

Abstract
Background
During early clinical development, prospective identification of a predictive biomarker and validation of an assay method may not always be feasible. Dichotomizing a continuous biomarker measure to classify responders also leads to challenges. We present a case study of a prospective–retrospective approach for a continuous biomarker identified after patient enrollment but defined prospectively before the unblinding of data. An analysis of the strengths and weaknesses of this approach and the challenges encountered in its practical application are also provided.
Methods
HERALD (NCT02134015) was a double-blind, phase 2 study in patients with non-small cell lung cancer (NSCLC) randomized to erlotinib with placebo or with high or low doses of patritumab, a monoclonal antibody targeted against human epidermal growth factor receptor 3 (HER3). While the primary objective was to assess safety and progression-free survival (PFS), a secondary objective was to determine a single predictive biomarker hypothesis to identify subjects most likely to benefit from the addition of patritumab. Although not identified as the primary biomarker in the study protocol, on the basis of preclinical results from 2 independent laboratories, expression levels of the HER3 ligand heregulin (HRG) were prospectively declared the predictive biomarker before data unblinding but after subject enrollment. An assay to measure HRG mRNA was developed and validated. Other biomarkers, such as epidermal growth factor receptor (EGFR) mutation status, were also evaluated in an exploratory fashion. The cutoff value for high vs. low HRG mRNA levels was set at the median delta threshold cycle. A maximum likelihood analysis was performed to evaluate the provisional cutoff. The relationship of HRG values to PFS hazard ratios (HRs) was assessed as a measure of internal validation. Additional NSCLC samples were analyzed to characterize HRG mRNA distribution.
Results
The subgroup of patients with high HRG mRNA levels (“HRG-high”) demonstrated clinical benefit from patritumab treatment with HRs of 0.37 (P = 0.0283) and 0.29 (P = 0.0027) in the high- and low-dose patritumab arms, respectively. However, only 102 of the 215 randomized patients (47.4%) had sufficient tumor samples for HRG mRNA measurement. Maximum likelihood analysis showed that the provisional cutoff was within the optimal range. In the placebo arm, the HRG-high subgroup demonstrated worse prognosis compared with HRG-low. A continuous relationship was observed between increased HRG mRNA levels and lower HR. Additional NSCLC samples (N = 300) demonstrated a similar unimodal distribution to that observed in this study, suggesting that the defined cutoff may be applicable to future NSCLC studies.
Conclusions
The prospective–retrospective approach was successful in clinically validating a probable predictive biomarker. Post hoc in vitro studies and statistical analyses permitted further testing of the underlying assumptions. However, limitations of this analysis include the incomplete collection of adequate tumor tissue and a lack of stratification. In a phase 3 study, findings are being confirmed, and the HRG cutoff value is being further refined.
ClinicalTrials.gov Number
Keywords: Biomarker, Patritumab, Erlotinib, Non-small cell lung cancer, HER3, Heregulin
Highlights
-
•
High heregulin levels predict benefit from patritumab treatment in patients with NSCLC.
-
•
A prospective–retrospective approach provisionally validated a predictive biomarker.
-
•
Post hoc analyses can be used to test underlying assumptions in biomarker validation.
-
•
The median may be a reasonable initial cutoff for a unimodal continuous biomarker.
1. Introduction
Ideally, predictive biomarkers are prospectively specified in clinical trials in oncology. However, this path may not always be feasible due to various challenges encountered during clinical development, including difficulties in early identification of a biomarker, or development and validation of an appropriate assay method. In addition, setting a cutoff for a continuous biomarker can be problematic, and the best approach is still an open area of debate (Altman et al., 1994, Jiang et al., 2007, Fridlyand et al., 2013, Hall et al., 2014). Use of a prospective–retrospective approach that is applied to a single predictive biomarker hypothesis has the advantage of avoiding the issue of a high false-positive rate due to multiple comparisons when multiple biomarker hypotheses are evaluated on an equal footing in an exploratory fashion (Beckman et al., 2011); however, there have been few reports published detailing the real-world implementation of a prospective–retrospective approach for biomarker identification.
Herein, we present a case in which a continuous biomarker for a targeted therapy, patritumab, was clinically validated using a prospective–retrospective approach (Beckman et al., 2011, Simon, 2005) during a phase 2 study. We also review the challenges faced and the strengths and weaknesses of the approach taken.
Members of the human epidermal growth factor receptor family (HER; i.e., epidermal growth factor receptor [EGFR], HER1, HER2, HER3, and HER4) have been implicated in oncogenesis (Liu and Kern, 2002, Hsieh and Moasser, 2007). Elevated expression levels of HER3 and its ligand heregulin (HRG) are found in many solid tumors, including non-small cell lung cancer (NSCLC) (Yi et al., 1997, al Moustafa et al., 1999, Müller-Tidow et al., 2005). In most cases, HER3-containing heterodimers are formed following conformational changes in HER3 induced by HRG binding (Carraway et al., 1994, Alroy and Yarden, 1997), leading to the activation of signaling pathways important for oncogenesis (Fig. 1) (Gullick, 1996, Olayioye et al., 2000, Yarden and Sliwkowski, 2001, Atlas et al., 2003, Tsai et al., 2003, de Alava et al., 2007, Ueno et al., 2008, Hegde et al., 2013). High HRG expression has been associated with increased HER3 phosphorylation (Krane and Leder, 1996, Zhou et al., 2006) and phosphatidylinositol-3-kinase (PI3K)-mediated signaling (Prigent and Gullick, 1994, Soltoff et al., 1994, Schoeberl et al., 2010, Shames et al., 2013).
Fig. 1.

Patritumab and HER3 signaling pathway. (A) Binding of the ligand heregulin to HER3 induces a conformational change that allows for receptor dimerization of HER3 with HER family receptors, such as EGFR or HER2. (B) Heterodimerization initiates the transphosphorylation of HER3 at specific tyrosine residues by the kinase of its heterodimeric partner. (C) The phosphorylated tyrosine kinase residues create 6 direct docking sites for PI3K, leading to the activation of other molecules further downstream in the PI3K–Akt pathway. EGFR: epidermal growth factor receptor; HER: human epidermal growth factor receptor; PI3K: phosphatidylinositol-3-kinase.
Upregulation and reactivation of HER3 function as escape mechanisms from EGFR inhibition and may play a role in tumor resistance to EGFR-targeting tyrosine kinase inhibitors (Schoeberl et al., 2010, Xia et al., 2005, Sergina et al., 2007, Wheeler et al., 2008, Baselga and Swain, 2009, Garrett et al., 2011). Consistent with this theory, HRG has been found to reverse sensitivity to EGFR inhibitors in preclinical models (Motoyama et al., 2002). Complete and sustained HER3 and/or HRG inhibition may, therefore, inhibit HER3-dependent resistance to EGFR inhibition and produce a more complete blockade of signaling from HER family members (Xia et al., 2005, Sergina et al., 2007).
