

Abstract
Leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) is a novel mitochondrial disease caused by mutations in EARS2, which encodes the mitochondrial glutamyl-tRNA synthetase (mtGluRS). A distinctive brain MRI pattern is the hallmark of the disease.
A 6-year-old boy presented at 3 months with feeding difficulties and muscle hypotonia. Brain MRI, at 8 months, showed hyperintensity of the deep cerebral and cerebellar white matter, thalamus, basal ganglia, brainstem, and thin corpus callosum. From the second year of life onward, the child reported global clinical improvement, parallel to partial resolution of brain MRI pattern. However, the last neuroimaging assessment revealed novel lesions within the left caudate and pallidum nuclei. DNA genomic sequencing analysis identified a novel EARS2 mutation.
This case expands the clinical and neuroradiological phenotype of LTBL presenting intermediate clinical manifestations between the severe and milder forms of the disease and previously unreported brain MRI features.
Introduction
Leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) has been recently defined as a novel mitochondrial disease condition characterized by early onset of neurologic symptoms, a biphasic clinical course, and peculiar neuroimaging (Steenweg et al. 2012a).
LTBL is caused by mutations in EARS2 gene, encoding mitochondrial glutamyl-tRNA synthetase (mtGluRS) (Steenweg et al. 2012a).
We identified a novel EARS2 mutation in a patient whose clinical and brain MRI features expand the phenotypic spectrum of LTBL.
Case Report
A 6-year-old boy, only child of healthy unrelated parents, was born at term after uneventful pregnancy and normal delivery. Birth weight was 3,430 g. Perinatal period was normal (APGAR score 9/10). At age 3 months, he presented with feeding difficulties, vomiting, failure to thrive, and muscle hypotonia. At age 8 months, his neurological examination showed developmental delay, axial muscle hypotonia, brisk tendon reflexes in all limbs, poor head control, and inability of sitting unaided. Laboratory investigations showed increased levels of lactate in plasma (46.4 mg/dl; normal range 8–22 mg/dl) but not in urine. EEG was normal. Brain MRI showed symmetrical T2 hyperintensities with restricted diffusion of the cerebral deep white matter, thalami, midbrain, dorsal part of the pons and medulla oblongata, dentate nuclei, and cerebellar white matter. A periventricular white matter rim was spared. The corpus callosum was thin (Fig. 1a–e). Single-voxel proton MR spectroscopy (MRS) showed markedly increased lactate (Fig. 1f).
Fig. 1.
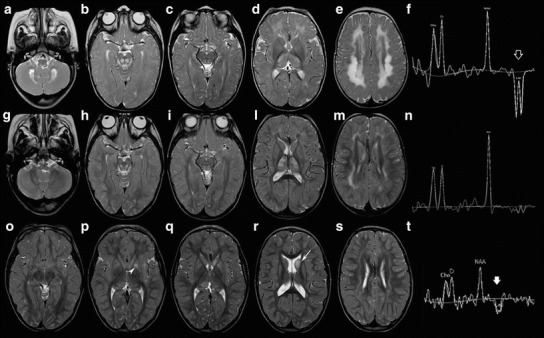
Brain MRI and MRS at age 8 months (a – f), 29 months (g – n), and 5 years and 10 months (o – t). Axial T2-weighted images show symmetrical T2 hyperintensity of the deep cerebral white matter, thalamus, basal ganglia, brainstem, cerebellar white matter, and dentate nuclei (a – e). MRS of the cerebral white matter shows markedly increased lactate (empty arrow, f). At age 29 months, axial T2-weightwed images show a substantial improvement of the signal abnormalities (g – m). MRS of the cerebral white matter reveals significantly reduced lactate (n). Three years later, axial T2-weighted images depict further improvement of these lesions (o – s). New signal abnormalities of the left caudate (arrow) and globus pallidus (arrowhead) are evident. MRS shows high lactate in the left caudate head (thick arrow, t)
At 10 months, a muscle biopsy revealed ragged-red and cytochrome c oxidase-negative fibers (Fig. 2), whereas spectrophotometric determination of respiratory chain complexes showed reduced activities of complexes I, III, and IV in the muscle homogenate.
