

Abstract
Targeted deletion of cytoskeletal muscle LIM protein (MLP) in mice consistently leads to dilated cardiomyopathy (DCM) after one or more months. However, next to nothing is known at present about the mechanisms of this process. We investigated whether diastolic performance including passive mechanics and systolic behavior are altered in 2-week-old MLP knockout (MLPKO) mice, in which heart size, fractional shortening and ejection fraction are still normal. Right ventricular trabeculae were isolated from 2-week-old MLPKO and wildtype mice and placed in an apparatus that allowed force measurements and sarcomere length measurements using laser diffraction. During a twitch from the unloaded state at 1 Hz, MLPKO muscles relengthened to slack length more slowly than controls, although the corresponding force relaxation time was unchanged. Active developed stress at a diastolic sarcomere length of 2.00 μm was preserved in MLPKO trabeculae over a wide range of pacing frequencies. Force relaxation under the same conditions was consistently prolonged compared with wildtype controls, whereas time to peak and maximum rate of force generation were not significantly altered. Ca2+ content of the sarcoplasmic reticulum (SR) and the quantities of Ca2+ handling proteins were similar in both genotypes. In summary, young MLPKO mice revealed substantial alterations in passive myocardial properties and relaxation time, but not in most systolic characteristics. These results indicate that the progression to heart failure in the MLPKO model may be driven by diastolic myocardial dysfunction and abnormal passive properties rather than systolic dysfunction.
Keywords: Cytoskeleton, Sarcomere, Myocardial contraction, Relaxation
1. Introduction
It is well established that targeted deletion of cytoskeletal proteins can lead to dilated cardiomyopathy (DCM) and heart failure in mice [1–4]. The homozygous deletion of muscle LIM protein (MLP) is one such case. MLP is primarily located at the Z-disc in striated muscle and in the spectrin-associated membrane complex of the costameres, which link myofibers laterally. It can also appear in the nucleus and was first identified as a regulator of muscle differentiation [5,6]. MLP contains two LIM domains, which are specialized cysteine- and histidine-rich protein–protein interaction domains. MLP knockout (MLPKO) mice show initial signs of DCM at 4 weeks, and the condition worsens to heart failure within several months. MLP interacts with the titin-binding protein T-cap, and mutations in the T-cap binding region of MLP have been found in a subset of patients with DCM.
The pathology of the MLPKO heart failure model has been well characterized in the adult, revealing disruption of the cytoarchitecture [1], increased passive myocardial stiffness [7], decreased ejection fraction [1] and alterations in Ca2+ handling [8,9]. However, the possible contributing or initiating factors of dilation and failure remain unclear. Diastolic causes may include altered passive properties or defects in relaxation, and systolic causes can involve defects in Ca2+ homeostasis or myofilament interaction, while combinations of diastolic and systolic causes are also conceivable.
We have previously identified two features of young MLPKO myocardium before the development of DCM, which indicate early changes in passive behavior: (1) decreased myocardial stiffness in papillary muscles from 2-week-old MLPKO mice and (2) a defect in the stretch-sensing apparatus in isolated cells from MLPKO neonates [10]. These results in conjunction lead us to suggest that the MLP/T-cap/titin molecular complex at the Z-disc may be implicated in mechanical signaling and regulation of passive myocardial properties. Titin may play a key role in this complex and in the development of heart failure in the MLPKO, since it constitutes over 80% of passive force during stretch [11], accounts for a substantial part of viscous forces [12,13] and appears to be involved in mechanical signaling as well (for review see Granzier and Labeit [14]). Direct measurements of sarcomere length can be used to further evaluate passive muscle mechanics in detail and possibly implicate diastolic dysfunction in the progression to DCM.
Interestingly, MLP deficient mice can be rescued by additionally deleting the gene encoding for phospholamban (PLB) [15], which regulates the sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA) activity, or by over-expression of a b-adrenergic receptor kinase inhibitor [16]. Both these methods are based on indirectly enhancing the activity of SERCA and thus improving Ca2+ handling by the SR during contraction and relaxation. Although the findings may appear to implicate contractile dysfunction as one of the primary defects of the disease, it is also possible that artificially improving SR function compensates for systolic dysfunction in the adult, whereas the initiating factor of DCM lies elsewhere.
The aim of this study was to investigate diastolic function, including passive properties and relaxation, as well as systolic function, including active stress and force generation characteristics, in young MLPKO hearts before the onset of DCM, to possibly identify the primary causes of the disease. We observed substantial alterations in passive behavior and relaxation time but no significant changes in systolic mechanics, suggesting diastolic dysfunction as the main contributor to DCM in this genetically engineered model.