Patritumab is a fully human anti-HER3 monoclonal antibody that demonstrates antitumor activity when used alone or with anti-EGFR inhibitors in preclinical cancer models (Freeman et al., 2009). Patritumab inhibits HRG-mediated HER3 signaling by binding to the extracellular domain of HER3 (thus preventing HRG from binding) and promoting the internalization and degradation of the receptor (Freeman et al., 2008, Treder et al., 2008a, Treder et al., 2008b).
Patritumab was assessed in the phase 2 HERALD study in which patients with advanced NSCLC were randomized to either patritumab in combination with erlotinib or placebo with erlotinib (von Pawel et al., 2014). While patritumab had been previously studied in a phase 1 study as a monotherapy in patients with solid tumors refractory to prior treatments (LoRusso et al., 2013), a predictive biomarker for response to patritumab had not yet been identified before initiation of the phase 2 study. Therefore, the phase 2 study had a secondary objective of defining a biomarker for a primary biomarker hypothesis (Beckman et al., 2011), based on preclinical data available after patient enrollment was completed in the clinical study and prior to data unblinding and statistical analysis (a prospective–retrospective approach (Simon, 2005)).
2. Materials and Methods
2.1. Study Design
Patients were eligible for enrollment in the HERALD study if they had histologically confirmed stage IIIB/IV NSCLC with measurable disease (per Response Evaluation Criteria in Solid Tumors guidelines, version 1.1) and documented disease progression or recurrence on at least 1 prior chemotherapy treatment. Other eligibility criteria have been previously presented (von Pawel et al., 2014). Approximately 215 patients were randomized to 1 of 3 arms: high-dose patritumab (18 mg/kg intravenously [IV] every 3 weeks [q3w]) with erlotinib (150 mg/day orally [PO]), low-dose patritumab (18 mg/kg IV loading dose, followed by 9 mg/kg q3w) with erlotinib (150 mg/day PO), or placebo with erlotinib (150 mg/day PO). Stratification factors include histology subtypes (adenocarcinoma vs. squamous vs. other) and best response to prior therapy (complete response or partial response vs. stable disease vs. progressive disease).
The primary objectives of the study were to assess safety and progression-free survival (PFS) in the intent-to-treat (ITT) population. A secondary objective was to identify and test a single primary predictive biomarker hypothesis. The study protocol was approved by the institutional review boards of the participating institutions. All patients provided written informed consent, including consent to provide tumor tissue to test for biomarkers predictive of patritumab response.
2.2. Biomarker Assays
Patients were required to provide a fresh tumor sample prior to treatment or to make available archived tumor tissue in order to have the potential predictive biomarkers for patritumab assessed. A blood sample for pharmacogenetic assessment was also collected pretreatment from each patient.
Mutations in the EGFR gene were analyzed in formalin-fixed paraffin-embedded (FFPE) tissue and ethylenediaminetetraacetic acid plasma samples using the Qiagen EGFR RGQ PCR Kit (Germantown, MD) on the Qiagen Rotor-Gene Q 5plex HRM (Germantown, MD) instrument. The method was validated, and analyses were performed by Covance Central Laboratory Services (Indianapolis, IN, and Geneva, Switzerland). The Qiagen EGFR RGQ PCR Kit detected mutations on exon 18 (G719A, G719S, G719 C), exon 20 (T790M, S768I), and exon 21 (L858R, L861Q), as well as exon 19 deletions and exon 20 insertions.
An immunohistochemistry (IHC) assay was developed and validated to measure HER3 expression in FFPE tissue by Mosaic Laboratories (Lake Forest, CA). HER3 expression was detected using a mouse anti-HER3 monoclonal antibody. Two lung cancer tissues were used as controls, and 2 cell lines (1 known to be negative and 1 known to be positive for HER3 expression) were used as quality control qualifiers. HER3 IHC staining was evaluated by a pathologist on a semiquantitative scale, and an H-score was calculated based on the percentage of cells staining at 4 intensity levels.
A validated quantitative sandwich immune assay was used to measure levels of the soluble p85 form of HER3 in serum. The sandwich assay used antibody reagents raised against p85 HER3 recombinant protein. Bound HER3 was detected with biotinylated mouse antihuman HER3 antibody followed by peroxidase-conjugated streptavidin, visualized with a tetramethylbenzidine substrate solution. The method was validated and samples were analyzed by Intertek Pharmaceutical Services (San Diego, CA).
HRG mRNA expression was evaluated using a quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay that was developed and validated by MolecularMD (Portland, OR). Total mRNA was extracted from FFPE tissue using Qiagen RNeasy FFPE (Germantown, MD), and cDNA was obtained from reverse transcription of the mRNA. Levels of mRNA from HRG and 3 reference genes were evaluated using qRT-PCR. The average PCR efficiency was within 90% to 110%, and linearity was ≥ 0.99. Intra-assay and interassay precision was evaluated with 6 different FFPE samples that were started from mRNA extraction from FFPE samples. The samples were analyzed by MolecularMD.
2.3. Prospective–Retrospective Approach for a Single Predictive Biomarker Hypothesis
The original intent in the HERALD study was to conduct a stratified, randomized phase 2 study that tested a single predictive biomarker hypothesis as a primary objective, as well as its efficacy in the full ITT population (Beckman et al., 2011). The single predictive biomarker hypothesis was required to avoid multiple statistical comparisons, which could contribute to false-positives and result in the inability to reproduce results in subsequent studies (Beckman et al., 2011, Simon, 2005).
At the start of the study, however, there were still a number of possible biomarker hypotheses and few validated assays available to measure potential analytes (e.g., HER3 expression and activation or HRG expression). We were therefore unable to declare the predictive biomarker as a primary end point in the clinical protocol. Instead a secondary objective was declared to define and test a predictive biomarker hypothesis to identify patient populations more likely to benefit from patritumab treatment. Therefore, the prospective–retrospective approach was used (Simon, 2005). The single predictive biomarker hypothesis was to be prospectively declared, in this case prior to the unblinding of the clinical data but after study initiation, and was to be tested regardless of the results from the ITT population analysis (Beckman et al., 2011). While this was a secondary objective, other predictive biomarkers were designated exploratory.
