strips for a total of 40 kVh using a Bio-Rad Protean isoelectric focusing cell. For the second-dimension SDS-PAGE, the focused immobilized pH gradient strips were equilibrated for 15 min each in equilibration buffer A (6 M urea, 2% SDS, 20% glycerol, 2% dithiothreitol, and 0.375 M Tris-HCl, pH 8.8) and then in equilibration buffer B (6 M urea, 2% SDS, 20% glycerol, 2.5% iodoacetamide, and 0.375 M Tris-HCl, pH 8.8). The equilibrated strips were loaded onto 15% Tris-glycine SDS-PAGE gels (Bio-Rad Criterion gels). Electrophoresis was conducted at 120 V. The mAb 8H4 were used for all the two-dimensional immunoblots, except for Fig. 3B, which was immunoreacted with mAb 3F4. Proteins on the SDS-PAGE gels were transferred onto polyvinylidene difluoride membranes at 70 V for 2 h at 4 °C. The membranes were blocked with 10% nonfat milk in TBST (150 mM NaCl, 0.1% Tween 20, 10 mM Tris-HCl, pH 7.6) overnight at 4 °C prior to incubation with either anti-PrP mAb 8H4 (1:10,000) or 3F4 (1:50,000). Following four washes with TBST, the polyvinylidene difluoride membranes were incubated with sheep anti-mouse Ig G conjugated with horseradish peroxidase (Amersham Biosciences). The PrP bands were visualized on Kodak X-Omat films by enhanced chemiluminescence (ECL Plus kit; Amersham Biosciences).
FIGURE 3. Two-dimensional maps of PK-resistant PrPSc in brains of CWD-affected cervids and sCJDMM1.

A and B, sCJDMM1; C and D, mule deer; E and F, white tail deer; G and H, elk. A, C, E and G, N-glycosidase F untreated; B, D, F and H, N-glycosidase treated. Three sets of spots are seen in the two-dimensional blots of all the N-glycosidase-untreated PrPSc species from both sCJD and CWD (A, C, E, and G). The spots formed by the cervid PrPSc extend more toward the basic part of the pH gradient (3.0–10.0) than those of sCJDMM1 PrPSc (compare panel A with C, E, and G). Note the relative overrepresentation of the diglycosylated and the underrepresentation of the unglycosylated form in cervid PrPSc as compared with sCJDMM1 PrPSc (A, C, E, and G). The deglycosylated PrPSc from the mule and white tail deer migrates toward the more basic part of the pH gradient (D and F), whereas the pH mobility of the deglycosylated elk PrPSc is more similar to that of the sCJDMM1 PrPSc (B and H). The position of molecular mass markers (in kDa) is indicated by dotted lines.
Conformational Stability Assay of PrPSc
Aliquots of brain homogenates were incubated with various concentrations of guanidine hydrochloride (GdnHCl) from 0 to 3.0 M (24, 25). GdnHCl stock solutions were prepared by dilution in water from an 8 M solution. Following incubation for 1 h at room temperature, all samples were precipitated in pre-chilled methanol at −20 °C for 2 h and centrifuged at 16,000×g for 20 min at 4 °C. Pellets were resuspended in lysis buffer and treated with 100 μg/ml PK for 1 h at 37 °C. The reaction was stopped by adding phenylmethylsulfonyl fluoride (3 mM) and gel loading buffer and boiled for 10 min. The samples were used for immunoblotting with 8H4.
Purification of PrPSc from CWD-affected Brains
Brain tissues (5–10 g) were used for purification of PK-resistant PrPSc core fragments according to the published method (26), as modified (27). After the final sedimentation of PK-digested PrPSc by ultracentrifugation, the pellets were denatured followed by deglycosylation with N-glycosidase F. PrPSc were purified further by micropreparative continuous elution SDS-PAGE (14% gel, Mini Prep Cell apparatus; Bio-Rad). Proteins were eluted at a flow rate of 70 μl/min and collected into 400-μl fractions. The fractions containing PrPSc as detected by immunoblotting with mAb 8H4 were pooled and lyophilized.
N-terminal Protein Sequencing
Purified PK-resistant PrPSc in the pooled fractions as described above were separated by 12% Tris-glycine SDS-PAGE mini-gels (Invitrogen), transferred onto Problott membranes (Applied Biosystems, Foster City, CA), and visualized by Coomassie Blue staining. N-terminal protein sequencing by automated Edman degradation was performed as described previously (22) at the ProSeq Microsequencing Facility (Boxford, MA) using the Applied Bio-systems 477A Protein Sequencer. N-terminal sequencing typically proceeded for 20–25 cycles. N-terminal sequences of PrPSc were determined by alignment of the experimentally determined amino acids at each cycle with the translated human PrP sequence (Swiss-Prot accession number P04156).