2. Methods
2.1. Trabecula mechanical testing
A total of 33 MLPKO mice and 43 isogenic wildtype control mice aged 12–17 days were used for this entire study. The experimental procedures were performed according to the guidelines of the University of Calgary and the University of California, San Diego. The mice were anesthetized using ether or isoflurane, and a cervical dislocation was performed. In one group of mice of both genotypes, heart and lung were excised and weighed. Lung dry weight was obtained after drying at 60 °C for 24 h. A second group was used for trabecula experiments, which were performed as described in detail in Stuyvers et al. for adult mice [17]. In short, the heart was quickly excised and retrogradely perfused with oxygenated hyperkalemic HEPES buffer solution at room temperature. The right ventricle was opened and a suitable trabecula identified which was preferably wide (>200 μm) but thin (<100 μm) and unbranched. The muscle was transferred to a small bath and mounted horizontally between a force transducer and a micromanipulator for muscle isometric twitch measurements. During an equilibration period of at least 1 h, the muscle was held at a medium stretch (SLpassive: ~ 2.1 μm) and was field stimulated at 1 Hz using 2 ms square pulses delivered by platinum electrodes. The entire experiment was performed while continuously superfusing the trabecula with oxygenated HEPES buffer solution (Ca2+ concentration: 2.0 mM) at room temperature. Sarcomere length was measured in the muscle center using laser diffraction as previously described [18,19].
Immediately after the equilibration period, the trabecula was completely unloaded, and SL was measured in the center of the muscle. For measurements of the stress–frequency relationship (SFR) and post-rest potentiation, the passive SL was kept constant at 2.00 μm. SFR data were obtained by randomly varying the stimulation frequency between 0.1 and 4 Hz. At each frequency, actively developed stress was allowed several minutes to stabilize before recording force. Post-rest potentiation was measured as the immediate force increase after reducing the stimulation frequency from the reference frequency of 1 Hz to 0.33, 0.20 or 0.10 Hz. The rest interval was thus varied between 3 and 10 s. On completion of the protocol, the muscle was loosened from the attachments. Width and thickness of the trabecula were measured under a dissection scope equipped with a reticle eyepiece for conversion of force to stress. In a third group of mice, trabeculae were isolated as described above and allowed to equilibrate for at least 30 min at 0.5 Hz and an approximate length of 90% of Lmax. Rapid cooling contractures were obtained by replacing the superfusate with ice-cold solution after a rest period of 15 s and compared with the steady-state twitch force at 0.5 Hz.
2.2. Data analysis
For every data point, 15–20 consecutive raw data cycles of the force and SL tracings were averaged using Matlab 6.5.0. SL tracings of shortening were also smoothed and normalized by the slack length. Subsequently, times to different fractions of shortening and relengthening were quantified. Force time-course parameters were determined after normalizing the averaged cycle by peak force. Finally, the relaxation time constant τ was determined by fitting a two-parameter exponential to the relaxation curve between the inflection point and the end of the cycle.
2.3. Immunoblotting of Ca2+ handling-related proteins
In order to investigate Ca2+ handling at the protein level, quantitative immunoblotting of cardiac SERCA, PLB and the Na+/Ca2+ exchanger was carried out in hearts from 2-week-old MLPKO and wildtype mice. The procedures used are described in detail in Harrer et al. [20]. Briefly, frozen ventricles from 2-week-old wildtype and MLPKO mice were homogenized with Polytron homogenizer (Blinkman Instruments, Westbury, NY) in 1.5 ml buffer containing 10 mM imidazole (pH 7.0), 300 mM sucrose, 1.0 mM dithiothreitol and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Protein concentration was determined by BCA protein assay kit (Pierce Biotechnology, Rockford, IL) using bovine serum albumin as a standard. Ten micrograms of proteins were separated by SDS-PAGE on 4–12% Bis–Tris gel (Invitrogen, Carlsbad, CA) and electrotransferred to PVDF membrane (BIO-RAD Laboratories, Hercules, CA). Blots were analyzed with antibodies against PLB (combined pentameric and monomeric forms, 1:1000, Upstate, Lake Placid, NY), Ser16-phosphorylated PLB (combined pentameric and monomeric forms, 1:3000, Upstate), SERCA-2 (1:1000, Affinity Bioreagents, Golden, CO) or Na+/Ca2+ exchanger 1 (1:100, Chemicon, Temecula, CA). Binding of the primary antibody was detected by peroxidase-conjugated secondary antibodies and visualized with the SuperSignal West Pico substrate kit (Pierce Biotechnology, Rockford, IL).
2.4. Statistical analysis
Data are expressed as mean ± S.E.M. Statistical comparisons were made by two-way ANOVA corrected for repeated measures followed by Fisher’s PLSD post-hoc analysis for all data obtained over a range of frequencies as well as the shortening and relengthening times from the unloaded state. Weights, unloaded SL and force time-course data were analyzed using unpaired two-tailed Student’s t-test. Differences of P < 0.05 were considered significant.
3. Results
Data from four of the MLPKO trabeculae were excluded from any active force analysis due to early spontaneous activity and triggered propagated contractions (or calcium waves). In all wildtype muscles, calcium waves did not set in for several hours and thus did not impede muscle viability during recording.
The age of the mice used was not statistically different between the groups (MLPKO: 13.8 ± 0.3 d, n = 11, wildtype: 14.5 ± 0.5 d, n = 14, P = 0.27). Body, heart and lung weights of 2-week-old MLPKO mice and wildtype controls are listed in Table 1. Heart weight and outer heart diameter, which was measured from an image taken from the freshly isolated heart immediately after excision, were not different between the genotype groups (P > 0.8, P > 0.9, respectively). Heart weight to body weight ratio was greater in the MLPKO mice (P < 0.001), which is clearly due to a smaller body weight compared with the controls. No difference was found in lung wet to dry weight ratio (P > 0.4), confirming that the MLPKO mice had not developed congestive heart failure at this age.