2.4. Statistical Analysis
The primary analysis for PFS used a stratified log-rank linear trend test for the dose–response relationship. This was followed by pairwise comparisons of each patritumab and erlotinib combination therapy and the control arm using the stratified log-rank test and accounting for the stratification factors at randomization. Kaplan–Meier curves were generated for PFS and used to calculate medians and 95% confidence interval (CI) for each treatment group.
Applying the prospective–retrospective approach, the key secondary analysis for PFS was performed for biomarker-positive patients following the same method described previously to compare the combination of patritumab plus erlotinib with the control arm. A biomarker-positive patient was prospectively defined as a patient with HRG mRNA expression above the cutoff, which was specified as the median delta threshold cycle (∆Ct) value in the blinded sample set.
Hazard ratios (HRs) at incremental cutoff values for HRG mRNA from the study were also assessed as an exploratory analysis. In assessing other potential HRG mRNA cutoff values, a maximum likelihood approach was used to identify optimal cutoff values (Altman et al., 1994). The continuous HRG mRNA values were dichotomized, and the cutoff value that resulted in the HR value with lowest P-value was selected as optimal based on this approach and compared with the provisional cutoff at the median.
To assess the distribution of HRG mRNA in a broader NSCLC population, 300 NSCLC tumor samples, in FFPE (n = 200; purchased from various commercial vendors) and as RNA (n = 100; provided by National Hospital Organization Kyushu Cancer Center, Fukuoka, Japan), were analyzed by MolecularMD using the validated qRT-PCR method.
Sensitizing mutations in EGFR are associated with improved clinical response to EGFR inhibitors (Paez et al., 2004, Pao et al., 2004, Sequist et al., 2008, Yang et al., 2008). To assess the potential for interaction with sensitizing EGFR mutations that may not have been detected in patients with unknown EGFR mutation status and could potentially confound the results (Polley et al., 2013), simulations (N = 10,000) were performed using re-sampling without replacement, assuming a 10% EGFR-sensitizing mutation prevalence based on published incidence rates of EGFR mutation in NSCLC patients and utilizing the EGFR mutation results from tissue samples within the study (Paez et al., 2004, Yang et al., 2008, Lynch et al., 2004) (i.e., out of 101 patritumab-treated patients with unknown EGFR status based on tissue alone, it was assumed that 10 patients would have sensitizing EGFR mutations). The HRs estimated for each scenario were summarized to characterize the potential effect that EGFR-sensitizing mutations could have on the HR observed in this study.
3. Results
3.1. Single Predictive Biomarker Hypothesis
At the start of the study there were still a number of possible predictive biomarker hypotheses and analytes. Therefore, we prospectively declared a single predictive biomarker hypothesis as a secondary objective without specifying the biomarker or measurement. Identification of the biomarker evolved during the course of the study.
3.2. Predictive Biomarker Selection
Since our target population was patients with NSCLC, we defined a predictive biomarker as one that could be measured in serum or in FFPE tumor tissue collected pretreatment to ensure the feasibility for future large scale studies and the applicability to clinical practice.
HRG expression was considered as a potential biomarker for patritumab efficacy. To assess HRG levels, a qRT-PCR method was developed. An IHC-based assay was evaluated but was found to lack selectivity for low vs. high levels of HRG in sample FFPE tissue slides. Assessment of HRG protein levels by Western blot was also considered, but it was determined that this approach would not be feasible for a large scale clinical study.
HER3 expression was also evaluated as a candidate biomarker. Patients with high levels of HER2 expression have higher rates of response to treatment with the HER2 inhibitor trastuzumab. It was hypothesized that a similar correlation might be observed between high levels of HER3 expression in tumors and improved response to patritumab treatment (Slamon et al., 1987, Mass et al., 2005). Therefore, an IHC assay was developed and validated to measure HER3 expression in FFPE tissue. While activated HER3 (evaluated by levels of phosphorylated HER3 [pHER3]) was also considered potentially relevant, it was not possible to validate an IHC method for assessing pHER3 in FFPE tissue. An assay method for soluble HER3 was also developed and validated.
After the HERALD study was initiated, results from preclinical studies measuring HER3, pHER3, and HRG levels by Western blot assay determined that HRG protein expression was the most relevant biomarker for predicting response to patritumab (Schneider et al., 2014, Yonesaka et al., 2014). In 50 tumor cell lines, HRG and HER3 expression levels were found to be variable and not correlated with each other. When cell lines were categorized into those with high HRG expression (HRG-high) and low HRG expression (HRG-low) using a cutoff equal to the median of the blinded sample data, HRG-high cell lines were significantly more sensitive to patritumab treatment compared with HRG-low cell lines, both in vitro (P = 0.002) and in tumor xenograft models. Protein expression analysis provided a sensitivity of 83.3% and specificity of 100% for correlating HRG-positive cell lines with sensitivity to patritumab treatment in vitro and a sensitivity of 71.4% and specificity of 100% in tumor xenograft models. In addition, HRG-high cell lines were observed to have activated HER3 and AKT, whose levels were reduced with patritumab treatment, and specific knockdown of HRG expression using siRNA led to reduced sensitivity to patritumab treatment. In contrast, levels of HER3 expression or activation were not found to correlate with sensitivity to patritumab treatment.
These findings led to the amendment of the statistical analysis plan of the HERALD study to state that HRG would be the primary predictive biomarker for the study and to specify that PFS in this single subgroup would be evaluated irrespective of the outcome in the ITT population. As there was no clinically feasible assay measuring HRG protein expression, HRG mRNA was employed as a surrogate. The hypothesis thus stated that patients with higher HRG mRNA levels in their tumors would derive greater benefit from treatment with patritumab. The cutoff between HRG-high and HGR-low subgroups was prospectively defined as the median measured on blinded samples before database lock.
3.3. HRG Levels and Efficacy in the HERALD Study
In total, 215 patients were randomized in the HERALD study; 212 patients received at least 1 dose of study treatment and were included in the ITT population. FFPE samples were collected in all patients, but only 103 patients provided a sufficient tumor sample for HRG mRNA measurement (101 patients in the ITT population). HRG biomarker distribution from the HERALD study is shown in Fig. 2. The median ∆Ct value (first, third quartile) was 3.9 (2.7–5.0) and was used as the cutoff value between HRG-high and HRG-low subgroups, as predefined in the statistical analysis plan. Patient demographics and baseline disease characteristics were generally similar between the ITT population and the subset of HRG-high patients (n = 51), although a higher proportion of patients in the ITT population had unknown EGFR mutation status (70.3% vs. 54.9%, respectively) (Supplemental Table S1). Among patients with known EGFR mutation status, 6.6% of patients in the ITT population and 8.7% of patients in the HRG-high subgroup had EGFR-sensitizing mutations. Patient demographics and baseline disease characteristics were also similar between the HRG-high and HRG-low patients (n = 40; data not shown).