RESULTS
Histopathology of CWD
The cerebral cortex and basal ganglia of the elk with CWD show minimal fine spongiform degeneration and astrogliosis with focal distribution. The spongiform degeneration with astrogliosis is more prominent in the thalamus where it forms clusters of coarse vacuoles. Fine spongiosis, often in small clusters, is present in the molecular layer of the cerebellum, dorsal nuclei of the pons, and in the substantia gelatinosa of the spinal cord (Fig. 1A). Occasional large neurons in various nuclei of the pons show a vacuole (Fig. 1A). No kuru or multicore plaques are detected. Neuronal loss and astrogliosis are minimal except for the molecular layer of the cerebellum, which shows rarefaction of granule cells with no indication of apoptosis.
FIGURE 1. Histopathology and immunohistochemistry of PrPSc in the brain stem of elk affected by CWD.

A, prominent spongiform degeneration with an intra-neuronal vacuole (arrow) (Pons; hematoxylin and eosin). B, immunohistochemistry demonstrates amyloid plaque-like PrPSc deposition (Pons; mAb 8H4).
Immunohistochemistry
PrPSc immunostaining of the elk brain tissue affected by CWD shows rounded granular aggregates of various sizes and densities corresponding to the so-called plaque-like pattern (Fig. 1B). Occasionally small plaque-like formations are arranged in short rows or the immunostain is linear, consistent with staining of cell processes. When the plaque-like deposits are confluent and loose, the immunostaining may become similar to the synaptic pattern. Occasional focal areas of coarse PrPSc immunostaining pattern apparently associated with coarse spongiosis are also present. In addition, there is occasional staining around intra-parenchymal vessels. The PrPSc immunostaining is consistently present in the cerebral cortex, basal ganglia, and thalamus. In the cerebellum the immunostaining is present in both molecular and granule cell layers as well as in the dentate nucleus. In the pons it is widespread over gray structures, whereas in the spinal cord it is generally confined to the dorsal part of the dorsal horns.
Detection and Typing of PrPSc in CWD
Brain samples from a total of seven mule deer, one white tail deer, and six elk affected by CWD tested positive for PK-resistant PrPSc on immunoblots. PrPSc characteristics of CWD-affected animals were compared with those of sporadic CJD cases associated with either PrPSc type 1 or type 2 (using nomenclature of Parchi et al. (28)). Representative results from three mule deer, one white tail deer, and three elk samples are shown in Fig. 2. The bands of the unglycosylated form of PK-resistant PrPSc species from mule deer, white tail deer, and elk all migrate to ~21 kDa and match the gel mobility of human PrPSc type 1, but not type 2 (Fig. 2, A and B). However, the PrPSc associated with all three species of cervids differs significantly in the ratio of the three glycoforms from the human PrPSc type 1 associated with sCJD affecting subjects who were homozygous for methionine at codon 129 (sCJDMM1, Ref. 16) (Fig. 2C). In the PrPSc type 1 associated with sCJDMM1, the monoglycosylated form is predominant, accounting for 44–45% of all the glycoforms, whereas the diglycosylated and unglycosylated account for 21–24 and 31–35%, respectively (16). In contrast, both elk and mule deer PrPSc species show the prevalence of the diglycosylated, representing 47–50% of the total, whereas the mono-glycosylated and unglycosylated forms represent ~30–33 and 20%, respectively (Fig. 2C). This PrPSc glycoform ratio of CWD is similar to that of BSE and variant CJD (20).
FIGURE 2. Immunoblots and glycoform ratios of PK-resistant PrPSc from CWD-affected elk and deer and sCJD associated with PrPSc types 1 and 2.

Human brain tissue was obtained from subjects with sCJDMM1 and sCJDMM2 according to the classification of Parchi et al. (16). Brain homogenates were treated with PK, followed by SDS/PAGE (12% gel) and immunoblotting using mAb 8H4. Enzymatic deglycosylation of PK-treated samples was performed using N-glycosidase F. A, the PK digest equivalent to 10 μl of 10% brain homogenates was loaded on SDS/PAGE gels. The PK-resistant PrPSc was detected as a triplet of bands representing diglycosylated (●), monoglycosylated (○), and unglycosylated (□) forms. B, following deglycosylation with N-glycosidase F, mainly unglycosylated (□)PrPSc was detected. PK-resistant PrPSc in CWD co-migrates with human PrPSc type 1 as demonstrated by the alignment of the unglycosylated bands (□) in panels A and B. C, three specimens of CWD-affected elk and mule deer were subjected to immunoblotting as in panel A except that the amount of the PK digest loaded was adjusted to similar intensity of PrPSc signals to assist the glycoform analysis by densitometry. The immunoblots were examined by densitometry to determine the ratio of diglycosylated (Diglyc), monoglycosylated (Monoglyc), and unglycosylated (Unglyc) forms of PrPSc expressed as percentage of the total (mean ± S.D.). Data for sCJDMM1 were obtained from Parchi et al. (16).