Table 1.
Weights and dimensions in 2-week-old MLPKO and wildtype mice
| Body weight (g) | Heart diameter (mm) | Heart weight (mg) | Heart weight/body weight (mg g−1) | Lung wet/dry weight | |
|---|---|---|---|---|---|
| Wildtype | 8.70 ± 0.22 | 4.77 ± 0.12 | 57.2 ± 1.8 | 6.58 ± 0.11 | 4.39 ± 0.16 |
| MLPKO | 6.84 ± 0.31 | 4.76 ± 0.08 | 56.5 ± 2.4 | 8.30 ± 0.16 | 4.59 ± 0.17 |
| P-value | P < 0.001 | NS | NS | P < 0.001 | NS |
Values are mean ± S.E.M.
3.1. Shortening from the unloaded length during a muscle isometric twitch
Fig. 1A shows the average time-course of sarcomere shortening from the unloaded length during a muscle isometric twitch at a stimulation rate of 1 Hz (wildtype: n = 8, MLPKO: n = 4, unless otherwise stated). The resting unloaded SL was significantly shorter in MLPKO trabeculae than in wildtype muscles (1.847 ± 0.008 μm in wildtype, n = 9, vs. 1.790 ± 0.015 μm in MLPKO, n = 6, P < 0.005). The amount of maximum shortening in percent of slack length was not significantly different between MLPKO and wildtype muscles (MLPKO: 5.95 ± 0.65%, wildtype: 6.96 ± 0.57%, P > 0.3). To analyze the time-course of shortening, the average time required for different amounts of relative shortening and relengthening was measured (Fig. 1B, C). Although the times for 1–4% shortening were consistently lower in the MLPKO than wildtype muscles, this difference was not statistically significant (P > 0.6), and time to maximum shortening was not statistically different between the groups either (MLPKO: 136.0 ± 31.2 ms, wildtype: 177.8 ± 13.6 ms, P = 0.18). The statistical power for these shortening characteristics was too low (<30%), however, to conclude with certainty that there was no relevant difference. In contrast, MLPKO trabeculae relengthened significantly more slowly than the control muscles (P = 0.04), while the relaxation time of the corresponding force time-course was not significantly different (P > 0.1, data not shown).
Fig. 1.

(A) Average time-course of shortening from unloaded length in both genotypes. (B) Time to different amounts of shortening expressed as percent shortening, and (C) time to different amounts of relengthening from peak shortening in MLPKO and wildtype muscles. The asterisk indicates a significant difference (P < 0.05) between the genotypes.
3.2. Twitch behavior during stretch
Fig. 2 shows the frequency-dependent behavior of trabeculae from both genotypes at a moderate stretch (MLPKO: n = 5, wildtype: n = 9). The figure is divided into systolic parameters (left panels) and diastolic parameters (right panels). In both genotypes, we observed a flat SFR in the range between 0.1 and 1 Hz (Fig. 2A). At higher frequencies, the SFR is generally positive, although at 3–4 Hz, active stress is decreased. There was no statistical difference, however, between the genotype groups. The maximum rate of force development (maximum dF/dt) increased (Fig. 2B), and time to peak force decreased with frequency in both genotypes (Fig. 2C). This behavior is to be expected, since the twitch shortens with frequency. We observed little difference between MLPKO and wildtypes in systolic stress and both characteristics of the systolic twitch dynamics (P = 0.59, P = 0.26, P = 0.82, respectively). Statistical power of these systolic parameters was sufficiently high (>90%) to suppose that the existing differences are most likely too small to be considered relevant in the development of cardiomyopathy (assuming physiologically relevant differences of 2.5 kPa, 2 ms−1 and 10 ms, respectively).
Fig. 2.

Systolic (A–C) and diastolic (D–F) behavior of MLPKO (○) and wildtype trabeculae (●) at a passive sarcomere length of 2.00 μm over the entire range of frequencies tested. (A) Absolute active developed stress. (B) Maximum dF/dt (maximum rate of force development), normalized by peak force. (C) Time from stimulus to peak force. (D) Diastolic stress (difference from minimum stress of all frequencies). (E) Minimum dF/dt (maximum rate of relaxation), normalized by peak force. (F) Relaxation time constant τ, computed from a fitted exponential to the relaxation time-course after the inflection point. The asterisks denote significant differences (P < 0.05) between the genotype groups.
The diastolic stress is plotted against frequency in Fig. 2D. Δ stress denotes the difference from the minimum stress occurring in the entire range of frequencies. In both genotypes, diastolic stress equaled minimum stress at low frequencies but increased at higher frequencies. In MLPKO this increase was greater than in controls, although over the entire range of frequencies this difference did not reach statistical significance (P > 0.4). Minimum dF/dt (maximum rate of relaxation, Fig. 2E) was consistently lower in MLPKO across the entire range of frequencies (P < 0.05). In addition, the relaxation time constant (Fig. 2F) was significantly elevated in MLPKO (P < 0.0005). The steep decrease in τ with frequency, however, shows that the MLPKO muscles do demonstrate frequency-dependent acceleration of relaxation. In fact, relative acceleration of relaxation from 0.1 to 4 Hz was somewhat greater in MLPKO than wildtypes (61% vs. 48% acceleration, respectively), although this difference was not significant.