Fig. 2.
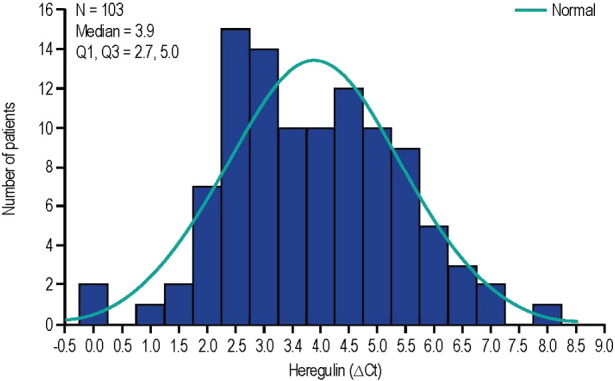
Distribution of HRG mRNA in the HERALD study. ΔCt: median delta threshold cycle; Q: quartile.
No PFS benefit was observed for patritumab plus erlotinib compared with placebo plus erlotinib in the ITT population, with HRs (95% CI) of 0.98 (0.67–1.42) in the high-dose arm and 0.77 (0.52–1.13) in the low-dose arm (Supplemental Table S2). HRG-high patients treated with patritumab and erlotinib, however, had significantly improved PFS compared with patients treated with erlotinib alone, with HRs (95% CI) of 0.37 (0.16–0.85) in the high-dose arm and 0.29 (0.13–0.66) in the low-dose arm (von Pawel et al., 2014). In HRG-low patients, no PFS benefit was observed, which were similar to results from the ITT population, with HRs (95% CI) of 0.91 (0.39–2.09) and 0.92 (0.39–2.22) in the high- and low-dose arms, respectively. In addition to the evidence suggesting that a high level of HRG expression may be predictive of clinical benefit from patritumab treatment, an exploratory analysis of PFS in HRG-high and HRG-low patients in the patritumab and placebo arms suggested that a high level of HRG expression may also be a negative prognostic factor in patients treated with single-agent erlotinib treatment (Fig. 3). HRG-high patients treated with placebo plus erlotinib demonstrated poorer PFS than HRG-low patients treated with placebo plus erlotinib.
Fig. 3.
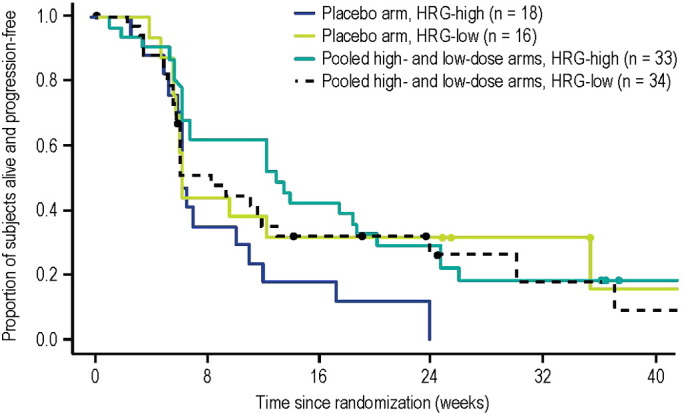
Progression-free survival in HRG-high and HRG-low patients in placebo and patritumab arms. HRG: heregulin.
HER3 protein expression in tissue and soluble HER3 levels were also evaluated, and, in contrast to results with HRG presented previously, no correlation was observed between HER3 or soluble HER3 levels and clinical benefit (data not shown). Since EGFR results were not available for all patients, simulations were performed to assess whether an imbalance in patients with sensitizing EGFR mutations could have biased our results. The results of these simulations (N = 10,000, each representing probability weighted possible distributions of the sensitizing mutations in the patients with unknown EGFR mutation status) showed that the distribution of possible HRs still resulted in a significant clinical response for patritumab (upper limit of HR < 0.45, upper limit of P-value < 0.03); therefore, it is unlikely that our results are an artifact from unbalanced undetected EGFR mutations (Supplemental Fig. S1).
3.4. HRG Distribution
HRG distribution was also assessed in FFPE and RNA samples for patients with NSCLC. The distribution shown in Fig. 4 shows a Gaussian distribution with a median ∆Ct value (first, third quartile) of 4.1 (2.9–5.6), similar to what was observed in the HERALD study population. In particular, there is no clear evidence of a bimodal distribution that might have given evidence of a natural cutoff at the intersection of 2 separate Gaussian curves as suggested by Shames et al. in squamous cell head and neck cancer (Shames et al., 2013).
Fig. 4.
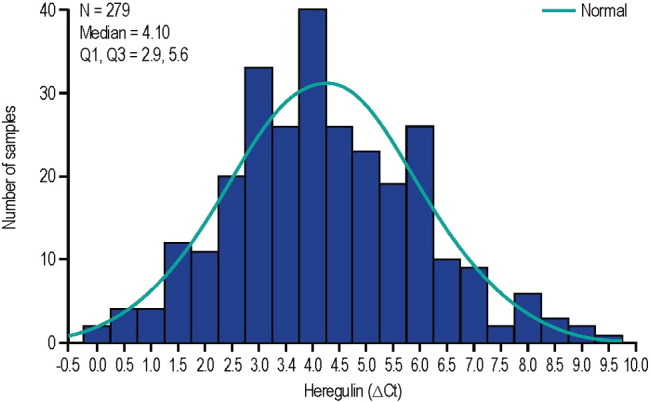
Distribution of HRG mRNA in non-small cell lung cancer samples. ΔCt: median delta threshold cycle; Q: quartile.
HRG demonstrated a continuous relationship between increased HRG mRNA expression (i.e., lower ∆Ct) and greater clinical benefit in terms of PFS (Table 1), internally validating HRG as a predictive biomarker for clinical benefit with patritumab treatment (Simon et al., 2009). While the median HRG mRNA value was the prespecified cutoff for this clinical study, the potential for optimum HRG cutoff values was also explored with maximum likelihood methods. Because this analysis suggested a broad optimum range of values consistent with the initial cutoff of 3.9, that value was retained. Further refinement of the cutoff value for HRG-high vs. HRG-low subgroups will be considered during development of a companion diagnostic and after further clinical assessment in the initial part of the 2-part phase 3 study (see below).
Table 1.