Two-dimensional Immunoblot
We further compared the PK-resistant PrPSc species associated with CWD of mule deer, white tail deer, and elk with that of sCJDMM1 using two-dimensional gel electrophoresis. All gel profiles show three rows of well resolved spots. However, the molecular mass, representation, and number of these spots vary between the cervid and sCJDMM1 samples (Fig. 3). In CWD preparations not treated with N-glycosidase, the upper row migrates between ~29–30 kDa as compared with the upper row in sCJD preparations that is detectable between ~27 and 29.5 kDa. Furthermore, the upper row includes at least 8 spots, which migrate between pH 8.3 and 5.3 of the gradient in the CWD preparations, whereas there are 9 spots in sCJDMM1 that migrate between pH 6.7 and 5.0. The middle row includes at least 9 spots between pH 9.0 and 5.7 in CWD and at least 8 spots between pH 7.0 and 5.0 in sCJDMM1. The molecular mass range of the middle row also is different, being ~27.5–25.0 kDa in CWD and ~26.5–24 kDa in sCJDMM1. The third row also differs as it includes at least three spots of 21.5–22.5 kDa distributed between pH 9.0 and 7.0 in CWD, whereas at least four spots of ~22 kDa are seen in sCJDMM1 that migrate between pH 8.0 and 6.7. The two-dimensional gel also confirms that, although in sCJDMM1 the middle and lower rows are much more prominent than the upper row, the opposite happens in CWD, where the upper row is much better represented than the other two. The profiles of the PK-resistant PrPSc core proteins obtained following deglycosylation by N-glycosidase confirm that the PrPSc unglycosylated core proteins of sCJDMM1 and CWD have similar molecular weights (Fig. 3, B, D, F, H). However, in CWD, deglycosylated PrPSc species are more basic than that in sCJDMM1. This difference is especially noticeable between sCJDMM1 and mule deer and white tail deer PrPSc preparations. Whereas in sCJDMM1 PrPSc distributes over the ~5.8–8.5 pH gradient with the major spots located between pH 5.9 and 6.8, in CWD PrPSc migrates between pH 6.5 and 9.5 with conspicuous spots located at pH 8.5–9.5 (Fig. 3, B, D, F). The number of the spots is also different with six detectable spots in sCJDMM1 and 5 and 7 in the mule and white tail deer. The PK-resistant PrPSc core of elk CWD displays a pH gradient distribution between 5.8 and 8.5, similar to that of sCJDMM1, but the spots appear to form a quite different pattern (Fig. 3, B and H).
Conformational Stability Assay of PrPSc
Conformational stability assay has been used to distinguish different prion strains based on the concentration of GdnHCl needed to denature the PK-resistant PrPSc present in the individual strains. GdnHCl-dependent PrPSc denaturation is determined by the fraction of PrPSc that is rendered PK sensitive following GdnHCl treatment (24, 25, 29). The GdnHCl concentration required to make half of the total PrPSc sensitive to PK digestion (GdnHCl ½) is used as a measurement of the relative conformational stability of PrPSc. The GdnHCl ½ was ~1.62 ± 0.01 M (n = 3) for PrPSc from CWD-affected elk and ~1.84 ± 0.17 M (n = 4) for PrPSc from sCJDMM1 (Fig. 4), but the difference was not statistically significant (p = 0.12, two-tail t-test). Our data indicate that there is no significant difference in the conformational stability between the two PrPSc species.
FIGURE 4. Conformational stability assay of PrPSc.

A and B, Western blots of PK-resistant PrPSc recovered from brain homogenates (A, cerebral cortex from CWD-affected elk; B, frontal cortex from a case of sCJDMM1) following treatment with increasing concentrations of GdnHCl. C, the levels of PK-resistant PrPSc from CWD (filled diamond) or sCJDMM1 (filled square) as a function of increasing concentrations of GdnHCl. Data obtained by densitometry of Western blots probed with mAb 8H4, as shown in panels A and B, are the average of independent experiments with samples from three elk with CWD and four human brains with sCJDMM1 (mean ± S.D.).
N-terminal Sequencing
Additional experiments have been carried out to further characterize the protein core of the PK-resistant PrPSc fragment associated with CWD and compare it with those of human PrPSc type 1. In two elk affected by CWD, we detected two major coexisting N-terminal sequences of PrPSc following treatment with PK (Fig. 5A), as previously observed in PrPSc type 1 associated with CJD (22). The N termini of the two fragments are at residues Gly-82 and Gly-78 in both CWD (Fig. 5A) and sCJDMM1 (Fig. 5B). In contrast, the N termini of the two major PK-resistant fragments of the PrPSc associated with sCJDVV1 are at residues Gly-86 and Gly-82 (ref 22). These findings confirm that the main PK cleavage sites of PrPSc are the same in CWD and sCJDMM1 but different from those of sCJDVV1.