3.3. Post-rest potentiation and rapid cooling contracture
Although at steady state, trabeculae did not show a substantial increase in force with decreased frequency, they did demonstrate typical post-rest behavior: Fig. 3A shows a representative transient force development of a trabecula from a young knockout mouse after decreasing the stimulation frequency from 1 to 0.2 Hz (or after 5 s of rest). After the first full rest interval, an initial increase in stress compared with 1 Hz was observed (post-rest potentiation). In the following beats the developed stress gradually weakened and eventually reached a steady-state level. The initial increase in force is dependent on the rest interval. Fig. 3B illustrates the average post-rest potentiation of force in the two genotypes as a function of rest time. While the post-rest potentiation was positive in all cases and increased with rest interval in both groups, it was significantly lower in MLPKO than in wild-type controls (P = 0.02, MLPKO n = 6, wildtype n = 7).
Fig. 3.

Post-rest potentiation and rapid cooling contracture. (A) Representative post-rest stress recording from a knockout muscle. The pacing frequency was switched from 1 to 0.2 Hz. The length of the first rest interval (shaded area) varied somewhat due to manual switch of frequency. (B) Average maximum post-rest potentiation in MLPKO (○) and wildtype trabeculae (●) at a passive sarcomere length of 2.00 μm. Post-rest potentiation is shown for different rest intervals. The asterisks indicate a significant difference (P < 0.05) between the genotypes. (C) Representative rapid cooling measurement in an MLPKO trabecula. The force drop occurred due to temporary drainage of the bath. The muscle contracted with exposure to ice-cold solution due to Ca2+ release from the SR. Peak force occurred after 2.5 s, after which the muscle slowly relaxed. (D) Amplitude of rapid cooling contracture relative to the steady-state developed force at 0.5 Hz. The ratio was slightly greater than one and not different between the genotypes, suggesting similar SR Ca2+ content.
Fig. 3C illustrates a representative measurement of a rapid cooling contracture in an MLPKO trabecula. After the stimulation was switched off, the bath was drained, causing the sharp drop in force. Refilling the bath with ice-cold solution restored the force and caused the trabecula to contract due to Ca2+ release from the SR. The contracture reached its peak typically after 2–4 s and declined to the resting state over a period of 15 s. The amplitude of contracture relative to the steady-state twitch force at 0.5 Hz was similar in both genotypes, as shown in Fig. 3D (P = 0.8, MLPKO n = 9, wildtype n = 11).
3.4. Expression of Ca2+ handling-related proteins
To possibly explain the differences in relaxation rate and post-rest potentiation, we also investigated whether changes in protein levels of PLB, Ser16-phosphorylated PLB, SERCA, and the Na+/Ca2+ exchanger occur in the 2-week-old MLPKO heart. Independent measurements in freshly isolated hearts from six young wildtype and seven MLPKO mice indicated that the quantities of all four proteins were similar in both genotypes (Fig. 4, PLB: P = 0.9, PLB-p: P = 0.2, SERCA: P = 0.8, Na+/Ca2+ exchanger: P = 0.6 from densitometry).
Fig. 4.
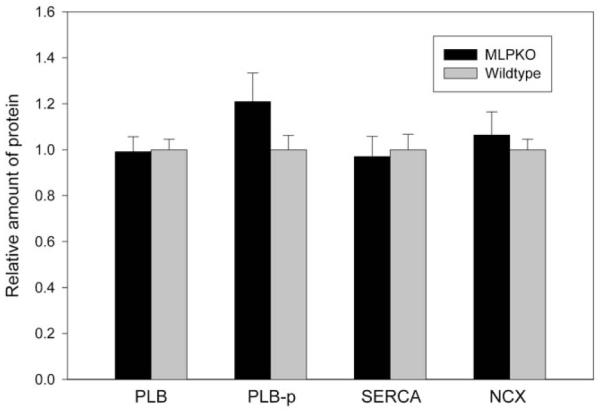
Densitometry data obtained from Western blots of the Ca2+ handling-related proteins phospholamban (PLB), Ser16-phosphorylated PLB (PLB-p), SERCA-2 and the Na+/Ca2+ exchanger (NCX). No statistical difference was detected in the quantity of any of these proteins between the MLPKO and wildtype controls (MLPKO n = 7, wildtype n = 6).
4. Discussion
We investigated the unloaded shortening time-course, systolic and diastolic stresses, post-rest potentiation, rapid cooling contractures as well as quantities of Ca2+ handling proteins in young MLPKO mice, to determine whether systolic or diastolic changes are already present in young MLPKO myocardium and may thus contribute to the later progression to DCM.
4.1. Systolic mechanics
We did not find differences in the SFR, time to peak stress and maximum rate of force generation between the genotype groups. This suggests that myofilament interaction and the ability to develop systolic stress may not be inherently impaired in young MLPKO myocardium.