Fig. 2.
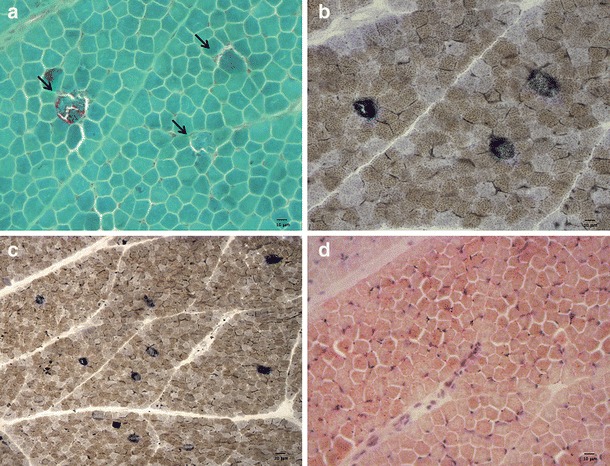
Muscle biopsy. (a) Trichrome staining showing three ragged-red fibers (arrows). (b) Combined COX–SDH staining (magnification 20×) confirms the same ragged-red fibers as COX-negative–SDH-positive fibers. (c) Combined COX–SDH staining (magnification 10×) highlighting several scattered COX-negative–SDH-positive fibers. (d) Oil red O staining showing lipid accumulation
The patient was treated with riboflavin (10 mg/day), thiamine (100 mg/day), and CoQ10 (100 mg/day).
At 14 months, neurological examination showed normal head circumference, mild axial muscle hypotonia, increased muscle tone in lower limbs, and brisk tendon reflexes in all limbs, but the child had acquired the ability of sitting without support and of standing with support.
Over the following 2 years, the clinical course showed significant improvement with the child being able to walk with support at the age of 21 months and without aid at 24 months. Nonetheless, the Griffiths developmental scale performed at 21 months showed a global development age of 11 months. At 29 months, neurological examination confirmed the presence of pyramidal signs with autonomous toe walking and widened base gait; the Griffiths developmental scale showed a development age of 15 months.
A brain MRI at age 30 months revealed striking improvement of the signal abnormalities and marked reduction of lactate in single-voxel proton MRS. Restricted diffusion persisted only in the thalami and posterior part of the pons (Fig. 1g–n).
During the following years, the patient showed progressive improvement of motor functions and never suffered from seizures. Blood lactate values progressively lowered from 46.4 mg/dl at 8 months to 33.4 mg/dl at 14 months, 32.5 mg/dl at 21 months, 33.3 mg/dl at 4 years, 19.4 mg/dl at 5 years, and 16.7 mg/dl at 6 years. Cardiac (including ECG and echocardiography), funduscopy, and hearing assessments were normal, as well as sensory and motor nerve conduction velocity studies. The last brain MRI, performed at the age of 5 years and 10 months, showed further improvement, although new areas of T2 hyperintensity without diffusion restriction were present within the head of the left caudate nucleus and pallidum, with a lactate peak on multi-voxel proton MRS (Fig. 1o–t). At this time the child showed mild generalized muscle weakness and unsupported toe walking gait with widened base.
At 6 years, cognitive assessment (WISC-IV) revealed a highly disharmonic profile with the following results: verbal comprehension index (VCI 92), perceptual organization index (POI 58), processing speed index (PSI 56), and working memory index (WMI 64) resulting into total intelligence quotient (IQ 58), i.e., mild mental retardation.