FIGURE 5. Determination of N-terminal PK cleavage sites of PrPSc in CWD and CJD by Edman sequencing.
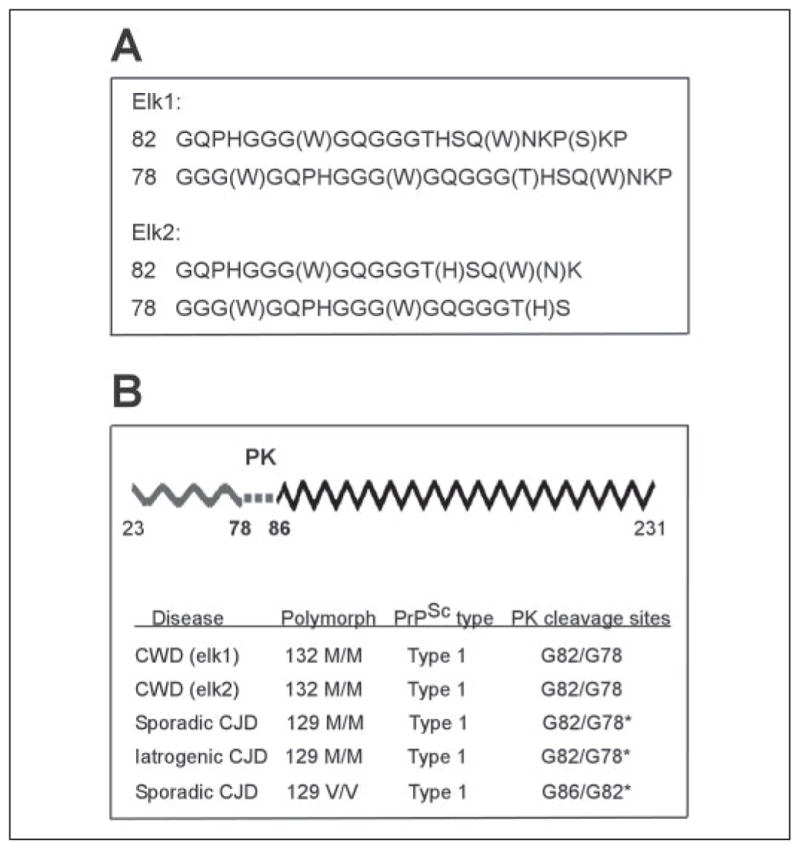
A, Edman sequencing of PK-resistant PrPSc purified from brains of two elk was performed according to the procedure described by Parchi et al. (22). B, schematic presentation of the full-length PrP polypeptide inclusive of residues 23–231 (upper). Dotted line signifies the PK cleavage sites in PrPSc. Wavy and zigzagged lines indicate PK-sensitive and PK-resistant regions, respectively. Comparison of the N-terminal PK cleavage sites of PrPSc in CWD and sCJD MM1, sCJDVV1, and iatrogenic CJDMM1 (lower). *, data from Parchi et al. (22). The two major PK-resistant fragments of CWD PrPSc share the N termini with sCJDMM1 and iatrogenic CJDMM1 but not with sCJDVV1.
PrPSc from the Hunter Cases
Immunoblots of brain homogenates from five hunter cases who had a genotype of MM and carried PrPSc type 1 show gel migration of the PK-resistant PrPSc fragment at 21 kDa, indistinguishable from that of sCJDMM1 and CWD (Fig. 6A). However, the glycoform ratio of each of these cases matches that of sCJDMM1 (Fig. 6B), but not CWD (Fig. 2C).
FIGURE 6. Immunoblots and glycoform ratios of PrPSc from CJDMM1 subjects with possible exposure to CWD.