Hazard ratios for various cutoff values of HRG based on data from the HERALD study.
| Cutoff for HRG-high (∆Ct) group | Number of events | HR (pooled patritumab dose vs. placebo) for PFS in HRG-high group | Log-rank P-value |
|---|---|---|---|
| 2.7 (first quartile) | 24 (18) | 0.180 | 0.0039 |
| 3.0 | 33 (24) | 0.177 | 0.0009 |
| 3.5 | 46 (36) | 0.283 | 0.0009 |
| 3.9 (median) | 51 (41) | 0.324 | 0.0013 |
| 4.5 | 65 (50) | 0.490 | 0.0190 |
| 5.0 (third quartile) | 76 (58) | 0.561 | 0.0429 |
ΔCt: median delta threshold cycle; HR: hazard ratio; HRG: heregulin; PFS: progression-free survival.
4. Discussion
After study initiation, a validated assay method to measure HRG mRNA expression from FFPE tissue was developed to assess a single primary biomarker hypothesis, which stated that patients with high HRG mRNA expression levels would more likely benefit from the addition of patritumab. The results confirmed that patients with advanced NSCLC and high levels of HRG mRNA expression showed significant clinical benefit for treatment with patritumab plus erlotinib compared with patients receiving erlotinib monotherapy. Interestingly, levels of HRG mRNA expression also appear to be a prognostic biomarker in patients who were treated with erlotinib monotherapy. This is consistent with molecular studies suggesting that HRG upregulation is a resistance mechanism to tyrosine kinase inhibitors and chemotherapy (Hegde et al., 2013, Zhou et al., 2006, Baselga and Swain, 2009, Xia et al., 2013).
Given the challenges faced at the onset of the study, the use of a prospective–retrospective approach for this phase 2 study was valuable in provisionally validating HRG mRNA expression levels as a predictive biomarker for patritumab efficacy. These challenges included the possibility of having to evaluate multiple biomarkers and the existence of few validated assays at the start of the study. Preclinical study results became available during the clinical phase 2 study and showed that HRG protein expression is predictive of preclinical patritumab efficacy in cell lines and mouse tumor xenograft models. The finding that HRG—but not HER3 expression—is a predictive biomarker for patritumab clinical efficacy is consistent with the relationship between the HER3 receptor and its ligand and the mechanism of action underlying patritumab activity. Activation of the HER3 pathway appears to be driven by increased HRG expression (Ueno et al., 2008, Zhou et al., 2006, Schoeberl et al., 2009, Schoeberl et al., 2010, Shames et al., 2013), which is associated with therapeutic resistance to a variety of agents, including those inhibiting the PI3K pathway (Zhou et al., 2006, Xia et al., 2013, Sato et al., 2013). These observations indicate that HRG activity is a key regulator of HER3 signaling and support that patritumab inhibits HRG-mediated HER3 activation (Freeman et al., 2008, Treder et al., 2008a, Treder et al., 2008b). Additionally, high HRG expression may result in a decrease in the apparent levels of membrane surface HER3 detectable through IHC owing to the internalization of the receptor (Hettmann et al., 2010). Low levels of this membrane surface receptor were not well detected by an IHC assay method, which may also explain why HER3 expression levels appear uncorrelated with patritumab efficacy in our study.
Strengths of this analysis include that the biomarker assay was analytically validated, the data were from a randomized clinical trial, and the hypothesis was prospectively stated prior to data unblinding (Khleif et al., 2010). The fact that a single biomarker was identified minimized the potential for Type I error, which can be encountered with multiple comparisons (Beckman et al., 2011). Internal validation was provided through exploratory analyses, which showed that decreasing ∆Ct values (i.e., increasing HRG mRNA expression levels) showed a trend for improved clinical benefit (i.e., lower HR) for patients treated with patritumab plus erlotinib compared with placebo plus erlotinib.
The maximum likelihood analysis showed that the median ΔCt value was within the range of optimal ΔCt values resulting in the lowest P-value for the HRs. An initial provisional choice of the median value appears to be a reasonable approach for continuous biomarkers with unimodal distributions, where the cutoff value is not already defined, since it provides the largest sample size for subgroup comparisons. However, subsequent validation and iterative refinement of any provisional cutoff against clinical data will always be required as a key component of companion diagnostic development for continuous biomarkers (Fridlyand et al., 2013). Importantly, the distribution of HRG mRNA observed in the HERALD study was unimodal and similar to the distribution observed in commercial samples; thus, it is likely to be representative of patients with NSCLC. The effort to translate this research assay method to a companion diagnostic assay is also an important consideration after the identification of a predictive biomarker. Optimizing an assay method for these purposes may result in changes to the defined cutoff value, the amount of tumor tissue required, and other logistical aspects of clinical trial conduct.
Despite samples being obtained from most patients, only 103 samples (i.e., < 50% of samples) could ultimately be analyzed for HRG mRNA, resulting in relatively small numbers of patients in the HRG-high and HRG-low subgroups (Khleif et al., 2010). The protocol allowed patients to enter the study if they had an available specimen, allowing them to begin therapy promptly. But it is clear from our results that a quality check of the specimens should have been required before enrollment, despite the possible resultant delays in beginning therapy. As the AACR-FDA-NCI Cancer Biomarkers Collaborative states, absence of high-quality biospecimens is one of the most significant roadblocks to developing and validating biomarkers (Mandrekar and Sargent, 2009). The tumor samples were collected from patients prior to treatment and prior to the selection of the primary biomarker hypothesis or the validation of the HRG assay method. Therefore, it was impossible to stratify the patient population by HRG status. In the absence of stratification and with fewer than half of the tumor samples ultimately being suitable for biomarker measurement, the sample size was not sufficiently large enough to assure that confounding factors did not bias the results (Patterson et al., 2011). In this case, simulations to assess the impact of potential interactions for various significant factors, such as undetected EGFR mutations, were an important mechanism to further qualify the robustness of clinical results when a predictive biomarker was not used in stratification. A large percentage of patients in the HERALD study had an unknown EGFR mutation status (70.3%), and sensitizing EGFR mutations were less prevalent (6.6%) than might be expected among patients with known EGFR mutation status and based on the published literature (~ 10% incidence rate (Paez et al., 2004, Yang et al., 2008)). This suggests the possibility that the group of patients with unknown EGFR mutation status may have had a high percentage of sensitizing EGFR mutations, potentially leading to an imbalance in EGFR sensitizing mutations in the HRG-high subgroup between the arms of the HERALD study. However, potential imbalances in EGFR mutation status were simulated based on tissue results and found to have been unlikely to have biased the results. Further, the patient demographics and disease characteristics of the HRG-high subgroup were similar to that of the overall ITT population. Finally, EGFR mutation status was also assessed by plasma measurements (data not shown), reducing the percentage of patients with unknown EGFR mutation status to approximately 30%, with no apparent imbalance in EGFR-sensitizing mutations. Nonetheless, the presence of confounding interactions cannot be fully dismissed.
