

Abstract
To determine how a substitutionally inert metal can play a catalytic role in the metalloenzyme nitrile hydratase (NHase), a reactive five-coordinate CoIII thiolate complex ([CoIII(S2Me2N3(Pr,Pr))](PF6) (1)) that resembles the active site of cobalt containing nitrile hydratase (Co NHase) was prepared. This was screened for reactivity, by using low-temperature electronic absorption spectroscopy, toward a number of biologically relevant “substrates”. It was determined 1 will react with azide, thiocyanate, and ammonia, but is unreactive toward nitriles, NO, and butyrate. Substrate-bound 1 has similar spectroscopic and structural properties as [CoIII(ADIT2)](PF6) (2). Complex 2 is a six-coordinate CoIII complex containing cis-thiolates and imine nitrogens, and has properties similar to the cobalt center of Co NHase. Substrate binding to 1 is reversible and temperature-dependent, allowing for the determination of the thermodynamic parameters of azide and thiocyanate binding and the rates of ligand dissociation. Azide and thiocyanate bind trans to a thiolate, and with similar entropies and enthalpies (thiocyanate: ΔH = −7.5 ± 1.1 kcal/mol, ΔS = −17.2 ± 3.2 eu; azide: ΔH = −6.5 ± 1.0 kcal/mol, ΔS = −12.6 ± 2.4 eu). The rates of azide and thiocyanate displacement from the metal center are also comparable to one another (kd = (7.22 ± 0.04) × 10−1 s−1 for thiocyanate and kd = 2.14 ± 0.50) × 10−2 s−1 for azide), and are considerably faster than one would expect for a low-spin d6 six-coordinate CoIII complex. These rates are comparable to those of an analogous Fe(III) complex, demonstrating that Co(III) and Fe(III) react at comparable rates when in this ligand environment. This study therefore indicates that ligand displacement from a low-spin CoIII center in a ligand environment that resembles NHase is not prohibitivly slow so as to disallow catalytic action in nonredox active cobalt metalloenzymes.
Nitrile hydratases (NHases) are mononuclear, noncorrinoid cobalt (III) and non-heme iron (III) metalloenzymes that are found in a variety of soil bacteria and catalyze the hydration of nitriles to amides.1 NHases have been utilized in a variety of applications including industrial-scale acrylamide synthesis,2 asymmetric amide synthesis,3 and toxic waste degradation.4 Initial spectroscopic studies showed the metals are low spin (S = 0 for cobalt and S = 1/2 for iron), and ligated by two to three cysteinates, three nitrogens, and a water molecule.5–10 X-ray crystal structures of the iron-containing NHase refined this picture, revealing the metal-ion is ligated by three cysteinates, two amide nitrogens (from the peptide backbone), and water or NO (Figure 1).5–12 The more recent crystal structure was performed at higher resolution then the earlier structure (1.7 vs 2.65 Å) and was of NO inactivated NHase.12 It showed two of the cysteinates were posttranslationally modified to either a sulfinate or a sulfenate (Figure 1b); however, their importance in catalysis has yet to be demonstrated. Nitrile hydration is proposed to occur through one of two mechanisms: water attack of a metal-bound nitrile, or a metal-bound hydroxide attacking a nitrile.11
Figure 1.
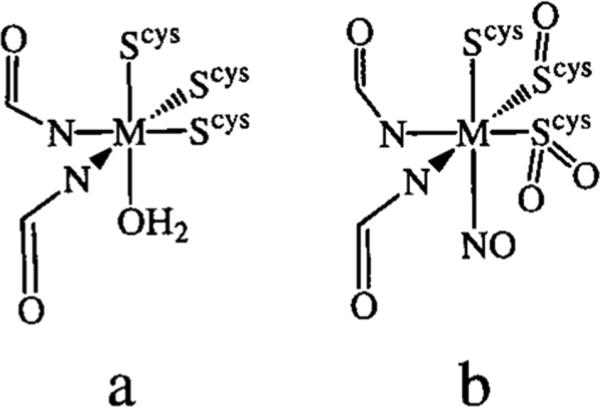
(A) Active site of NHase as determined by X-ray crystal-lography (2.65 Å resolution).11 (B) Active site of NO-inactivated NHase as determined by X-ray crystallography (1.7 Å resolution).12
Although there is no crystal structure available for cobalt-containing NHase (Co NHase), a comparison of its gene sequence with iron-containing NHase (Fe NHase) reveals that the two different forms of the enzyme have a high sequence homology.13 The active-site regions are nearly identical, with only one noncoordinating residue differing between the two forms. Such high sequence homologies suggest that the cobalt center is ligated in a fashion identical to iron. Further evidence that cobalt ligation is identical to that of iron was recently provided by showing that replacement of the Fe3+ ion in Fe NHase from Rhodococcus sp. n-771 with Co3+ produced an enzyme with an absorption spectrum and catalytic properties that are identical to that of Co NHase from Pseudomonas putida NRRL-18668.14
Fe NHase can be deactivated in the dark and reactivated by exposing the enzyme to light.7,12,15–21 This is caused by NO binding to the active site in the dark, followed by photocleavage of the Fe–NO bond, which reactivates the enzyme. Co NHase has never been observed to display this behavior. It is unknown why Co NHase does not appear to be deactivated by NO, but it is possible that the enzyme is not able to coordinate NO to the metal center. Butyrate was demonstrated to be a competitive inhibitor of Fe NHase when activity was lost whenever a butyrate buffer was used to solubilize the enzyme.22 ENDOR studies have since suggested that butyrate does not displace water and bind to the metal-site, instead, it merely rests in the active-site cavity, blocking NHase activity.23 The mechanism of butyrate inhibition of Co NHase is currently unknown.