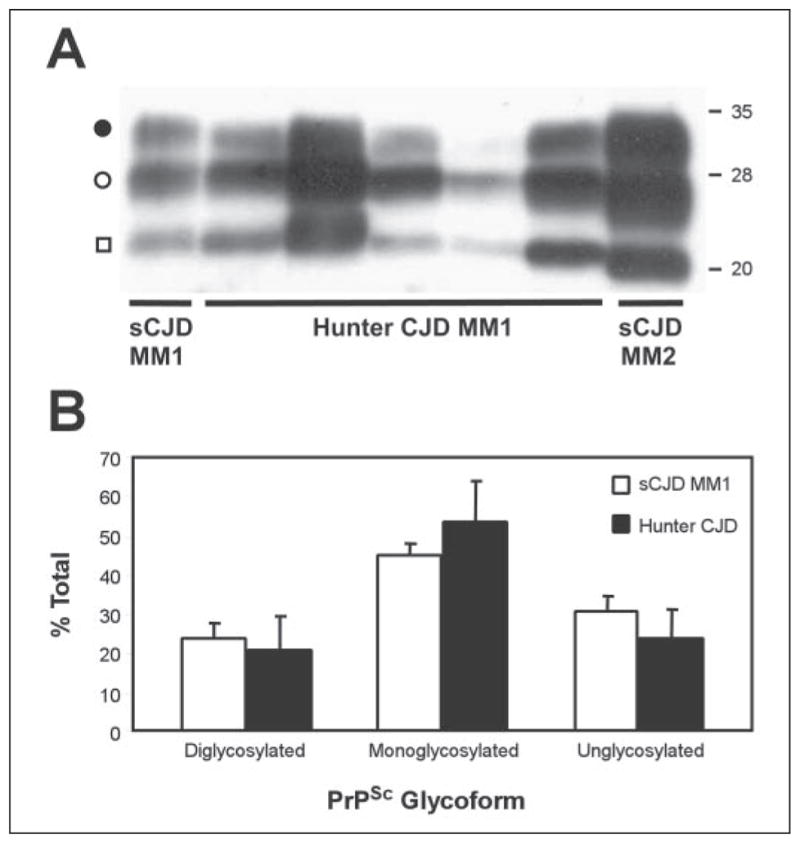
Five cases of CJD in subjects with MM1 and a history of potential exposure to CWD due to hunting and consumption of venison (hunter CJD MM1) were used for analysis. Control cases were obtained from subjects with sCJDMM1 and sCJDMM2 according to the classification of Parchi et al. (16). Brain homogenates were treated with PK, followed by SDS/PAGE (12% gel) and Western blotting using the mAb 3F4. A, immunoblot. The PrPSc bands detected represent diglycosylated (●), mono-glycosylated (○), and unglycosylated (□) forms. The three PrPSc bands of the hunter CJDMM1 are aligned with those of the sCJDMM1, but not with those of sCJDMM2, as especially shown by the band containing the unglycosylated form (□). B, glycoform ratio of PK-resistant PrPSc from the hunter CJDMM1 and sCJDMM1 cases. The ratios of diglycosylated, monoglycosylated, and unglycosylated forms of PrPSc in the hunter cases were determined by densitometry of the corresponding immunoblots and were averaged and expressed as percentage of the total (mean ± S.D.). Data for sCJDMM1 were obtained from Parchi et al. (16).
DISCUSSION
Consistent with previous studies, we observed that spongiform degeneration involving the neuropil as well as the cell body of neurons is the histopathological hallmark of CWD in free-ranging elk, whereas astrogliosis and neuronal loss are generally minimal. These lesions preferentially affect subcortical regions, especially the thalamus and the lower brain stem where some anatomical structures are preferentially affected. The immunostaining shows a PrPSc distribution in loose clusters of granules of different sizes, a pattern commonly defined as plaque like. Other patterns such as perivascular and perineuronal arrangements are occasionally seen. PrPSc has a wider distribution than the spongiform degeneration, so cerebral cortex and cerebellum that show minimal spongiform degeneration are relatively well immunostained for PrPSc.
The type and distribution of the histological lesions and of the PrPSc immunostaining in the cervid samples are different from those observed in human prion diseases. However, the lack of similarity between CWD and the human disease may be possible even if a subtype of CJD believed to be sporadic is actually acquired from CWD. Overwhelming evidence indicates that vCJD is caused by the transmission of BSE to humans (30, 31). Therefore, comparing BSE with vCJD may provide guidance on how to approach the issue of possible transmission of CWD to humans. The histopathology and the PrPSc immunohistochemistry of BSE are different from those of vCJD in the type and topography of the lesions as well as the pattern of the PrPSc immunostaining (32, 33). In BSE the lesions are most prominent in subcortical structures of the brain, especially basal ganglia, thalamus, hypothalamus, and selected regions of brain stem and spinal cord, whereas the cerebral and cerebellar cortices are minimally affected (33). Vacuole formation in the neuronal cell body is a prominent lesion in BSE, whereas kuru plaques are lacking (33). In contrast, vCJD is characterized by severe involvement of cerebral and cerebellar cortices and the presence of kuru plaques surrounded by vacuoles called florid plaques (32). Consistent with the different histopathology, PrPSc immunostaining in BSE is distributed as thick granules widespread in the neuropil but often with a perineuronal and perivascular distribution (33), whereas in vCJD the PrPSc immunohistochemistry is dominated by the intense staining of the kuru plaques. Contrary to the histopathology and immunohistochemistry, the characteristics of the PrPSc are fairly similar in BSE and vCJD (34): both PrPSc species are type 2 and have similar glycoform ratios (32). Assuming that if CWD is transmitted to humans it would follow the BSE-to-vCJD pattern of disease conversion in which histopathology and PrPSc immunostaining change but the basic characteristics of PrPSc are conserved, we have compared in detail the characteristics of the PrPSc associated with CWD with those of the PrPSc species associated with sCJD as well as with CJD in individuals who may have been exposed to CWD.