In addition, while a general biomarker hypothesis was stated in the protocol, the specific biomarker (i.e., HRG mRNA) and a description of the validated assay were defined only in the statistical analysis plan. An alternative approach would have been to amend the protocol once the preclinical data and assay became available and to formally elevate the single predictive biomarker hypothesis to a second primary end point (Beckman et al., 2011, Simon, 2005). As the HRG mRNA assay became available close to study completion, the protocol was not amended to specify the HRG subgroup analysis as the secondary primary end point due to the extra time and costs such an approach would have necessitated. However, this decision diminished the perceived credibility of the results in some instances, and the trade-off for omitting this administrative step may therefore have not been optimal, despite the development time it saved.
Due to the caveats in this prospective–retrospective approach and the potential for confounders in the observed effect size, a 2-part phase 3 study (HER3 Lung) will be conducted (NCT02134015) (Paz-Ares et al., 2014). Part A of the study will enroll patients with any level of HRG mRNA expression, and the statistical analysis will assess HRG-high and HRG-low biomarker groups for clinical benefit as measured by PFS to confirm the results of the HERALD study. The HRG cutoff will be further refined based on the clinical benefit observed in part A and in the HERALD study, using maximum likelihood methods (Altman et al., 1994, Jiang et al., 2007). This refined cutoff will then be applied in the pivotal part B, which will enroll HRG-high patients only, per revised criteria. Within part B, HRG-high patients will be further stratified into 2 levels of HRG mRNA expression. If part B is positive and the drug is registered, the information about these strata may be available to further inform physician and patient choices.
5. Conclusions
While early identification of a predictive biomarker before study initiation would have been ideal, this prospective–retrospective approach still allowed the clinical validation of the predictive biomarker HRG for patritumab. Further ex vivo studies conducted to assess the biomarker distributions on NSCLC tissues assisted in our understanding of how to extrapolate these data to other NSCLC populations. In addition, the exploratory analyses that allowed internal validation of the biomarker by assessing alternative cutoff values and the impact of potential confounding by various factors were useful to qualify the observed results. Although not implemented in this study, based on this experience, we recommend that future prospective–retrospective studies (1) be amended to specify the predictive biomarker hypothesis as a primary objective in the clinical study protocol when the specific predictive biomarker is identified and (2) ensure a process for the evaluation of viable tumor samples at the time of enrollment. For instance, slides from collected FFPE tissue could be stained with hematoxylin and eosin and evaluated for sufficient tumor tissue immediately upon receipt, which could preclude the problems encountered with tumor sample quality. Patients with inadequate samples would not be eligible for the clinical study. The turnaround time for this evaluation would have to be rapid, however, to avoid patients having to wait for therapy.
Conflicts of Interest
J.M., D.F., W.F., S.C., K.N., Z.T., C.C., and R.B. are/were employees of and own stock in Daiichi Sankyo. R.B. has served as a consultant for the Cancer Institute of New Jersey and owns stock in Johnson and Johnson. M.S., S.B., J.R., and J.B. are employees of U3 Pharma. T.H. is an employee of Complix. J.vP. has served as a consultant or advisor for Daiichi Sankyo, Novartis, Pfizer, and Vertex.
Author Contributions
D. F., T.H., Z.T., C.C., and R.B. designed the study. D.F., W.F., T.H., M.S., S.B., J. R., J. B., K.N., and J.vP. collected the data. J. M., D.F., T.H., M.S., S.B., J. R., J. B., K.N., S. C., Z.T., C.C., and R.B. analyzed and interpreted the data. J.M., D.F., W.F., T.H., M.S., S.B., J. R., J. B., K.N., S.C., Z.T., J.vP., C.C., and R.B. wrote the manuscript.
Funding
This study was funded by Daiichi Sankyo, Inc.
Acknowledgments
We thank MolecularMD (Portland, OR) for the expediency with which they developed and validated the rtPCR assay method for the measurement of HRG mRNA in FFPE samples. National Hospital Organization Kyushu Cancer Center kindly provided high-quality RNA samples of tumors for our analysis. Third-party writing assistance was provided by BlueMomentum, a division of KnowledgePoint360, an Ashfield company, and supported by Daiichi Sankyo, Inc.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2015.02.005.
Appendix A. Supplementary Data
Supplementary material.
References
- al Moustafa A.E., Alaoui-Jamali M., Paterson J., O'Connor-McCourt M. Expression of P185erbB-2, P160erbB-3, P180erbB-4, and heregulin alpha in human normal bronchial epithelial and lung cancer cell lines. Anticancer Res. 1999;19:481–486. [PubMed] [Google Scholar]
- Alroy I., Yarden Y. The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand–receptor interactions. FEBS Lett. 1997;410:83–86. doi: 10.1016/s0014-5793(97)00412-2. [DOI] [PubMed] [Google Scholar]
- Altman D.G., Lausen B., Sauerbrei W., Schumacher M. Dangers of using “optimal” cutpoints in the evaluation of prognostic factors. J. Natl. Cancer Inst. 1994;86:829–835. doi: 10.1093/jnci/86.11.829. [DOI] [PubMed] [Google Scholar]
- Atlas E., Cardillo M., Mehmi I., Zahedkargaran H., Tang C., Lupu R. Heregulin is sufficient for the promotion of tumorigenicity and metastasis of breast cancer cells in vivo. Mol. Cancer Res. 2003;1:165–175. [PubMed] [Google Scholar]
- Baselga J., Swain S.M. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- Beckman R.A., Clark J., Chen C. Integrating predictive biomarkers and classifiers into oncology clinical development programmes. Nat. Rev. Drug Discov. 2011;10:735–748. doi: 10.1038/nrd3550. [DOI] [PubMed] [Google Scholar]