Although two crystal structures are now available for NHase, a number of issues concerning spectroscopic properties, metal coordination environment, and the mechanism of catalysis have yet to be adequately addressed. Recently, a number of coordinatively saturated and unsaturated model compounds have been synthesized aimed at understanding these properties.21,24–34 Low-spin Co(III) is not typically utilized in nonredox active enzymes that require ligand displacement from the metal center.35 This is probably a consequence of low-spin Co(III) being a substitution inert metal center. Despite this, the cobalt form of NHase displays higher activity than Fe NHase.2 Recently we reported the synthesis of a five-coordinate cobalt compound ([CoIII(S2Me2N3(Pr,Pr))](PF6) (1) (Figure 2) that was found to be reactive toward both thiocyanate and ammonia.36 Furthermore, both thiocyanate and ammonia binding were found to be reversible. Reported herein are the reactivity of 1 toward a number of NHase substrates, inactivators, and inhibitors. Also presented are the results of kinetic and thermodynamic studies of azide and thiocyanate binding to, and dissociation from, 1. In addition, the synthesis and spectroscopic properties of a six-coordinate complex ([CoIII(ADIT2)](PF6) (2)) that mimics the active site of NHase is presented.
Figure 2.
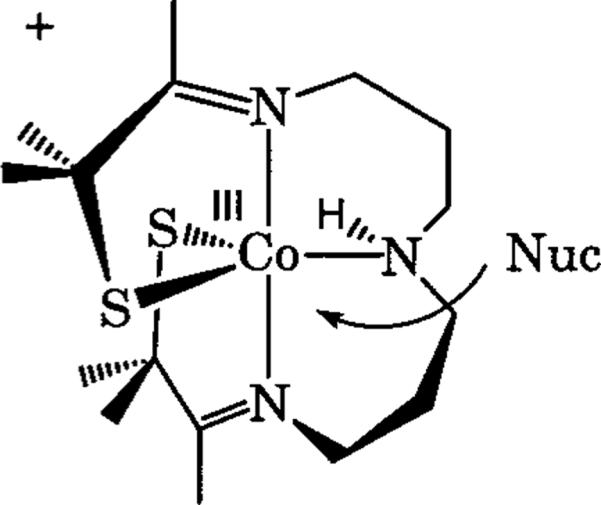
[CoIII(S2Me2N3(Pr,Pr))](PF6) (1) depicting the open-site where a nucleophile could bind.
Experimental Section
General Methods
All manipulations were performed with Schlenk-line techniques or in a glovebox under an atmosphere of dinitrogen. Chemical reagents obtained through commercial vendors were of the highest purity available and used without further purification. Solvents were purified through standard methods and degassed prior to use.37 3-Methyl-3-mercapto-2-butanone was prepared through literature methods.25 IR spectra were recorded using a Perkin-Elmer 1720 FTIR. NMR spectra were obtained using a Bruker WM-500 spectrometer and recorded at 20 °C unless otherwise noted. Chemical shifts (δ) are reported in parts per million (ppm) and are referenced to the residual protio solvent peak. UV/vis-near-IR spectra were recorded on a Hewlett-Packard 8450 spectrometer. Elemental analyses were performed by Canadian Microanalytical Service Ltd. (Delta, BC), Atlantic Microlab Inc. (Norcross, GA), and Galbraith Labs (Knoxville, TN).
[CoIIIS2Me2N3(Pr,Pr)](PF6) (1)
[CoIII(S2Me2N3(Pr,Pr))](PF6) (1) was prepared as previously described,33 except it was recrystalized three times from acetonitrile/diethyl ether, instead of once. Yield: 23%. 1H NMR (CD2Cl2) 37.0 (bs), 25.6 (bs), 17.2 (bs), 15.9 (bs), 14.2 (bs), 3.3 (bs), 0.4 (bs), −1.2 (bs), −3.7 (bs), −5.1 (bs), −8.8 (bs), −12.6 (bs).
[CoIII(ADIT2)](PF6) (2)
Sodium hydroxide (84 mg, 2.10 mmol) was dissolved in 5 mL of methanol and 236 mg of 3-methyl-3-mercapto-2-butanone was added to the stirred hydroxide solution. After 10 min, the thiolate solution was added in 1-mL portions to a stirred solution of cobalt(II) chloride hexahydrate (238 mg, 1.00 mmol) in 10 mL of methanol. A brick red precipitate formed instantly. After 30 min, 120 mg of ethylenediamine (2.00 mmol) was added in 5 mL of methanol. The solution turned a deep purple, became homogeneous after 5 min, and was stirred overnight. Ferrocinium hexaflurophosphate (350 mg, 1.05 mmol) was then added in 5 mL of acetonitrile and the solution became a magenta color after 10 min. The solution was stirred for an additional 12 h and filtered, then the solvent was removed by vacuum. Ferrocene was extracted from the product with diethyl ether resulting in a brown powder. This was dissolved in 2 mL of acetonitrile and diethyl ether (20 mL) was cautiously layered on top. The two layers were allowed to slowly diffuse together at −35 °C, and after 5 days [CoIII[ADIT2]](PF6) (2) had formed as dark brown crystals (507 mg, 57% yield). 1H NMR (MeCN-d3): 3.39 (4 H, m, CH2), 2.96 (4 H, m, CH2), 2.84 (4H, bs, NH2), 2.04 (6 H, s, CH3), 1.59 (6H, s, CH3), 1.38 (6H, s, CH3). Electronic absorption spectrum (CH2Cl2): λmax (εM) 261 (sh), 279 (16 000), 356 (sh), 472 nm (2200). IR (KBr pellet) ν (cm−1): 1641 (imine), 1578 (imine). Anal. Calcd for CoC14H30N4S2PF6: C, 32.19; H, 5.79; N, 10.72. Found: C, 32.22; H, 5.69; N, 9.68
[CoIII(S2Me2N3(Pr,Pr))(NCS)] (3)
Tetrakis(n-butyl)ammonium thio cyanate (477 mg, 1.59 mmols) was dissolved in 1 mL of acetonitrile. 1 (450 mg, 0.795 mmols) in 2 mL of acetonitrile was slowly added dropwise to the thiocyanate solution. The solution was left undisturbed at room temperature for 24 h, at which time 6 mL of diethyl ether was cautiously layered overtop, followed by cooling to −35 °C. After 48 h [CoIII(S2Me2N3(Pr,Pr))(NCS)] (3) had formed as dark pink crystals (126 mg, 35% yield). 1H NMR (CD2Cl2): 3.98 (1H, bs, NH2), 3.12 (4H, m, CH2), 2.45 (4H, bs, CH2), 2.21 (3H, s, CH3), 1.98 (3H, s, CH3), 1.65 (6H, bs, CH3), 1.23 (6H, bs, CH3), 0.86 (4H, m, CH2). Electronic absorption spectra (CH2Cl2): λmax (εM) 230 (18 700), 262 (15 200), 286 (16 500), 361 (sh), 501 (1000), 628 nm (sh). (MeOH) λ (εM): 282 nm (17 000). IR (KBr pellet) ν (cm−1): 1611 (imine), 2107 (thiocyanate). Anal. Calcd for CoC17H31N4S3: C, 45.72; H, 6.99; N, 12.54. Found: C, 45.76; H 6.95; N, 12.79.