It is striking that we found a flat SFR at lower frequencies than 1 Hz in both genotypes, since this stands in contrast to the steeply negative SFR in this range of frequencies found recently in adult mice by Stuyvers et al. [17]. It is likely that the flat relationship at low frequencies is due to the young age of the mice used. In rats, the SR develops within the first 4–5 weeks after birth [21–23], and the Ca2+ transient is mainly generated by Ca2+ current through sarcolemmal channels at this developmental stage, mostly bypassing the SR. This is supported by the finding that in the first week after birth in rats, expression of the Na+/Ca2+ exchanger is higher than in adults [24]. If this was also the case in young mice, the Na+/Ca2+ exchanger may be dominating over the SERCA pump and would extrude more calcium at low frequencies than can be resequestered by the SR, consequently depleting the SR of calcium during each contraction and leading to low active stresses at steady state. In any case, this behavior was found independent of the genotype.
At frequencies above 1 Hz, we mainly found a positive SFR, as reported before under the same conditions in trabeculae from adult mice [17]. Above 3–4 Hz, active stress began to decline in both genotypes. This may have two reasons. Firstly, the oxygen demand of the muscle likely rose above the oxygen supply by diffusion especially in relatively thick muscles, leading to a hypoxic core, as suggested previously by Stuyvers et al. [17]. In fact, the MLPKO muscles were thicker on average than the wildtype muscles, and although this difference was not statistically significant (122 ± 26 vs. 85 ± 7 μm, P > 0.1), it may account for the small difference in SFR at high frequencies between the genotypes. Secondly, the time between stimuli likely became too short for complete relaxation, leading to an increase in cytosolic Ca2+ concentration at end-diastole, which explains the observed slight increase in end-diastolic stress at high frequencies as well as the decrease in active stress.
4.2. Diastolic mechanics
Our results show that young MLPKO trabeculae relengthened more slowly to their resting state after contraction from an unloaded length than the wildtype controls, while the corresponding force time-course is not altered accordingly. This suggests that the slower relengthening, as well as slightly faster shortening, may not result from altered calcium handling or myofilament interaction, but rather may be a reflection of abnormal passive visco-elastic material properties. Given the importance of titin in static and dynamic passive myocardial mechanics [14,25] as well as its indirect association with MLP, it stands to reason that altered titin function may underlie differences in passive properties. We propose a visco-elastic model and a wormlike chain model of titin function to investigate this idea further (see Section 4.5). The analysis shows that the observed changes in sarcomere length, shortening time-course and passive stiffness in the MLPKO can all be explained by possible titin dysfunction.
In our previous study, we found that a mechanism leading to cardiomyopathic changes in the myocardium of MLPKO mice may stem from an inherent inability to sense mechanical stretch stimuli with defects in the cardiac Z-disc/titin macromolecular complex [10]. Data from the current study suggest that the alterations in passive muscle mechanics in the MLPKO mouse may also directly contribute to chamber dilation, an increase in wall stress and the deterioration of cardiac function. We speculate that the stretch-sensing defect and newly-identified abnormal mechanostructural properties may both contribute to the disease process in this cardiomyopathy mouse model, and both abnormalities may be explained by the indirect association of MLP with titin via T-cap.
We also observed a significant impairment of force relaxation in stretched MLPKO muscles compared with control, in contrast to the normal force time-course in the unloaded state. Both the maximum rate of relaxation and the relaxation time constant were significantly altered over the entire range of frequencies tested. Relaxation time is thought to be regulated mainly by SERCA activity [26,27] and myofilament properties (reviewed in Hunter [28]). Quantitative immunoblotting showed similar quantities of SERCA, PLB, phosphorylated PLB and the Na+/Ca2+ exchanger in both genotypes at this early age. It appears, therefore, that the activity of the major Ca2+ handling proteins may be excluded as a source of prolonged relaxation. Instead, myofilament properties themselves may be a rate-limiting factor of relaxation, particularly under our experimental conditions of low temperatures, low frequencies and higher stretches [28]. This may explain why the MLPKO muscles showed prolonged relaxation only at a moderate stretch but not in the unstretched state. Furthermore, the structural sarcomeric disruption that is characteristic for the young MLPKO, together with our supposition of normal Ca2+ cycling by the major Ca2+ handling proteins, supports the idea that altered passive recoil properties of the myofilaments may produce the prolonged relaxation in MLP deficiency. On the other hand, the substantially lower post-rest potentiation in the MLPKO muscles seems to suggest that SR Ca2+ cycling may be impaired, since post-rest potentiation is often regarded as a measure of SR Ca2+ load [29,30]. However, it is also plausible that most of the Ca2+ responsible for post-rest force entered the cytoplasm through the L-type channel, since at this early age the SR may not be fully developed. To further investigate the relative contribution of SR Ca2+ cycling and myofilament properties in prolonged relaxation, we also performed rapid cooling in the trabeculae, which causes a sudden release of calcium from the SR and appears to be a more reliable measure of SR Ca2+ content [31–33]. Although most investigators report a long-lasting contracture with rapid cooling in larger species, a quick relaxation within seconds, as we have observed, seems to be characteristic for mouse trabeculae [34]. In our study, the amplitude of the rapid cooling contracture relative to the steady-state twitch force were similar in both genotypes, further supporting the view that SR Ca2+ cycling may not be altered in MLP deficiency. However, direct Ca2+ measurements in MLPKO myocytes would be necessary to completely resolve this issue, because we cannot rule out the possibilities that Ca2+ buffering may be affected by MLP disruption or that Ca2+ currents may also be indirectly altered, since MLP’s binding partner T-cap is associated with a stretch-regulated potassium channel [35].