- Carraway K.L., III, Sliwkowski M.X., Akita R., Platko J.V., Guy P.M., Nuijens A., Diamonti A.J., Vandlen R.L., Cantley L.C., Cerione R.A. The erbB3 gene product is a receptor for heregulin. J. Biol. Chem. 1994;269:14303–14306. [PubMed] [Google Scholar]
- de Alava E., Ocaña A., Abad M., Montero J.C., Esparís-Ogando A., Rodriguez C.A., Otero A.P., Hernández T., Cruz J.J., Pandiella A. Neuregulin expression modulates clinical response to trastuzumab in patients with metastatic breast cancer. J. Clin. Oncol. 2007;25:2656–2663. doi: 10.1200/JCO.2006.08.6850. [DOI] [PubMed] [Google Scholar]
- Freeman D., Ogbagabriel S., Rothe M., Radinsky R., Treder M. AACR Meeting Abstracts. 2008. Fully human anti-HER3 monoclonal antibodies (mAbs) have unique in vitro and in vivo functional and antitumor activities versus other HER family inhibitors; p. LB-21. [Google Scholar]
- Freeman D., Ogbagabriel S., Schneider M., Radinsky R., Hettman T. U3-1287 (AMG 888), a fully human anti-HER3 mAb, demonstrates in vitro and in vivo efficacy in NSCLC models. Mol. Cancer Ther. 2009;8:B171. [Google Scholar]
- Fridlyand J., Simon R.M., Walrath J.C., Roach N., Buller R., Schenkein D.P., Flaherty K.T., Allen J.D., Sigal E.V., Scher H.I. Considerations for the successful co-development of targeted cancer therapies and companion diagnostics. Nat. Rev. Drug Discov. 2013;12:743–755. doi: 10.1038/nrd4101. [DOI] [PubMed] [Google Scholar]
- Garrett J.T., Olivares M.G., Rinehart C., Granja-Ingram N.D., Sánchez V., Chakrabarty A., Dave B., Cook R.S., Pao W., McKinely E., Manning H.C., Chang J., Arteaga C.L. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullick W.J. The c-erbB3/HER3 receptor in human cancer. Cancer Surv. 1996;27:339–349. [PubMed] [Google Scholar]
- Hall J.A., Salgado R., Lively T., Sweep F., Schuh A. A risk-management approach for effective integration of biomarkers in clinical trials: perspectives of an NCI, NCRI, and EORTC working group. Lancet Oncol. 2014;15:e184–e193. doi: 10.1016/S1470-2045(13)70607-7. [DOI] [PubMed] [Google Scholar]
- Hegde G.V., de la Cruz C.C., Chiu C., Alag N., Schaefer G., Crocker L., Ross S., Goldenberg D., Merchant M., Tien J., Shao L., Roth L., Tsai S.P., Stawicki S., Jin Z., Wyatt S.K., Carano R.A., Zheng Y., Sweet-Cordero E.A., Wu Y., Jackson E.L. Blocking NRG1 and other ligand-mediated Her4 signaling enhances the magnitude and duration of the chemotherapeutic response of non-small cell lung cancer. Sci. Transl. Med. 2013;5:171ra18. doi: 10.1126/scitranslmed.3004438. [DOI] [PubMed] [Google Scholar]
- Hettmann T., Schneider M., Ogbagabriel S., Xie J., Juan G., Hartmann S., Radinsky R., Freeman D.J. U3-1287 (AMG 888), a fully human anti-HER3 mAb, inhibits HER3 activation and induces HER3 internalization and degradation. Cancer Res. 2010;70:LB-306. [Google Scholar]
- Hsieh A.C., Moasser M.M. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br. J. Cancer. 2007;97:453–457. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Freidlin B., Simon R. Biomarker-adaptive threshold design: a procedure for evaluating treatment with possible biomarker-defined subset effect. J. Natl. Cancer Inst. 2007;99:1036–1043. doi: 10.1093/jnci/djm022. [DOI] [PubMed] [Google Scholar]
- Khleif S.N., Doroshow J.H., Hait W.N. AACR-FDA-NCI Cancer Biomarkers Collaborative, AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin. Cancer Res. 2010;16:3299–3318. doi: 10.1158/1078-0432.CCR-10-0880. [DOI] [PubMed] [Google Scholar]
- Krane I.M., Leder P. NDF/heregulin induces persistence of terminal end buds and adenocarcinomas in the mammary glands of transgenic mice. Oncogene. 1996;12:1781–1788. [PubMed] [Google Scholar]
- Liu J., Kern J.A. Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am. J. Respir. Cell Mol. Biol. 2002;27:306–313. doi: 10.1165/rcmb.4850. [DOI] [PubMed] [Google Scholar]
- LoRusso P., Jänne P.A., Oliveira M., Rizvi N., Malburg L., Keedy V., Yee L., Copigneaux C., Hettmann T., Wu C.Y., Ang A., Halim A.B., Beckman R.A., Beaupre D., Berlin J. Phase I study of U3-1287, a fully human anti-HER3 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2013;19:3078–3087. doi: 10.1158/1078-0432.CCR-12-3051. [DOI] [PubMed] [Google Scholar]
- Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., Louis D.N., Christiani D.C., Settleman J., Haber D.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Mandrekar S.J., Sargent D.J. Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J. Clin. Oncol. 2009;27:4027–4034. doi: 10.1200/JCO.2009.22.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass R.D., Press M.F., Anderson S., Cobleigh M.A., Vogel C.L., Dybdal N., Leiberman G., Slamon D.J. Evaluation of clinical outcomes according to HER2 detection by fluorescence in situ hybridization in women with metastatic breast cancer treated with trastuzumab. Clin. Breast Cancer. 2005;6:240–246. doi: 10.3816/CBC.2005.n.026. [DOI] [PubMed] [Google Scholar]
- Motoyama A.B., Hynes N.E., Lane H.A. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 2002;62:3151–3158. [PubMed] [Google Scholar]
- Müller-Tidow C., Diederichs S., Bulk E., Pohle T., Steffen B., Schwäble J., Plewka S., Thomas M., Metzger R., Schneider P.M., Brandts C.H., Berdel W.E., Serve H. Identification of metastasis-associated receptor tyrosine kinases in non-small cell lung cancer. Cancer Res. 2005;65:1778–1782. doi: 10.1158/0008-5472.CAN-04-3388. [DOI] [PubMed] [Google Scholar]
- Olayioye M.A., Neve R.M., Lane H.A., Hynes N.E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez J.G., Jänne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J., Naoki K., Sasaki H., Fujii Y., Eck M.J., Sellers W.R., Johnson B.E., Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I., Singh B., Heelan R., Rusch V., Fulton L., Mardis E., Kupfer D., Wilson R., Kris M., Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.D., Cohen N., Karnoub M., Truter S.L., Emison E., Khambata-Ford S., Spear B., Ibia E., Sproule R., Barnes D., Bhathena A., Bristow M.R., Russell C., Wang D., Warner A., Westelinck A., Brian W., Snapir A., Franc M.A., Wong P., Shaw P.M. Prospective–retrospective biomarker analysis for regulatory consideration: white paper from the industry pharmacogenomics working group. Pharmacogenomics. 2011;12:939–951. doi: 10.2217/pgs.11.52. [DOI] [PubMed] [Google Scholar]
