

Abstract
Introduction: Niemann-Pick type C disease is a rare disorder caused by impaired intracellular lipid transport due to mutations in either the NPC1 or the NPC2 gene. Ninety-five % of NPC patients show mutations in the NPC1 gene. A much smaller number of patients suffer from NPC2 disease and present respiratory failure as one of the most frequent symptoms. Several plasma oxysterols are highly elevated in NPC1 and can be used as a biomarker in the diagnosis of NPC1.
Methods: Plasma cholestane-3β,5α,6β-triol was evaluated as biomarker for NPC2 by GC/MS and LC-MS/MS analysis. The diagnosis was confirmed by Sanger sequencing and filipin staining.
Results: We report three NPC2 patients with typical respiratory problems and a detailed description of the nature of the lung disease in one of them. All patients had elevated levels of plasma cholestane-3β,5α,6β-triol. In two of these patients, the positive oxysterol result led to a rapid diagnosis of NPC2 by genetic analysis. The phenotype of the third patient has been described previously. In this patient a cholestane-3β,5α,6β-triol concentration markedly above the reference range was found.
Conclusions: Measurement of plasma cholestane-3β,5α,6β-triol enables to discriminate between controls and NPC1 and NPC2 patients, making it a valuable biomarker for the rapid diagnosis not only for NPC1 but also for NPC2 disease.
The measurement of oxysterols should be well kept in mind in the differential diagnosis of lysosomal diseases, as the elevation of oxysterols in plasma may speed up the diagnosis of NPC1 and NPC2.
Keywords: Cholestane-3β,5α,6β-triol; Niemann-Pick type C disease; NPC2; Oxysterols; Pulmonary alveolar proteinosis
Introduction
Niemann-Pick type C disease (NPC; OMIM 257220; OMIM 607625) is a rare autosomal recessive lysosomal storage disorder affecting the intracellular trafficking of unesterified cholesterol and other lipids (for review, see Mengel et al. 2013). Two disease-causing genes have been identified, NPC1 (MIM#607623) and NPC2 (MIM#601015), encoding two proteins, that are involved in the transport of cholesterol and other lipids out of late endosomes and lysosomes. As a consequence, unesterified cholesterol accumulates within these compartments. NPC1 is a large membrane spanning glycoprotein mainly located in the late endosomes (Carstea et al. 1997; Higgins et al. 1999). NPC2 is a small soluble protein, binding nonesterified cholesterol with high affinity (Okamura et al. 1999; Storch and Xu 2009).
The majority of NPC patients have a defect in NPC1 (95%), while only 5% of the NPC cases are due to mutations in NPC2. To date, more than 300 mutations have been described in the NPC1 gene; about 20 mutations have been reported in the NPC2 gene. Most NPC2 mutations lead to a severe phenotype, frequently presenting with pronounced pulmonary involvement (Millat et al. 2001; Verot et al. 2007).
The onset of symptoms may vary from early infancy to late adulthood, and clinical manifestations are extremely heterogeneous. Systemic symptoms include isolated spleno- or hepatosplenomegaly in infancy or childhood and precede the onset of neurological signs. Characteristic neurological manifestations include saccadic eye movement abnormalities, cerebellar signs (ataxia, dystonia/dysmetria, dysarthria), gelastic cataplexy, and epileptic seizures. Pulmonary infiltration with foam cells is usually restricted to patients with early onset disease and is more frequent in patients with severe NPC2 mutations.
The current diagnostic screening methods for NPC include biochemical testing, such as filipin staining in fibroblasts, and measuring the plasma chitotriosidase activity followed by a mutation analysis of the NPC1 and NPC2 genes. No specific biomarker was available for NPC until Porter et al. (2010) demonstrated the usefulness of cholesterol oxidation products in human plasma as new biomarker for NPC1. In NPC1 cells, cholesterol accumulation is associated with oxidative stress (Reddy et al. 2006; Zampieri et al. 2009), resulting in increased non enzymatic oxidation of cholesterol in different tissues (Porter et al. 2010). Therefore, a small fraction of the cholesterol is oxidized to so-called oxysterols, which can be measured in EDTA plasma and serum by GC/MS and LC-MS/MS. In plasma of NPC1 patients, cholesterol oxidation products such as 7-ketocholesterol and cholestane-3β,5α,6β-triol are significantly elevated (Porter et al. 2010). In fact, the plasmatic levels of these oxysterols allow distinguishing between NPC1 patients and controls. Only one NPC2 patient has been detected after oxysterol analysis so far (Boenzi et al. 2014).