Since the MRI pattern and the muscle biopsy findings corresponded to those reported in leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL), after receiving written informed consent from the parents of the patient, in agreement with the Declaration of Helsinki and approved by the Ethical Committees of the Fondazione IRCCS Istituto Neurologico “C. Besta,” Milan, Italy, we analyzed the nucleotide sequence of the exons and exon–intron boundaries of EARS2 gene. We found a new homozygous variant (c.902G>C/p.Gly301Ala) in EARS2. The mutation was heterozygous in the healthy parents and absent in the public single-nucleotide polymorphism databases, including dbSNP (http://www.ncbi.nlm.nih.gov/ projects/SNP) and EVS (http://evs.gs.washington.edu/EVS), which altogether contain approximately 12,000 alleles. In addition, the p.Gly301Ala change scored very highly for likelihood to be deleterious according to ad hoc software for pathogenicity prediction (damaging for Polyphen2, p = 0.960; Panther, 0.95; MutPred, 0.969; SIFT and MutationTaster).
Discussion
Two distinct presentations and clinical courses have been reported in LTBL. The severe form is characterized by early-onset hypotonia, delayed psychomotor development, seizures, and persistent lactate elevation. In the mild form, clinical manifestations usually occur after the age of 6 months, with irritability and psychomotor regression, but clinical and biochemical improvement takes place from the second year of life without further clinical deterioration (Steenweg et al. 2012a, b; Talim et al. 2013). Respiratory chain enzyme activities in muscle have been found either reduced or normal in individual patients (Steenweg et al. 2012a). The clinical picture correlates well with the severity of neuroimaging. Although extensive symmetrical deep white matter abnormalities and signal changes of the thalami, brainstem, and cerebellar white matter together with increased lactate on MRS are the hallmark of LTBL (Steenweg et al. 2012a, b; Talim et al. 2013), significant improvement without new lesions and normalization of MRS have been observed in the mild form, whereas patients with the severe forms undergo progressive atrophy of the affected structures. However, long-term clinical and radiological follow-up has still to be completely elucidated. A rapidly progressive and fatal disease has recently been reported in an infant harboring a homozygous mutation in EARS2 in association with severe combined deficiency of respiratory chain complexes I and IV in skeletal muscle and dysgenesis in the posterior part of the corpus callosum at brain MRI (Talim et al. 2013).
Our patient carrying a new mutation in EARS2 showed a presentation of intermediate severity between the two aforementioned forms. Early-onset feeding difficulties, muscle hypotonia, and developmental delay were reflected by the severe brain MRI pattern detected at 3 months of age, which spontaneously improved from the second year of life onward, parallel to a slowly progressive clinical improvement, with the acquisition of motor milestones such as sitting and walking unaided in the second year of life. Interestingly, at 6 years, cognitive assessment revealed normal verbal index and impaired nonverbal functions, leading to a severely irregular neuropsychological profile with impossibility of obtaining a reliable total IQ. Indeed, although it was consistent with the diagnosis of mild mental retardation, the normality of verbal functioning allowed him to compensate the gaps in the performance field. It is likely that these data correlate with the documented neuroimaging improvement. Interestingly, we also documented asymptomatic lesions within the left caudate and pallidum nuclei during the most recent MRI, at age 5 years and 10 months; these lesions were not previously reported in LTBL. The appearance of new brain lesions, even with a transient and reversible pattern, has been described during the course of other mitochondrial disorders such as pyruvate dehydrogenase complex deficiency (Giribaldi et al. 2012). However, they typically occur during episodes of metabolic crisis that did not occur in our patient.
Mutations in different mitochondrial tRNA synthetases appear to be associated with a spectrum of syndromes characterized by striking tissue specificity, the heart (AARS2), the kidney (SARS2), or the peripheral nervous system (GARS2 and KARS2) (Konovalova and Tyynismaa 2013). The involvement of the brain reveals remarkable segmental involvement of specific tracts and nuclei in association with mutations in different genes: for instance, DARS2 mutations are typically linked to leukoencephalopathy with brainstem and spinal cord involvement and high lactate (LBSL) (Scheper et al. 2007), RARS2 mutations to pontocerebellar hypoplasia type 6 (PCH6) (Edvardson et al. 2007), and EARS2 to LTBL. The mechanistic basis of this specificity, particularly in the central nervous system, remains unexplained, being possibly related to difference in mitochondrial translation throughout organ development (Konovalova and Tyynismaa 2013; Scheper et al. 2007; Edvardson et al. 2007; Diodato et al. 2014). Recently, experimental studies have shown that cell-type-specific differences in the sensitivity to mutations may explain the selective vulnerability of specific white matter tracts in LBSL despite the ubiquitous distribution of the mitochondrial aspartyl-tRNA synthetase encoded by DARS2 (van Berge et al. 2012). A similar mechanism is postulated in PCH6 (Cassandrini et al. 2013) and might also apply to LTBL.