[CoIII(S2Me2N3(Pr,Pr))(N3)] (4)
[CoIII(S2Me2N3(Pr,Pr))(N3)] (4) was prepared in an analogous manner to 3 with use of 446 mg of tetrakis-(n-butyl)ammonium azide (1.59 mmols) and 450 mg of 1 (0.795 mmols) affording 157 mg of the title compound 4 as purple crystals (46% yield). 1H NMR (CD2Cl2): 4.02 (1H, bs, NH2), 3.17 (4H, m, CH2), 2.48 (4H, bs, CH2), 2.29 (3H, s, CH3), 1.93 (3H, s, CH3), 1.61 (6H, bs, CH3), 1.27 (6H, bs, CH3), 0.82 (4H, m, CH2). Electronic absorption spectra (CH2Cl2): λmax (εM) 230 nm (10 000), 269 nm (7400), 282 nm (7500), 349 nm (4700), 523 nm (1700). (MeOH) λ (εM): 282 nm (7800). IR (KBr pellet) ν (cm−1): 1612 (imine), 2023 (azide). Anal. Calcd for CoC16H31N6S2: C, 44.44; H, 7.13; N, 19.39. Found: C, 44.63; H, 7.26; N, 19.52.
Spectrophotometrically Monitored Substrate Binding To 1
A 0.100 mM (250 mL) solution of 1 in methylene chloride was prepared. Ten milliliters of this solution was diluted to between 0.075 and 0.050 mM, placed in a custom-built quartz dewar, and cooled to dry ice/acetone temperatures (−77 °C). The UV/vis spectrum was then monitored as 1 to 1000 equiv of substrate were titrated into the solution. Upon binding of substrate a band close to 285 nm grows in, while bands at 356, 445, and 525 nm corresponding to five-coordinate 1 disappear. In this manner, the affinity of 1 toward azide, thiocyanate, butyrate, nitriles, ammonia, and NO was evaluated.
Binding Studies
Both azide and thiocyanate binding studies were performed in methanol with freshly prepared solutions of 1. Bu4nN(N3) and Bu4nN(SCN) were used as the azide and thiocyanate sources. Equilibrium constants (Keq) were measured spectrophotometrically at five different temperatures in a custom-built quartz optical dewar using the appropriate cryogenic bath: dry ice/acetone (−77 °C), liquid nitrogen/acetonitrile (−41 °C), dry ice/ethylene glycol (−15 °C), ice (0 °C), and water (22-25 °C). The initial concentration of 1 ([1]0) varied between 0.125 and 0.073 mM. The azide or thiocyanate salts were added to the solution so the [N3−]0 to [1]0 (or [SCN−]0 to [1]0) ratios were between 40% to 80% of the bound cobalt complex. This range gave the least amount of error when calculating equilibrium constants (Keq). Solutions were maintained at constant ionic strength ([i] = 0.022 ± 0.001 M) using an appropriate amount of KPF6. All concentrations were then corrected for the changes in the volume of methanol as previously described.38 To minimize the error in the calculations of Keq, absorption spectra were recorded at the wavelength that gave the greatest difference between bound and unbound cobalt species (282 nm for both azide and thiocyanate). Molar extinction coefficients (ε) were determined in methanol for 3 and 4 by titrating in excess azide or thiocyanate, respectively. It was determined ε(3) = 17 000 M−1 cm−1 and ε(4) = 7800 M−1 cm−1 in methanol at 282 nm. These were found to vary insignificantly over the temperature range investigated. Equilibrium constants were then measured according to the methods of Drago using eqs 1 and 2.39,40 Thermodynamic parameters were then obtained for azide and thiocyanate binding using van't Hoff plots.
| (1) |
| (2) |
Where X is either azide or thiocyanate and 1X is azide- or thiocyanate-bound 1.
Kinetics of Azide and Thiocyanate Dissociation
A 0.800 mM stock solution of 1 in methanol was prepared. Ligand binding (kon) rates were then determined at room temperature by stopped-flow methods on an OLIS USA rapid-scanning monochromator, equipped with an OLIS-RSA detector, interfaced with a Dell Optiplex G1 personal computer. The stock solution of 1 was injected into methanolic solutions of either (Bun)4N3 or (Bun)4SCN. The concentrations of azide and thiocyanate were then increased until pseudo-first-order conditions were reached (~170 mM for azide and ~200 mM for thiocyanate). In this manner the rates for ligand dissociation (kd) could be determined by using eq 3.
| (3) |
Where Keq is the equilibrium binding constant at room temperature determined according to eq 2.