Sporadic prion disease can be classified into five distinct subtypes of sCJD and sporadic fatal insomnia, based on the methionine/valine polymorphic genotype at codon 129 of the PrP gene and the two conformers (types 1 and 2) of the brain PrPSc (16, 28). PrPSc types 1 and 2 are distinguished because, following treatment with PK, their protease-resistant and unglycosylated fragments migrate to ~21 and ~19 kDa on SDS-PAGE gels, respectively (28, 35). The distinct gel mobility is the result of the different size of the PrPSc fragments generated by the PK treatment, which cleaves the two PrPSc types at different sites, likely because the two PrPSc types have distinct conformations (22). Of the five subtypes of sCJD, two are associated with PrPSc type 1 (16). The first subtype includes patients who are homozygous for methionine (MM) or heterozygous methionine/valine (MV) at codon 129, identified as sCJDMM1 and sCJDMV1, respectively; the patients of the second subtype, identified as sCJDVV1, are homozygous valine at codon 129 (16). We have compared in detail deer and elk PrPSc only with the human PrPSc type 1 associated with sCJDMM1 because the gel mobility and N terminus following PK cleavage of the PrPSc associated with CWD are indistinguishable in elk, mule deer, and white tail deer and match most closely that of sCJDMM1, whereas they are quite different from that of human PrPSc type 2 associated with other subtypes of sCJD. Sequencing analysis shows that the two main N termini of the PK-resistant PrPSc associated with CWD in elk and with sCJDMM1 are residues 82 and 78 in both conditions, but slightly different from the N termini at residues 86 and 82 of the sCJDVV1-associated PK-resistant PrPSc fragment. The identical N termini of the two major PrPSc PK-resistant fragments indicate that both PrPSc species are cleaved by PK at the same sites and therefore their protein cores have conformations that are similar using this relatively crude criterion (22). The elk CWD PrPSc and the sCJDMM1 PrPSc species also have very similar characteristics according to the conformational stability assay, a test that assesses the stability of the PrPSc measured by the loss of PK-resistant PrPSc in molecules as a function of treatment with increasing amounts of the denaturant GdnHCl. However, substantial differences have emerged from the determination of the glycoform ratios and mobility in two-dimensional immunoblots. The ratios of the three bands representing diglycosylated, monoglycosylated, and unglycosylated forms of PK-resistant PrPSc show the overrepresentation of the diglycosylated band, which accounts for ~50% of the total PrPSc and the underrepresentation of the unglycosylated band, which accounts for ~20% of the total in both elk and mule deer PrPSc. The glycoform pattern and ratio of CWD-associated PrPSc is in basic agreement with an earlier report by Race et al. (36) although they observed much great variability among individual animals. This glycoform ratio is also similar to that of BSE and vCJD (20, 34, 36–38). We have recently demonstrated that the type, glycoform pattern, and ratio of elk PrPSc as reported here are reproducible upon transmission to cervidized transgenic mice (39). In contrast, the glycoform ratios observed in all forms of sCJD show an overrepresentation of the monoglycosylated form that accounts for ~50%, whereas between 20 and 30% each is accounted for by the other two forms. We found a similar ratio of the PrPSc glycoforms in several CJD-affected big game hunters who might have been exposed to CWD. These cases had the genotype, PrPSc type, and disease phenotype of sCJD, strongly suggesting that in these cases the CJD is likely sporadic and not acquired from the consumption of CWD-contaminated cervid meat.
Two-dimensional immunoblots of PK-resistant PrPSc from CWD and sCJDMM1 also demonstrate heterogeneity between the two profiles. The heterogeneity is likely to be in large part due to the difference in the type and amount of glycans in the two glycosylated isoforms present in PrPSc of sCJDMM1 and CWD. Although most of the heterogeneity between the glycosylated forms can be attributed to the different glycoform ratios, some is likely to be because of differences in electrical charges that probably reflect distinct characteristics of the glycans associated with PrPSc. However, heterogeneity persists even after deglycosylation, indicating that there is additional diversity between PrPSc from CWD and sCJDMM1, most likely related to the variations in amino acid sequence.
There are thirteen variant amino acids between human PrP residues 78–231 and corresponding sequences of deer and elk. The differences between elk and deer PrP sequences include one variant amino acid and three polymorphic amino acids. The effect of these variations in the PrP sequences is a change in the predicted pI values. It is predicted that in the region corresponding to human PrP-(78–231), deer and elk PrP will exhibit an increase in pI by 0.88 and 0.56, respectively, as compared with human PrP. Therefore, deer PrP is most basic, followed by elk PrP and human PrP. This is consistent with the result of two-dimensional immunoblots showing that deglycosylated PrPSc of deer migrated toward most basic pI, followed by that of elk and human. Other causes for heterogeneity may be related to the C-terminal glycolipid anchor that may contain various numbers of sialic acids or the presence of additional minor N-terminal cleavage sites, all of which may have contributed to some overlapping two-dimensional spots of human, elk, and deer PrPSc.