The heart weight to body weight ratio was higher in MLPKO than in wildtype mice. This could be an indication of hypertrophy or even cardiomyopathy. However, absolute heart weight was not different between the groups. In the case of heart failure even at this young age, one would expect the lung wet to dry weight ratio to be higher in the MLPKO due to water accumulation in the lungs. That this is not the case in MLPKO supports the view that no serious cardiac dysfunction is present in 2-week-old MLPKO mice. The penetrance of the cardiomyopathic phenotype was reported as almost 100% [1]. Therefore, we could be confident that the 2-week-old MLPKO mice used would have developed DCM at a later time.
4.3. Limitations
It is important to note that our experimental conditions differed from the in vivo environment in several aspects: Firstly, trabecula testing was performed in near absence of catecholamines. This was confirmed by adding 1 μM of the beta-blocker propranolol to the muscle superfusate, which had a negligible effect on developed force and relaxation rate. Secondly, all experiments were conducted at room temperature. Both catecholamines and higher temperature have a positive lusitropic effect, which may compensate for the impairment in relaxation time. Further experiments are necessary to clarify the possible involvement of MLP in relaxation.
The time-course of shortening in the MLPKO around the time of maximum shortening appeared flat and noisier than in the control muscles (average shown in Fig. 1A), such that maximum shortening was difficult to determine accurately. This may be associated with the characteristic misregistration of sarcomeres observed in MLPKO myocardium [1], which would likely increase the zeroth-order artifact in the laser diffraction pattern. Indeed, the diffraction pattern in MLPKO muscles was wider and often showed several peaks (data not shown), possibly representing groups of sarcomeres at slightly different lengths, giving rise to a noisier shortening time-course. As another possible explanation, the SL reaches such low levels in the MLPKO muscles that the thick filaments may even abut on the Z-discs (at approximately 1.69 μm), especially since the Z-discs are abnormally wide [10].
4.4. Summary
This study presents evidence that progression to DCM may be initiated by diastolic dysfunction rather than systolic function in the case of the MLPKO model. Systolic stress and force generation times were preserved in the MLPKO, suggesting that active force transmission is normal. Passive force transmission, as one main determinant of diastolic function, may play a key role in the progression of DCM, since we observed an altered shortening time-course from slack length, in addition to decreased stiffness. These changes may be mediated by the elastic properties of titin. In addition, relaxation time was prolonged in MLPKO mice, suggesting abnormal Ca2+ cycling or an alteration in myofilament properties. Since SR Ca2+ content and the amounts of Ca2+ handling proteins were not altered, it is likely that myofilament interaction may be responsible for the increase in relaxation time.
4.5. Appendix: modeling the visco-elastic behavior in the MLPKO sarcomere
To investigate the mechanical significance of the observed differences in shortening behavior, we considered a model of titin function proposed by Helmes et al. [25]. The giant protein titin has been identified as a major determinant of passive visco-elasticity in cardiac muscle. Since MLP is indirectly associated with titin through the titin-binding protein T-cap [10], a reduction of elastic stiffness of I-band titin may ensue from MLP deficiency and lead to decreased passive forces during stretch as well as decreased restoring forces during relengthening in MLPKO muscles. We used a mathematical visco-elastic model with four elements (Fig. 5A) to test the hypothesis that the difference in shortening behavior may be explained by the previously reported decrease in elastic stiffness [10]. k1 and η represent the elastic and viscous behavior, respectively, of the shortening muscle center, while k2 represents the stiffness of the lengthening muscle ends. The input force generated by the contractile element was simulated as the average force time-course measured in all muscles. k1 was chosen for both wildtype and MLPKO directly from the passive stress–strain relationship [10]. k2 and η were adjusted to match the measured wildtype shortening time-course (Fig. 5B) as far as possible. To reconstruct maximum shortening, the shortening data were fitted using a third order polynomial, and relengthening was approximated by an exponential in this plot. The prescribed k1 for the MLPKO, reduced by 27% compared with the wildtype, lead to slightly faster shortening and slower relengthening (Fig. 5C), as seen experimentally. Thus, the differences in shortening time-course in MLPKO myocardium are consistent with the reduction in elastic stiffness we previously reported.
Fig. 5.

Model of the shortening behavior from the unloaded length. (A) Four-element mathematical visco-elastic model of a contracting isolated muscle. The spring constant k1 and dashpot constant η represent the visco-elastic behavior of the shortening part of the muscle, spring constant k2 represents the lengthening muscle ends. The contractile force FA was prescribed using the average measured force. (B) Fitted experimental data of the time-course of shortening in wildtype and MLPKO trabeculae. (C) Model of a normal (wildtype) and an MLPKO shortening time-course. The two curves were produced using values for k1 taken directly from the passive stress–strain relationship. (D) Wormlike chain model of titin-based force in wildtype and knockout muscles, compared with the measured stress–strain relationship. For further details see text.