Methanolic solutions of 1 injected into methanol produced no observable change in the visible spectrum over the time the stopped-flow experiment was performed. Reduction in the concentration of 1 produced no significant change in the calculated kd values.
X-ray Crystal Studies
Crystals were submersed in mineral oil in a glovebox. Suitable crystals were selected, mounted on a glass capillary with super glue, and immediately placed under a stream of nitrogen. Data were collected at either −112 (4) or 22 °C (2) on a Nonius KappaCCD diffractometer. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares methods, and hydrogen atoms were located from difference maps refined with a riding model. Data were then refined using SHELEX-97,41 and the data was corrected by scaling and averaging using SCALEPACK42 on a Silicon Graphics O2. Crystallographic data for 2 and 4 are displayed in Table 1.
Table 1.
Crystal Data for [CoIII(ADIT2)](PF6)(2) and [CoIII(S2Me2N3(Pr,Pr))(N3)] (4)
| 2 | 4 | |
|---|---|---|
| formula | C16H34N5F6PS2Co | C16H31N6S2Co |
| fw | 564.50 | 430.52 |
| temp, K | 293(2) | 161(2) |
| unit cella | triclinic | monoclinic |
| space group | P1 | Cc |
| a, Å | 7.953(2) | 16.114(1) |
| b, Å | .209(3) | 10.927(1) |
| c, Å | 4.492(4) | 11.331(1) |
| α, deg | 94.82(1) | 90.0 |
| β, deg | 91.93(2) | 96.240(1) |
| γ, deg | 97.59(2) | 90.0 |
| V, Å3 | 1274.8(6) | 1983.3(3) |
| Z | 2 | 4 |
| σcalc, g/cm3 | 1.471 | 1.442 |
| Rb | 0.0804 | 0.0412 |
| R w | 0.1947 | 0.1125 |
| GOF | 1.045 | 1.142 |
In all cases: Mo Kα (λ = 0.71070 Å) radiation.
R = ∑∥Fo| – |Fc∥/∑|Fo|; Rw = [∑w(|Fo| – |Fc|)2/∑wFo2]1/2, where w–1 = [σ2count + (0.05F2)2]/4F2.
X-ray quality crystals of both 2 and 4 were grown in an analogous manner. The purified compounds were dissolved in a minimal amount of acetonitrile and diethyl ether (10 times the amount of acetonitrile used) was cautiously layered over the top of the acetonitrile layer. The two solvents were then allowed to slowly diffuse together at −30 °C over 7 days to afford X-ray quality crystals.
Results and Discussion
Screening the Binding Affinity of 1 toward Biologically Relevant “Substrates”
Five-coordinate [CoIII(S2Me2N3(Pr,Pr))]-(PF5) (1) was screened for reactivity with a number of different biologically relevant substrates using low-temperature UV/vis spectroscopy in methylene chloride. Complex 1 is paramagnetic (S=1), and has bands in the electronic absorption spectrum at 248, 356, 445, and 525 nm. Upon binding a sixth ligand, the bands at 356, 445, and 525 nm all diminish, and an intense absorption band is produced near 280 nm. It was determined that 1 will readily bind the following substrates: azide (NHase inhibitor),43 ammonia,36 and thiocyanate (nitrile mimic). Butyrate (NHase competitive inhibitor20), nitriles (NHase substrate1), and NO (NHase inactivator7,12,15–21) did not coordinate to the metal center of 1 under the conditions investigated.
Azide, thiocyanate, and ammonia bind to 1 reversibly, and binding was found to be both temperature- and solvent-dependent (Figure 3). The anionic ligands, azide and thiocyanate, will only bind to the metal center of 1 at low temperatures, high anion concentration, or in low dielectric solvents. Since binding is reversible and a large difference exists between bound and unbound species in the electronic absorption spectrum, the thermodynamics and kinetics of azide and thiocyanate binding can be readily investigated, and are discussed below. The neutral ligand ammonia was found to bind only at high concentrations and low temperatures, with complete binding detected only below −70 °C.36
Figure 3.

Variable-temperature electronic absorption spectrum of [CoIII(S2Me2N3(Pr,Pr))](PF6) (1) in methanol with 2 equiv of Bu4NN3 taken over a temperature range of 25 to −77 °C.
Butyrate is a competitive NHase inhibitor, and was found to be unreactive toward 1. The fact that butyrate will not bind to 1 suggests that Co(III) does not have an affinity for butyrate in this ligand environment. Analogous to the conclusions reached for butyrate inhibition of Fe NHase based on ENDOR, our work therefore suggests that butyrate inhibits Co NHase activity by merely resting in the active site, rather than coordinating to the metal center.23 It was also determined that NO will not bind to the cobalt center of 1, even under an atmosphere of NO. Instead what is observed is slow decomposition of 1, resulting in complete decomposition after 5 h at −77 °C. This is in stark contrast to the iron analogue of 1, [FeIII(S2Me2N3(Pr,Pr))](PF6), which will quantitatively bind NO, to afford an isolable complex.26 Similarly, Fe NHase binds NO, whereas Co NHase does not. This work suggests that the reason for this is that Co(III) does not have a high affinity for NO when it is in an environment resembling that of NHase.