In conclusion, the parallel study of CWD that naturally occurs in elk and deer and of prion disease occurring in humans with or without a history of potential exposure to CWD reveals substantial differences in the type and distribution of the histological lesions as well as in the pattern of PrP immunohistochemistry. Similar findings have been reported when natural BSE and vCJD were compared. However, in BSE and vCJD the basic characteristics of PrPSc, such as protein type based on the size of the PK-resistant PrPSc fragment, and the glycoform ratio are similar. This is consistent with the notion that BSE PrPSc serves as template for the human PrPSc associated with vCJD. In contrast, although the size of the PK-resistant PrPSc fragment of CWD matches most closely that of sCJDMM1, the glycoform ratio of CWD PrPSc is different from that of the PrPSc associated with sCJDMM1 as well as all non-familial human prion diseases except for vCJD. Furthermore, CJDMM1 cases potentially exposed to CWD also had the same glycoform ratio as the other sporadic cases. These findings suggest that PrPSc associated with CJDMM1 (as well as sCJDMV1 and sCJDVV1)3 in several individuals (15) who were potentially exposed to CWD is not derived from the CWD PrPSc and therefore in these individuals CJD was not acquired from CWD. This conclusion is in agreement with the results of a recent experimental study on the transmissibility of elk CWD to various transgenic mice (39). Although CWD could be transmitted to transgenic mice expressing the elk PrP in a PrP knock-out background following an incubation of 118–143 days, none of the transgenic mice expressing the human PrP manifested prion disease during their lifespan following intracranial inoculation of CWD-affected brain homogenate, indicating that there is a substantial species barrier for transmission of elk CWD to humans. A recent report has shown that CWD can be transmitted to squirrel monkeys, a non-human primate (40). However, squirrel monkeys are very permissive to prions of different species, including sheep scrapie to which humans are resistant. Hence, squirrel monkeys may not be used as a suitable model of human transmission of CWD. Although at present transmission of CWD to humans may be doubtful, should it occur it is likely that the PrPSc associated with this human prion disease may have a glycoform ratio similar to that of CWD. However, the resulting histopathology and PrP immunostaining patterns cannot be predicted. It is therefore important that the study of cases affected by non-familial CJD includes detailed analysis of the PrPSc characteristics.
Acknowledgments
We thank L. Ma and W. Wang for technical assistance and D. Kofskey for histological and immunohistochemical staining.
Footnotes
This work was supported in part by United States Department of Agriculture Grant 2002-35201-12608 (to S. G. C.), Department of Defense Grant DAMD17-03-1-0283 (to S. G. C.), Centers for Disease Control and Prevention Contract UR8/CCU515004-01 (to P. G.), and National Institutes of Health Grant AG-14359 (to S. G. C. and P. G.).
The abbreviations used are: CWD, chronic wasting disease; CJD, Creutzfeldt-Jakob disease; vCJD, variant CJD; PrPSc, the disease-associated, scrapie isoform of prion protein; BSE, bovine spongiform encephalopathy; PK, proteinase K; GdnHCl, guanidine hydrochloride; mAb, monoclonal antibody; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