In conclusion, our study expands the clinical and neuroradiological phenotype of LTBL. Further studies are needed to define the phenotypic spectrum of this novel disorder and to better understand the factors influencing mitochondrial translation in different tissues.
Acknowledgments
We wish to thank Paolo Broda and Annagloria Incontrera for technical assistance.
The financial supports of Telethon Italy (Grant no. GUP09004) and of Pierfranco and Luisa Mariani Foundation, Italy, are gratefully acknowledged.
Take-Home Message
In this report, we present the first long-term clinical and neuroradiological follow-up of a LTBL/EARS2 patient, showing previously unreported brain MRI features.
Compliance with Ethics Guidelines
Roberta Biancheri, Eleonora Lamantea, Mariasavina Severino, Daria Diodato, Marina Pedemonte, Denise Cassandrini, Alexandra Ploederl, Federica Trucco, Chiara Fiorillo, Carlo Minetti, Filippo M. Santorelli, Massimo Zeviani, and Claudio Bruno declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Author Contributions
Study concept and design: Biancheri and Bruno
Acquisition of data: Biancheri, Lamantea, Severino Diodato, Cassandrini, Pedemonte, Trucco, Fiorillo, Ploederl, and Bruno
Analysis and interpretation of data: Biancheri, Lamantea, Santorelli, Zeviani, and Bruno
Drafting of the manuscript: Biancheri, Zeviani, and Bruno
Critical revision of the manuscript for important intellectual content: Minetti, Santorelli, Zeviani, and Bruno
Obtained funding: Zeviani
Administrative, technical, and material support: Bruno
Study supervision: Biancheri, Lamantea, and Bruno
Footnotes
Competing interests: None declared
Contributor Information
Claudio Bruno, Email: claudio2246@gmail.com.
Collaborators: Johannes Zschocke
References
- Cassandrini D, Cilio MR, Bianchi M, et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis. 2013;36:43–53. doi: 10.1007/s10545-012-9487-9. [DOI] [PubMed] [Google Scholar]
- Diodato D, Ghezzi D, Tiranti V. The mitochondrial aminoacyl tRNA synthetases: genes and syndrome. Int J Cell Biol. 2014;2014:787956. doi: 10.1155/2014/787956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giribaldi G, Doria-Lamba L, Biancheri R, et al. Intermittent-relapsing pyruvate dehydrogenase complex deficiency: a case with clinical, biochemical, and neuroradiological reversibility. Dev Med Child Neurol. 2012;54:472–476. doi: 10.1111/j.1469-8749.2011.04151.x. [DOI] [PubMed] [Google Scholar]
- Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. 2013;108:206–211. doi: 10.1016/j.ymgme.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- Steenweg ME, Vanderver A, Ceulemans B, et al. Novel infantile-onset leukoencephalopathy with high lactate level and slow improvement. Arch Neurol. 2012;69:718–722. doi: 10.1001/archneurol.2011.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talim B, Pyle A, Griffin H, et al. Multisystem fatal infantile disease caused by a novel homozygous EARS2 mutation. Brain. 2013;136:e228. doi: 10.1093/brain/aws197. [DOI] [PubMed] [Google Scholar]
- van Berge L, Dooves S, van Berkel CG, et al. Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem J. 2012;44:955–962. doi: 10.1042/BJ20110795. [DOI] [PubMed] [Google Scholar]