Synthesis and Properties of [CoIII(ADIT2)](PF6) (2), [CoIII(SMe2N3(Pr,Pr))(SCN)] (3), and [CoIII(S2Me2N3(Pr,Pr))(N3)] (4)
[CoIII(ADIT2)](PF6) (2) was synthesized through a “onepot” Schiff-base condensation on CoCl2 followed by oxidation with ferrocinium. The sodium thiolate of 3-methyl-3-mercapto-2-butanone was added to a methanolic solution of CoCl2 followed by the addition of 2 equiv of ethylenediamine. The purple solution was then oxidized with ferrocenium hexafluorophosphate. Extraction of ferrocene and recrystalization from acetonitrile and diethyl ether produced 2 as dark brown crystals in good yield.
Selected bond lengths for 2 are listed in Table 2. EXAFS-determined bond lengths for Co NHase are 2.20 Å for the Co–S bonds and 1.95 Å for the Co–N bonds.44 An X-ray crystal structure solved for 2 shows similar S–Co (mean bond length = 2.21(0.01) Å) and N–Co (mean bond length=1.95(0.03) Å) bond lengths. These are shorter than the typical average bond length for low-spin Co(III).45 The cobalt center of complex 2 is in a distorted octahedral environment (Figure 4) with each ADIT ligand in a mer configuration. A comparison between the electronic absorption spectrum of 2 and that of substrate-bound 1 reveals they are essentially identical, and appear to be characteristic of six-coordinate CoIII in this ligand environment. The absorption spectrum of 2 (Figure S-4) is essentially featureless except for an intense absorption band at 279 nm (ε = 16 000 M−1 cm−1). This is similar to the absorption spectrum of Co NHase from Rhodocaccus rhodochorus J1, which is essentially featureless except for a shoulder around 280 nm.44
Table 2.
Comparison of Selected Metrical Parameters for Six-Coordinate [CoIII(ADIT2)](PF6)(2), [CoIII(S2Me2N3(Pr,Pr))(SCN)] (3), [CoIII(S2Me2N3(Pr,Pr))(N3)] (4), and Five-Coordinate [CoIIIS2Me2N3(Pr,Pr)](PF6)(1)a
| 2 | 3 | 4 | 1 | |
|---|---|---|---|---|
| Co–S(1) | 2.232(3) | 2.218 (2) | 2.216 (2) | 2.162 (22) |
| Co–S(2) | 2.226(3) | 2.231(2) | 2.223(2) | 2.158(2) |
| Co–N(1) | 1.905(7) | 1.956(7) | 1.944(5) | 1.923(4) |
| Co–N(2) | 1.906(7) | 1.962(4) | 1.946(5) | 1.923(4) |
| Co–N(3) | 2.013(7) | 2.070(4) | 2.122(5) | 2.060(5) |
| Co–N(4) | 2.011(7) | 1.974(4) | 2.066(5) | N/A |
| S(1)–Co–S(2) | 93.6(1) | 91.3(1) | 93.6(2) | 126.8(1) |
| N(1)–Co–N(2) | 177.6(3) | 174.6(2) | 174.0(2) | 179.2(2) |
| S(1)–Co–N(3) | 174.0 (2) | 174.8(2) | 173.5(2) | 117.3(1) |
| S(2)–Co–N(4) | 171.9(2) | 177.6(2) | 175.0(2) | N/A |
| N(1)–Co–S(1) | 96.8(2) | 92.4(2) | 91.8(2) | 92.2(1) |
| N(1)–Co–S(2) | 85.9(2) | 84.6(2) | 84.5(2) | 87.0(1) |
| N(2)–Co–S(1) | 85.6(2) | 85.8(2) | 85.1(2) | 87.4(1) |
| N(2)–Co–S(2) | 94.2(2) | 91.6(2) | 90.5(2) | 92.2(1) |
| N(3)–Co–N(4) | 90.9(2) | 96.9(4) | 85.4(2) | N/A |
Bond lengths are in angstroms and angles are in degrees.
Figure 4.

ORTEP of six-coordinate [CoIII(ADIT2)]+ (2) showing 50% probability ellipsoids and the atom-labeling scheme. All H atoms have been omitted for clarity.
The synthesis and characterization of [CoIII(S2Me2N3(Pr,Pr))(SCN)] (3) was previously communicated elsewhere,36 so a detailed description will not be presented here. [CoIII(S2Me2N3(Pr,Pr))(N3)] (4) was prepared in a manner analogous to 3. Five-coordinate 1 was dissolved in a minimal amount of acetonitrile and added to the appropriate alkylammonium azide (4) or thiocyanate (3) salt. After sitting undisturbed at room temperature for 1 day, diethyl ether was layered over the top of the solution, and the resulting six-coordinate complex [CoIII(S2Me2N3(Pr,Pr))(N3)] (4) or [CoIII(S2Me2N3(Pr,Pr))(SCN)] (3) was obtained as a crystalline solid (Scheme 1).
Scheme 1.
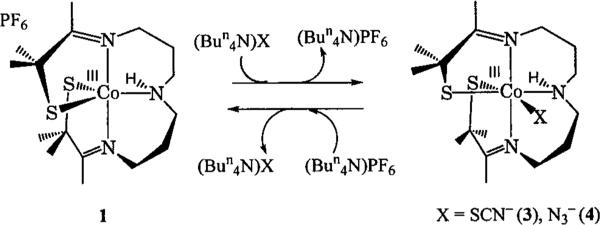
The X-ray crystal structure of 4 shows that it contains Co-(III) in a distorted octahedral geometry, with azide bound to a site that is trans to a thiolate (S(2), Figure 5) in a bent configuration. Selected bond lengths and angles for 4 are presented in Table 2. Upon binding azide, the Co–S bond trans to the binding site lengthens from 2.16(2) Å in 1 to 2.22(1) Å in 4. This is shorter than the bond length found in most Co-(III)–thiolate complexes, but is identical to the length of the Co-S bond trans to thiocyanate in 3.36 The average N-N bond lengths for the bound azide in 4 are identical to the bond lengths of free azide, just as the C–S and C–N bond lengths of free thiocyanate are identical to bound thiocyanate in 3. A comparison of the mean Co–S bond lengths in 2, 3, and 4 shows they are all identical at 2.22 Å and similar to the EXAFS-determined bond lengths of Co NHase. The mean Co–N distances of 2 (1.95 Å), 3 (2.01 Å), and 4 (2.00 Å) are also comparable to the EXAFS-determined distance of Co NHase.