- Paz-Ares L., von Pawel J., Moritz B., Mendell J., Jin X., Copigneaux C., Beckman R. Phase (Ph) 3 study of patritumab (P) plus erlotinib (E) in EGFR wild-type subjects with advanced non-small cell lung cancer (NSCLC) Ann. Oncol. 2014;25:iv426–iv470. [Google Scholar]
- Polley M.Y., Freidlin B., Korn E.L., Conley B.A., Abrams J.S., McShane L.M. Statistical and practical considerations for clinical evaluation of predictive biomarkers. J. Natl. Cancer Inst. 2013;105:1677–1683. doi: 10.1093/jnci/djt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent S.A., Gullick W.J. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Yashiro M., Takakura N. Heregulin induces resistance to lapatinib-mediated growth inhibition of HER2-amplified cancer cells. Cancer Sci. 2013;104:1618–1625. doi: 10.1111/cas.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M., Blum S., Wenzl C., Bange J., Ogbagabriel S., Freeman D.J., Ruhe J. Heregulin expression level as a predictive biomarker for patritumab efficacy. J. Clin. Oncol. 2014;32:2618. [Google Scholar]
- Schoeberl B., Pace E.A., Fitzgerald J.B., Harms B.D., Xu L., Nie L., Linggi B., Kalra A., Paragas V., Bukhalid R., Grantcharova V., Kohli N., West K.A., Leszczyniecka M., Feldhaus M.J., Kudla A.J., Nielsen U.B. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci. Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- Schoeberl B., Faber A.C., Li D., Liang M.C., Crosby K., Onsum M., Burenkova O., Pace E., Walton Z., Nie L., Fulgham A., Song Y., Nielsen U.B., Engelman J.A., Wong K.K. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist L.V., Martins R.G., Spigel D., Grunberg S.M., Spira A., Jänne P.A., Joshi V.A., McCollum D., Evans T.L., Muzikansky A., Kuhlmann G.L., Han M., Goldberg J.S., Settleman J., Iafrate A.J., Engelman J.A., Haber D.A., Johnson B.E., Lynch T.J. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- Sergina N.V., Rausch M., Wang D., Blair J., Hann B., Shokat K.M., Moasser M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames D.S., Carbon J., Walter K., Jubb A.M., Kozlowski C., Januario T., Do A., Fu L., Xiao Y., Raja R., Jiang B., Malekafzali A., Stern H., Settleman J., Wilson T.R., Hampton G.M., Yauch R.L., Pirzkall A., Amler L.C. High heregulin expression is associated with activated HER3 and may define an actionable biomarker in patients with squamous cell carcinomas of the head and neck. PLoS ONE. 2013;8:e56765. doi: 10.1371/journal.pone.0056765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005;23:7332–7341. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- Simon R.M., Paik S., Hayes D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009;101:1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon D.J., Clark G.M., Wong S.G., Levin W.J., Ullrich A., McGuire W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Soltoff S.P., Carraway K.L., III, Prigent S.A., Gullick W.G., Cantley L.C. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treder M., Hartmann S., Ogbagabriel S., Borges E., Green L., Kang J., Radinsky R., Rothe M., Freeman D. AACR Meeting Abstracts. 2008. Fully human anti-HER3 monoclonal antibodies (mAbs) inhibit oncogenic signaling and tumor cell growth in vitro and in vivo; p. LB-20. [Google Scholar]
- Treder M., Ogbagabriel S., Moor R., Schulze-Horsel U., Hettmann T., Rothe M., Radinsky R., Freeman D. Fully human anti-HER3 mAb U3-1287 (AMG 888) demonstrates unique in vitro and in vivo activities versus other HER family inhibitors in NSCLC models. Eur. J. Cancer Suppl. 2008;6:99. [Google Scholar]
- Tsai M.S., Shamon-Taylor L.A., Mehmi I., Tang C.K., Lupu R. Blockage of heregulin expression inhibits tumorigenicity and metastasis of breast cancer. Oncogene. 2003;22:761–768. doi: 10.1038/sj.onc.1206130. [DOI] [PubMed] [Google Scholar]
- Ueno Y., Sakurai H., Tsunoda S., Choo M.K., Matsuo M., Koizumi K., Saiki I. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int. J. Cancer. 2008;123:340–347. doi: 10.1002/ijc.23465. [DOI] [PubMed] [Google Scholar]
- von Pawel J., Tseng J., Dediu M., Schumann C., Moritz B., Mendell-Harary J., Jin X., Feng W., Copigneaux C., Beckman R.A. Phase 2 HERALD study of patritumab (P) with erlotinib (E) in advanced NSCLC subjects (SBJs) J. Clin. Oncol. 2014;32:8045. [Google Scholar]
- Wheeler D.L., Huang S., Kruser T.J., Nechrebecki M.M., Armstrong E.A., Benavente S., Gondi V., Hsu K.T., Harari P.M. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W., Gerard C.M., Liu L., Baudson N.M., Ory T.L., Spector N.L. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene. 2005;24:6213–6221. doi: 10.1038/sj.onc.1208774. [DOI] [PubMed] [Google Scholar]
- Xia W., Petricoin E.F., III, Zhao S., Liu L., Osada T., Cheng Q., Wulfkuhle J.D., Gwin W.R., Yang X., Gallagher R.I., Bacus S., Lyerly H.K., Spector N.L. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2 + breast cancer models. Breast Cancer Res. 2013;15:R85. doi: 10.1186/bcr3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.H., Yu C.J., Shih J.Y., Chang Y.C., Hu F.C., Tsai M.C., Chen K.Y., Lin Z.Z., Huang C.J., Shun C.T., Huang C.L., Bean J., Cheng A.L., Pao W., Yang P.C. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J. Clin. Oncol. 2008;26:2745–2753. doi: 10.1200/JCO.2007.15.6695. [DOI] [PubMed] [Google Scholar]
- Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yi E.S., Harclerode D., Gondo M., Stephenson M., Brown R.W., Younes M., Cagle P.T. High c-erbB-3 protein expression is associated with shorter survival in advanced non-small cell lung carcinomas. Mod. Pathol. 1997;10:142–148. [PubMed] [Google Scholar]
- Yonesaka K., Kawakami H., Kaneda H., Okamoto I., Hirotani K., Nishio K., Nakagawa K. The expression level of HER3 ligand heregulin mRNA as a predictive biomarker for anti-HER3 antibody patritumab combined with erlotinib in non-small cell lung cancer. J. Clin. Oncol. 2014;32:e19082. [Google Scholar]
- Zhou B.B., Peyton M., He B., Liu C., Girard L., Caudler E., Lo Y., Baribaud F., Mikami I., Reguart N., Yang G., Li Y., Yao W., Vaddi K., Gazdar A.F., Friedman S.M., Jablons D.M., Newton R.C., Fridman J.S., Minna J.D., Scherle P.A. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.