Since passive visco-elastic behavior is largely determined by titin, we further tested whether titin dysfunction in the MLPKO may explain both the decrease in stiffness and the decrease in SL. We utilized a wormlike chain model of titin previously developed [36,37] with contour lengths and persistence lengths of normal titin taken from the literature. The MLPKO model was based on the assumption that the tight interaction between actin and titin near the Z-disc is disturbed, since it was observed that SL and stiffness are decreased after complete actin extraction from single myocytes [36]. If we assume a partial detachment of titin from actin, thus increasing the contour length of the free titin segment and decreasing the contour length of the attached segment by the amount according to the observed decrease in SL, the new configuration shows a decrease in stiffness quantitatively consistent with our experimental results (Fig. 5D). Titin-based force and measured stress were plotted against strain using an estimated scaling factor based on the finding that, in mice, titin is responsible for >80% of stress at low strains [11]. The proposed model of titin-based passive dysfunction is also consistent with the possibility that active force transmission may be normal in the case of MLP deficiency: while in the absence of cross-bridge activity, I-band titin is largely responsible for passive forces during stretch above and contraction below slack length, cross-bridge formation could bypass the function of I-band titin, and active forces may be transmitted directly from the contractile filaments to the Z-disc.
Acknowledgments
This work was supported by the National Institutes of Health (grants HL64321, HL43026, HL53773), the National Science Foundation (grant BES-0086482) and the Canadian Institutes of Health Research. H. ter Keurs is supported as a Medical Scientist by the Alberta Heritage Foundation for Medical Research.
References
- [1].Arber S, Hunter JJ, Ross J, Jr., Hongo M, Sansig G, Borg J, et al. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88(3):393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- [2].Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, et al. Disruption of the sarcoglycan–sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98(4):465–74. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- [3].Pashmforoush M, Pomies P, Peterson KL, Kubalak S, Ross J, Jr., Hefti A, et al. Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med. 2001;7(5):591–7. doi: 10.1038/87920. [DOI] [PubMed] [Google Scholar]
- [4].Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol. 1997;29(8):2107–24. doi: 10.1006/jmcc.1997.0446. [DOI] [PubMed] [Google Scholar]
- [5].Arber S, Halder G, Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79(2):221–31. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- [6].Kong Y, Flick MJ, Kudla AJ, Konieczny SF. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol. 1997;17(8):4750–60. doi: 10.1128/mcb.17.8.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Omens JH, Usyk TP, Li Z, McCulloch AD. Muscle LIM protein deficiency leads to alterations in passive ventricular mechanics. Am J Physiol Heart Circ Physiol. 2002;282(2):H680–H687. doi: 10.1152/ajpheart.00773.2001. [DOI] [PubMed] [Google Scholar]
- [8].Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ, et al. Cellular and functional defects in a mouse model of heart failure. Am J Physiol Heart Circ Physiol. 2000;279(6):H3101–H3112. doi: 10.1152/ajpheart.2000.279.6.H3101. [DOI] [PubMed] [Google Scholar]
- [9].Su Z, Yao A, Zubair I, Sugishita K, Ritter M, Li F, et al. Effects of deletion of muscle LIM protein on myocyte function. Am J Physiol Heart Circ Physiol. 2001;280(6):H2665–H2673. doi: 10.1152/ajpheart.2001.280.6.H2665. [DOI] [PubMed] [Google Scholar]
- [10].Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111(7):943–55. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- [11].Wu Y, Cazorla O, Labeit D, Labeit S, Granzier H. Changes in titin and collagen underlie diastolic stiffness diversity of cardiac muscle. J Mol Cell Cardiol. 2000;32(12):2151–62. doi: 10.1006/jmcc.2000.1281. [DOI] [PubMed] [Google Scholar]
- [12].Kulke M, Fujita-Becker S, Rostkova E, Neagoe C, Labeit D, Manstein DJ, et al. Interaction between PEVK-titin and actin filaments: origin of a viscous force component in cardiac myofibrils. Circ Res. 2001;89(10):874–81. doi: 10.1161/hh2201.099453. [DOI] [PubMed] [Google Scholar]
- [13].Linke WA, Fernandez JM. Cardiac titin: molecular basis of elasticity and cellular contribution to elastic and viscous stiffness components in myocardium. J Muscle Res Cell Motil. 2002;23(5–6):483–97. doi: 10.1023/a:1023462507254. [DOI] [PubMed] [Google Scholar]