Figure 5.

ORTEP of six-coordinate [CoIII(S2Me2N3(Pr,Pr))(N3)] (4) showing 50% probability ellipsoids and the atom-labeling scheme. All H atoms, except N(3)–H, have been omitted for clarity.
In contrast to five-coordinate 1, which is paramagnetic (S = 1), six-coordinate 2, 3, and 4 are all diamagnetic (S = 0). If one monitors substrate binding by 1H NMR, the paramagnetically shifted peaks of 1 collapse into diamagnetic signals upon binding substrate. Substrate binding can be followed by variable-temperature NMR spectroscopy. As was demonstrated earlier with thiocyanate,36 azide binding to 1 can only be detected by 1H NMR spectroscopy at low temperatures in MeOH-d4, or in methylene chloride. In MeOH-d4, as the temperature is lowered, the paramagnetic spectrum disappears at −20 °C (Figure S-3). Below −20 °C an intermediate species that cannot be assigned to either 1 or 4 is produced. This species persists at temperatures as low as −65 °C. Below this temperature a broad signal (line width of roughly 5 ppm) is produced. At −78 °C this signal expands into a spectrum that is identical to the spectrum of intact 4 obtained in methylene chloride. This process is reversible throughout the temperature range investigated.
Binding and Dissociation of Azide and Thiocyanate to and from 1
Binding of azide and thiocyanate was determined to be reversible in methanol, allowing us to determine the thermodynamics of azide and thiocyanate binding (Figure 3). Thermodynamic parameters were determined according to the methods of Drago and co-workers.39,40 Equilibrium constants were determined at five different temperatures for both azide and thiocyanate, and were then analyzed by using van't Hoff plots (Table 3 and Figure S-5). For thiocyanate binding in MeOH, ΔH was determined to be −7.5 ± 1.1 kcal/mol and ΔS was determined to be −17.2 ± 3.2 eu. The thermodynamic parameters for azide binding in MeOH were determined to be −6.5 ± 1.0 kcal/mol and −12.6 ± 2.4 eu for ΔH and ΔS, respectively. The free energy of azide or thiocyanate binding is close to zero at room temperature, as indicated by the relatively small Keq. A possible explanation for this relatively weak binding in MeOH is that it takes energy to de-solvate the anion before it can bind to the metal center of 1, making the unbound five-coordinate state lower in energy than it would be with a naked anion. The entropies are negative, consistent with an associative process;46 however, they are not as negative as one would expect for an associative process. This is most likely due to the fact that the anion reactants desolvate during the formation of neutral 3 or 4 thereby providing an entropic driving force that slightly offsets the disfavorable entropy of ligand binding.
Table 3.
Equilibrium Constants (Keq) Observed for Azide and Thiocyanate Binding to [CoIII(S2Me2N3(Pr,Pr))](PF6)(1) at Ambient Temperature (22 and 25 °C for azide and thiocyanate, respectiveley), 0 °C, –15 °C, –41 °C, and –77 °C
| azide (M–1) | thiocyanate (M–1) | |
|---|---|---|
| ambient temp | 75.5 ± 7.5 | 50 ± 5 |
| 0 °C | 280 ± 30 | 140 ± 14 |
| –15 °C | 580 ± 50 | 480 ± 40 |
| –41 °C | 3000 ± 300 | 4000 ± 400 |
| –77 °C | 29000 ± 3000 | 29000 ± 300 |
The thermodynamic parameters of azide binding to [FeIII(S2Me2N3(Pr,Pr))](PF6) were previously determined to be similar to those for azide binding to 1.25 This is despite the fact that azide is binding to two different metal ions which should have vastly different properties. Two conclusions could be drawn from this. One explanation is that these numbers mainly reflect the desolvation of the anions from methanol. However, once the anion is desolvated, one would still expect there to be a large enthalpy in favor of substrate-bound 1. Therefore one may be tempted to reach a different conclusion. Because there is little difference in going from a Co(III) (in 1) to Fe(III) (in [FeIII(S2Me2N3(Pr,Pr))](PF6)), these results seem to indicate that the ligand environment itself exerts a large influence over the chemistry taking place at the metal center. This is supported by the kinetics discussed below. Likewise, the protein of NHase appears to play a similar role since Fe and Co NHase appear to have similar reactivities, with the exception of NO binding.
The kinetics of azide and thiocyanate dissociation were also investigated. The rates for anion binding (kon) to the metal center of 1 were measured by stopped-flow methods at high anion concentration in methanolic solutions. From these, and the room-temperature equilibrium binding constants (Keq), it was possible to calculate rate constants for azide and thiocyanate dissociation from 1 (kd). Thiocyanate was found to dissociate from 1 with a rate of (7.22 ± 0.04) × 10−1 s−1. Azide dissociation occurred at a slower rate with kd = (2.14 ± 0.50) × 10−2 s−1. Both of these rates are faster than would be expected for a low-spin six-coordinate Co(III) complex, and are comparable to those of an analogous Fe(III) system,34 demonstrating that Co(III) and Fe(III) react at comparable rates when in this ligand environment. After an extensive review of the literature, we were only able to locate one system where ligand dissociation occurs faster from Co(III).47 Chin and co-workers measured kd of phosphates from a dinuclear [Co2III(tacn)2] complex. For these systems the rate of phosphate dissociation ranged from (3.76 ± 0.05) × 102 M−1 s−1 to (3.00 ± 0.3) × 10−5 M−1 s−1. Dissociation of azide or thiocyanate from 1 in MeOH falls within this range under the conditions examined.