S. G. Chen and P. Gambetti, unpublished data.
References
- 1.Williams ES, Young S. J Wildl Dis. 1980;16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB, Williams E, Laplanche JL, Shinagawa M. In: Prion Biology and Diseases. 2. Prusiner S, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. pp. 545–594. [Google Scholar]
- 3.Miller MW, Williams ES. Curr Top Microbiol Immunol. 2004;284:193–214. doi: 10.1007/978-3-662-08441-0_8. [DOI] [PubMed] [Google Scholar]
- 4.Williams ES. Clin Lab Med. 2003;23:139–159. doi: 10.1016/s0272-2712(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 5.Spraker TR, Zink RR, Cummings BA, Wild MA, Miller MW, O’Rourke KI. Vet Pathol. 2002;39:110–119. doi: 10.1354/vp.39-1-110. [DOI] [PubMed] [Google Scholar]
- 6.Spraker TR, Zink RR, Cummings BA, Sigurdson CJ, Miller MW, O’Rourke KI. Vet Pathol. 2002;39:546–556. doi: 10.1354/vp.39-5-546. [DOI] [PubMed] [Google Scholar]
- 7.Liberski PP, Guiroy DC, Williams ES, Walis A, Budka H. Acta Neuropathol (Berl) 2001;102:496–500. doi: 10.1007/s004010100417. [DOI] [PubMed] [Google Scholar]
- 8.Williams ES, Young S. Vet Pathol. 1993;30:36–45. doi: 10.1177/030098589303000105. [DOI] [PubMed] [Google Scholar]
- 9.Peters J, Miller JM, Jenny AL, Peterson TL, Carmichael KP. J Vet Diagn Investig. 2000;12:579–582. doi: 10.1177/104063870001200618. [DOI] [PubMed] [Google Scholar]
- 10.Sigurdson CJ, Spraker TR, Miller MW, Oesch B, Hoover EA. J Gen Virol. 2001;82(Pt 10):2327–2334. doi: 10.1099/0022-1317-82-10-2327. [DOI] [PubMed] [Google Scholar]
- 11.Miller MW, Williams ES. Nature. 2003;425:35–36. doi: 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- 12.Bunk S. PLoS Biol. 2004;2:E121. doi: 10.1371/journal.pbio.0020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams ES, Miller MW. Rev Sci Tech. 2002;21:305–316. doi: 10.20506/rst.21.2.1340. [DOI] [PubMed] [Google Scholar]
- 14.Bartz JC, Marsh RF, McKenzie DI, Aiken JM. Virology. 1998;251:297–301. doi: 10.1006/viro.1998.9427. [DOI] [PubMed] [Google Scholar]
- 15.Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Emerg Infect Dis. 2004;10:977–984. doi: 10.3201/eid1006.031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 17.Will RG. Br Med Bull. 2003;66:255–265. doi: 10.1093/bmb/66.1.255. [DOI] [PubMed] [Google Scholar]
- 18.Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, MacKenzie J, Knight R, Will RG, Ironside JW. Ann Neurol. 2004;55:851–859. doi: 10.1002/ana.20127. [DOI] [PubMed] [Google Scholar]
- 19.Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. Proc Natl Acad Sci U S A. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. Nature. 1997;389:448–450. 526. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 21.Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS. Proc Natl Acad Sci U S A. 1998;95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Proc Natl Acad Sci U S A. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. J Biol Chem. 2003;278:40429–40436. doi: 10.1074/jbc.M308550200. [DOI] [PubMed] [Google Scholar]
- 24.Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Proc Natl Acad Sci U S A. 2004;101:1380–1385. doi: 10.1073/pnas.0307825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolton DC, Bendheim PE, Marmorstein AD, Potempska A. Arch Biochem Biophys. 1987;258:579–590. doi: 10.1016/0003-9861(87)90380-8. [DOI] [PubMed] [Google Scholar]
- 27.Chen SG, Parchi P, Brown P, Capellari S, Zou W, Cochran EJ, Vnencak-Jones CL, Julien J, Vital C, Mikol J, Lugaresi E, Autilio-Gambetti L, Gambetti P. Nat Med. 1997;3:1009–1015. doi: 10.1038/nm0997-1009. [DOI] [PubMed] [Google Scholar]
- 28.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 29.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 30.Scott M, Peretz D, Ridley RM, Baker HF, DeArmond SJ, Prusiner SB. In: Prion Biology and Diseases. 2. Prusiner S, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. pp. 434–482. [Google Scholar]
- 31.Will RG, Alpers MP, Dormont D, Schonberger LB. In: Prion Biology and Diseases. 2. Prusiner S, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. pp. 629–671. [Google Scholar]
- 32.Ironside JW. Brain Pathol. 1996;6:379–388. doi: 10.1111/j.1750-3639.1996.tb00869.x. [DOI] [PubMed] [Google Scholar]
- 33.Wells GA, Wilesmith JW. Brain Pathol. 1995;5:91–103. doi: 10.1111/j.1750-3639.1995.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 34.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 35.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 36.Race RE, Raines A, Baron TG, Miller MW, Jenny A, Williams ES. J Virol. 2002;76:12365–12368. doi: 10.1128/JVI.76.23.12365-12368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Monaco S, Caramelli M. Proc Natl Acad Sci U S A. 2004;101:3065–3070. doi: 10.1073/pnas.0305777101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parchi P, Capellari S, Chen SG, Petersen RB, Gambetti P, Kopp N, Brown P, Kitamoto T, Tateishi J, Giese A, Kretzschmar H. Nature. 1997;386:232–234. doi: 10.1038/386232a0. [DOI] [PubMed] [Google Scholar]
- 39.Kong Q, Huang S, Zou WQ, Vanegas D, Wang M, Wu D, Yuan J, Zheng M, Bai H, Deng H, Chen K, Jenny AL, O’Rourke K, Belay ED, Schonberger LB, Petersen RB, Sy MS, Chen SG, Gambetti P. J Neurosci. 2005;25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marsh RF, Kincaid AE, Bessen RA, Bartz JC. J Virol. 2005;79:13794–13796. doi: 10.1128/JVI.79.21.13794-13796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]