- [14].Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94(3):284–95. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- [15].Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, et al. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99(3):313–22. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- [16].Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr., et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;95(12):7000–5. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stuyvers BD, McCulloch AD, Guo J, Duff HJ, ter Keurs HE. Effect of stimulation rate, sarcomere length and Ca(2+) on force generation by mouse cardiac muscle. J Physiol. 2002;544(Pt 3):817–30. doi: 10.1113/jphysiol.2002.024430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kentish JC, ter Keurs HE, Ricciardi L, Bucx JJ, Noble MI. Comparison between the sarcomere length–force relations of intact and skinned trabeculae from rat right ventricle. Influence of calcium concentrations on these relations. Circ Res. 1986;58(6):755–68. doi: 10.1161/01.res.58.6.755. [DOI] [PubMed] [Google Scholar]
- [19].van Heuningen R, Rijnsburger WH, ter Keurs HE. Sarcomere length control in striated muscle. Am J Physiol. 1982;242(3):H411–H420. doi: 10.1152/ajpheart.1982.242.3.H411. [DOI] [PubMed] [Google Scholar]
- [20].Harrer JM, Kiss E, Kranias EG. Application of the immunoblot technique for quantitation of protein levels in cardiac homogenates. Biotechniques. 1995;18(6):995–8. [PubMed] [Google Scholar]
- [21].Vornanen M. Excitation–contraction coupling of the developing rat heart. Mol Cell Biochem. 1996;163–164:5–11. doi: 10.1007/BF00408635. [DOI] [PubMed] [Google Scholar]
- [22].Gomez JP, Potreau D, Raymond G. Intracellular calcium transients from newborn rat cardiomyocytes in primary culture. Cell Calcium. 1994;15(4):265–75. doi: 10.1016/0143-4160(94)90066-3. [DOI] [PubMed] [Google Scholar]
- [23].Bassani RA, Bassani JW. Contribution of Ca(2+) transporters to relaxation in intact ventricular myocytes from developing rats. Am J Physiol Heart Circ Physiol. 2002;282(6):H2406–H2413. doi: 10.1152/ajpheart.00320.2001. [DOI] [PubMed] [Google Scholar]
- [24].Koban MU, Moorman AF, Holtz J, Yacoub MH, Boheler KR. Expressional analysis of the cardiac Na–Ca exchanger in rat development and senescence. Cardiovasc Res. 1998;37(2):405–23. doi: 10.1016/s0008-6363(97)00276-9. [DOI] [PubMed] [Google Scholar]
- [25].Helmes M, Trombitas K, Granzier H. Titin develops restoring force in rat cardiac myocytes. Circ Res. 1996;79(3):619–26. doi: 10.1161/01.res.79.3.619. [DOI] [PubMed] [Google Scholar]
- [26].Bassani RA, Mattiazzi A, Bers DM. CaMKII is responsible for activity-dependent acceleration of relaxation in rat ventricular myocytes. Am J Physiol. 1995;268(2 Pt 2):H703–H712. doi: 10.1152/ajpheart.1995.268.2.H703. [DOI] [PubMed] [Google Scholar]
- [27].Li L, Chu G, Kranias EG, Bers DM. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am J Physiol. 1998;274(4 Pt 2):H1335–H1347. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- [28].Hunter WC. Role of myofilaments and calcium handling in left ventricular relaxation. Cardiol Clin. 2000;18(3):443–57. doi: 10.1016/s0733-8651(05)70155-7. [DOI] [PubMed] [Google Scholar]
- [29].Pieske B, Maier LS, Schmidt-Schweda S. Sarcoplasmic reticulum Ca2+ load in human heart failure. Basic Res Cardiol. 2002;97(Suppl 1):I63–I71. doi: 10.1007/s003950200032. [DOI] [PubMed] [Google Scholar]
- [30].Maier LS, Bers DM, Pieske B. Differences in Ca(2+)-handling and sarcoplasmic reticulum Ca(2+)-content in isolated rat and rabbit myocardium. J Mol Cell Cardiol. 2000;32(12):2249–58. doi: 10.1006/jmcc.2000.1252. [DOI] [PubMed] [Google Scholar]
- [31].Bers DM, Bassani RA, Bassani JW, Baudet S, Hryshko LV. Paradoxical twitch potentiation after rest in cardiac muscle: increased fractional release of SR calcium. J Mol Cell Cardiol. 1993;25(9):1047–57. doi: 10.1006/jmcc.1993.1117. [DOI] [PubMed] [Google Scholar]
- [32].Hryshko LV, Stiffel V, Bers DM. Rapid cooling contractures as an index of sarcoplasmic reticulum calcium content in rabbit ventricular myocytes. Am J Physiol. 1989;257(5 Pt 2):H1369–H1377. doi: 10.1152/ajpheart.1989.257.5.H1369. [DOI] [PubMed] [Google Scholar]
- [33].Bluhm WF, Lew WY. Sarcoplasmic reticulum in cardiac length-dependent activation in rabbits. Am J Physiol. 1995;269(3 Pt 2):H965–H972. doi: 10.1152/ajpheart.1995.269.3.H965. [DOI] [PubMed] [Google Scholar]
- [34].Stull LB, Leppo MK, Marban E, Janssen PM. Physiological determinants of contractile force generation and calcium handling in mouse myocardium. J Mol Cell Cardiol. 2002;34(10):1367–76. doi: 10.1006/jmcc.2002.2065. [DOI] [PubMed] [Google Scholar]
- [35].Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, et al. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001;313(4):775–84. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- [36].Granzier H, Kellermayer M, Helmes M, Trombitas K. Titin elasticity and mechanism of passive force development in rat cardiac myocytes probed by thin-filament extraction. Biophys J. 1997;73(4):2043–53. doi: 10.1016/S0006-3495(97)78234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Trombitas K, Freiburg A, Centner T, Labeit S, Granzier H. Molecular dissection of N2B cardiac titin’s extensibility. Biophys J. 1999;77(6):3189–96. doi: 10.1016/S0006-3495(99)77149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]