These models have shown that ligand displacement from a low-spin six-coordinate d6 Co(III) center is not impossible. Rapid ligand displacement from 3 and 4 may be promoted by the thiolate that is trans to the binding site. Previous studies by Deutch and Corrano have shown that thiolates have a strong trans labilizing effect.48,49 Furthermore, the only published NHase model that catalyzes nitrile hydration is a Co(III) model that contains a thiolate trans to the open site.27 Thus, it would appear that the thiolate trans to the open site may dictate the reactivity properties of these complexes. NHase has a cysteinate trans to a vacant site where substrate would bind. The utilization of a trans cysteinate could aid the enzyme in catalyzing the hydration of a nitrile regardless of the mode of action. In the case where a bound hydroxide attacks the nitrile, the labilizing effect could aid in the initial displacement of hydroxide from the metal center. In the case of C–N bond activation of a bound nitrile, the labilizing effect of a trans thiolate could aid in the dissociation of the amide formed in the reaction.
Supplementary Material
Acknowledgment
The authors wish to thank Mr. Dirk Schweitzer and Mr. Henry Jackson for thoughtful discussions. We would also like to thank Dr. Gordon Rice for assistance with stopped-flow measurements, Professor James Mayer for use of his stopped-flow instrument (UW/NIH Grant GM50422), and Mr. James Law for assistance with low-temperature NMR studies. Support from the NIH (Grant GM 45881) is gratefully acknowledged.
Footnotes
Supporting Information Available: Crystallographic data for compounds 4 (Tables 1–4) and 2 (Tables 5–8), kinetic traces for azide and thiocyanate dissociation from 3 and 4 (Figures S-1 and S-2), variable-temperature 1H NMR for 4 (Figure S-3), and the UV/vis spectrum of 2-4 NH3 bound 1 (Figure S-4) and van't Hoff plots (Figure S-5) (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kobyashi MS. Nat. Biotechnol. 1998;16:733–736. doi: 10.1038/nbt0898-733. [DOI] [PubMed] [Google Scholar]
- 2.Kobyashi M, Nagasawa T, Yamada H. Tibtech. 1992;10:402–408. doi: 10.1016/0167-7799(92)90283-2. [DOI] [PubMed] [Google Scholar]
- 3.Maddrell SJ, Turner NJ, Crosby J. Tetrahedron Lett. 1996;37:6001–6004. [Google Scholar]
- 4.Wyatt JM, Knowles CJ. Int. Biodeterior. Biodegrad. 1995;35:227–248. [Google Scholar]
- 5.Sugiura Y, Kuwahara J, Nagasawa T, Yamada H. J. Am. Chem. Soc. 1987;109:5848–5850. [Google Scholar]
- 6.Nelson MJ, Jin H, Turnser MI, Jr., Grove G, Scarrow RC, Breanna BA, Que L., Jr. J. Am. Chem. Soc. 1991;113:7072–7073. [Google Scholar]
- 7.Nagamune T, Honda J, Kobyashi Y, Sasabe H, Endo I, Ambe F. Hyperfine Interact. 1992;71:1271–1274. [Google Scholar]
- 8.Jin H, Turner IM, Nelson MJ, Gurbiel RJ, Doan PE, Hoffman BM. J. Am. Chem. Soc. 1993;115:5290–5291. [Google Scholar]
- 9.Brennan BA, Cummings JG, Chase DB, Turner MI, Jr., Nelson M. J. Biochemistry. 1996;35:10068–10077. doi: 10.1021/bi960163t. [DOI] [PubMed] [Google Scholar]
- 10.Scarrow RC, Brennan BA, Nelson M. J. Biochemistry. 1996;35:10078–10088. doi: 10.1021/bi960164l. [DOI] [PubMed] [Google Scholar]
- 11.Hauge W, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y. Structure. 1997;5:691–699. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 12.Nagashima SN, Naoshi D, Tsujimura M, Takio K, Odaka M, Yohda M, Kamiya N, Endo I. Nat. Struct. Biol. 1998;5:347–351. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]
- 13.Payne MS, Wu S, Fallon RD, Tudor G, Stieglitz B, Turner IM, Jr., Nelson MJ. Biochemistry. 1997;36:5447–5454. doi: 10.1021/bi962794t. [DOI] [PubMed] [Google Scholar]
- 14.Nojiri M, Nakayama H, Odaka M, Yohda M, Takio K, Endo I. FEBS Lett. 2000;465:173–177. doi: 10.1016/s0014-5793(99)01746-9. [DOI] [PubMed] [Google Scholar]
- 15.Nagamune T, Kurata H, Makoto H, Honda J, Koike H, Ikeuchi M, Inoue Y, Hirata A, Endo I. Biochem. Biophys. Res. Commun. 1990;168:437–442. doi: 10.1016/0006-291x(90)92340-6. [DOI] [PubMed] [Google Scholar]
- 16.Nagamune T, Honda J, Cho W, Kamiya N, Teratini Y, Hirata A, Sasbe H, Endo I. J. Mol. Biol. 1991;220:221–222. doi: 10.1016/0022-2836(91)90006-r. [DOI] [PubMed] [Google Scholar]
- 17.Tsujimura M, Masafumi O, Nagashima S, Yohda M, Endo I. J. Biochem. 1996;119:407–413. doi: 10.1093/oxfordjournals.jbchem.a021256. [DOI] [PubMed] [Google Scholar]
- 18.Odaka M, Noguchi T, Nagashima S, Yohda M, Yabuki S, Hoshino M, Inoue Y, Endo I. Biochem. Biophys. Res. Commun. 1996;221:146–150. doi: 10.1006/bbrc.1996.0560. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi T, Hoshino M, Tsujimura M, Odaka M, Inoue Y, Endo I. Biochemistry. 1996;35:16777–16781. doi: 10.1021/bi961562r. [DOI] [PubMed] [Google Scholar]
- 20.Odaka M, Fujii K, Hoshino M, Noguchi T, Tsujimura M, Nagashima S, Yohda M, Nagamune T, Inoue Y, Endo I. J. Am. Chem. Soc. 1997;119:3785–3791. [Google Scholar]
- 21.Scarrow RC, Stickler BS, Ellison JJ, Shoner SC, Kovacs JA, Cummings JC, Nelson MJ. J. Am. Chem. Soc. 1998;120:9237–9245. [Google Scholar]
- 22.Kopf A, Bonnet D, Artuad I, Pétré D, Mansuy D. Eur. J. Biochem. 1996;240:239–244. doi: 10.1111/j.1432-1033.1996.0239h.x. [DOI] [PubMed] [Google Scholar]
- 23.Doan PE, Gurebiel RJ, Cummins JC, Nelson MJ, Hoffman BM. J. Inorg. Biochem. 1999;74:116. [Google Scholar]
- 24.Shoner SC, Barnhart D, Kovacs JA. Inorg. Chem. 1995;34:4517–4518. [Google Scholar]
- 25.Ellison JJ, Niestedt A, Shoner SC, Barnhart D, Cowen JA, Kovacs JA. J. Am. Chem. Soc. 1998;120:5691–5700. [Google Scholar]
- 26.Schweitzer D, Ellison JJ, Shoner SC, Lovell S, Kovacs JA. J. Am. Chem. Soc. 1998;120:10996–10997. [Google Scholar]
- 27.Noveron JC, Olmstead MM, Mascharak PK. J. Am. Chem. Soc. 1999;121:3553–3554. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 28.Noveron JC, Herradora R, Olmstead MM, Mascharak PK. Inorg. Chim. Acta. 1999;285:269–276.26. [Google Scholar]
- 29.Artaud I, Chatel S, Chauvin AS, Bonnet D, Kopf MA, Leduc P. Coord. Chem. ReV. 1999;190:577–586. [Google Scholar]
- 30.Heinrich L, Li Y, Vaissermann J, Chottard G, Chottard J-C. Angew. Chem., Int. Ed. Engl. 1999;38:3526–3527. doi: 10.1002/(sici)1521-3773(19991203)38:23<3526::aid-anie3526>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 31.Tyler LA, Noveron JC, Olmstead MM, Mascharak PK. Inorg. Chem. 2000;39:357–362. doi: 10.1021/ic990794m. [DOI] [PubMed] [Google Scholar]
- 32.Jackson HL, Shoner SC, Cowen JA, Lovell S, Barnhart D, Kovacs JA. Inorg. Chem. 2000:39. doi: 10.1021/ic001271d. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kung IY, Schweitzer D, Shearer J, Jackson HL, Taylor WD, Lovell S, Kovacs JA. J. Am. Chem Soc. 2000;122:8299–8300. [Google Scholar]
- 34.Schweitzer D, Shearer J, Rittenberg DK, Shoner SC, Ellison JJ, Loloee R, Lovell S, Barnhart D, Kovacs JA. doi: 10.1021/ic0109187. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. p. 104. [Google Scholar]
- 36.Shearer J, Kung IY, Lovell S, Kovacs JA. Inorg. Chem. 2000;39:4998–4999. doi: 10.1021/ic0005689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrin DD, Armarego WLF, Perrin DR. Purification of Laboratory Chemicals. 2nd ed. Pergamon Press; Elmsford, NY: 1980. [Google Scholar]
- 38.Dean JA, editor. Lange's Handbook of Chemistry. Chapter 10. 12th ed. Mcgraw-Hill; New York: 1979. pp. 127–128. [Google Scholar]
- 39.Guidry RM, Drago RS. J. Am. Chem. Soc. 1973;95:6645. doi: 10.1021/ja00801a020. [DOI] [PubMed] [Google Scholar]
- 40.Epley TD, Drago RS. J. Am. Chem. Soc. 1969;91:2883. [Google Scholar]
- 41.Sheldrick GM. SHELEX97. University of Gottingen; Germany: 1997. [Google Scholar]
- 42.Otinowski Z, Minor W. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Nagasawa T, Ryuno K, Yamada H. Biochem. Biophys Res. Commun. 1986;139:1305–1312. doi: 10.1016/s0006-291x(86)80320-5. [DOI] [PubMed] [Google Scholar]
- 44.Brennan BA, Alms G, Nelson MJ, Durney LT, Scarrow RC. J. Am. Chem. Soc. 1996;118:9194–9195. [Google Scholar]
- 45.Based on a survey of the Cambridge Crystallographic Data Base. Average low-spin Co-S bond length: 2.24 Å. Average low-spin Co-N bond length: 2.05 Å.
- 46.In methylene chloride it was shown the rates of formation of azide and thiocyanate bound 1 were first order in both 1 and thiocyanate or azide, indicative of an associative process (data not shown).
- 47.Williams NH, Cheung W, Chin J. J. Am. Chem. Soc. 1998;120:8079–8087. [Google Scholar]
- 48.Elder RC, Kennard GJ, Payne MD, Deutsch E. Inorg. Chem. 1978;17:1296–1303. [Google Scholar]
- 49.Higgs TC, Ji D, Czernuszewicz RS, Matzanke BF, Schunemann V, Trautwein AX, Helliwell M, Ramierez W, Carrano CJ. Inorg. Chem. 1998;37:2383–2392. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.