We report three NPC2 patients with elevated plasma cholestane-3β,5α,6β-triol levels; in two of them a genetic analysis was initiated after positive oxysterol results, leading to a rapid diagnosis of NPC2. The clinical phenotype of the first patient gives new insights into the physiology of the lung disease in NPC2. The third NPC2 patient has already been described by Griese et al. (2010). We show that this patient also had an elevated cholestane-3β,5α,6β-triol plasma concentration.
Materials and Methods
LC-MS/MS
The concentration of cholestane-3β,5α,6β-triol was determined in 50 μl plasma. Cholestane-3β,5α,6β-triol D7 (TRC) was used as internal standard. The analytes were derivatized into dimethylglycine esters based on the method of Jiang et al. (2011). The chromatographic separation was performed on a column Symmetry C18 (2.1 × 50 mm 3.5 μm) using a linear gradient of water and acetonitrile (pH 3; 1 mM ammonium formate). The mass spectrometer Waters Xevo TQ MS was used as detector, and quantification was based on an 8-point calibration curve. The cutoff value for the LC-MS/MS method was 30 ng/ml.
GC/MS
10 ng of d7-cholestane-3β,5α,6β-triol (Santa Cruz) as an internal standard (IS) was added to 100 μL of plasma or serum. Alkaline saponification and lipid extraction were performed using the method of Klansek et al. (1995) with some modifications: plasma or serum was subjected to alkaline saponification with potassium hydroxide/isopropanol followed by extraction of the free triol with carbon tetrachloride and derivatization with N-Methyl-N-(trimethylsilyl)trifluoroacetamide/1-methylimidazole (19:1 v/v). 1 μL was analyzed isothermal at 280°C by gas chromatography-mass spectrometry (GC/MS) using a Shimadzu QP2010Plus with an Rtx-200MS-column (Restek, 30 m, 0.25 mm, 0.5 μm). For quantification, the triols were monitored with ions m/z 403 and m/z 410 in EI-SIM-mode. Concentrations were calculated from the linear response range of standard curve established for cholestane-3β,5α,6β-triol/IS (Porter et al. 2010). The cutoff value for the GC/MS method was 50 ng/ml.
Mutation Analysis
All exons of NPC1 (NM_000271) and NPC2 (NM_006432) and their flanking intronic sequences were amplified by PCR and analyzed by Sanger sequencing. Primer sequences and PCR conditions are available upon request. Putative mutations were confirmed by sequencing duplicate PCR products and by the DNA analysis from parents.
Filipin Staining
Intracellular accumulation of unesterified cholesterol was analyzed in cultured fibroblasts by filipin staining as previously described (Blanchette-Mackie et al. 1988).
Chitotriosidase Activity
The chitotriosidase activity was measured as described earlier (Hollak et al. 1994). Plasma samples were diluted 1:10, 1:20, 1:40, and 1:80 with demineralized water before incubation. The reaction was stopped with 2 ml ethylenediamine (Fluka 03550) after 15 min. The product of the enzymatic reaction, fluorescent 4-methylumbelliferone, was measured using a spectral fluorophotometer at 360 and 450 nm. The enzyme activity was expressed in nmol/h/ml (normal 100 nmol/h/ml).
Results
Case Reports
Patient 1
The first patient was born at term (38 + 4 gestational weeks) with a weight of 3,360 g, a length of 51 cm, and a head circumference of 35 cm. Pregnancy and postpartum adaptation were uneventful. There was no history of neonatal cholestasis. The girl was the second child of healthy parents, both of German origin. The family history was unremarkable; the older brother of the patient is healthy.
The patient was first admitted to a hospital at 10 weeks of age for further workup for failure to thrive and respiratory difficulties. Furthermore, the patient was found to have mild hepatosplenomegaly. There was a rapid progression of respiratory insufficiency with recurrent atelectasis. From the age of 4 months on, continuous artificial ventilation was required. Repeated bronchoalveolar lavage did not improve the respiratory situation. At 5 months of age, lateral thoracotomy with biopsy and partial resection of segments 5 and 9 of the right lung was performed. However, the respiratory situation did not improve. At 7 months of age, the patient was transferred to a specialized hospital for further workup. CT scans revealed ground glass-shading of both lungs in CT scan (Fig. 1a), and pathological surfactant protein composition (protein C deficiency) similar to alveolar proteinosis was found in bronchoalveolar lavage fluid (Fig. 1c). Alveolae were filled with fine granular material and foam cells (Fig. 1b, d). Abdominal ultrasound confirmed mild hepatosplenomegaly (liver in anterior axillary line 9.2 cm, spleen 7 cm), both with normal parenchyma. There were no signs of cholestasis.
Fig. 1.
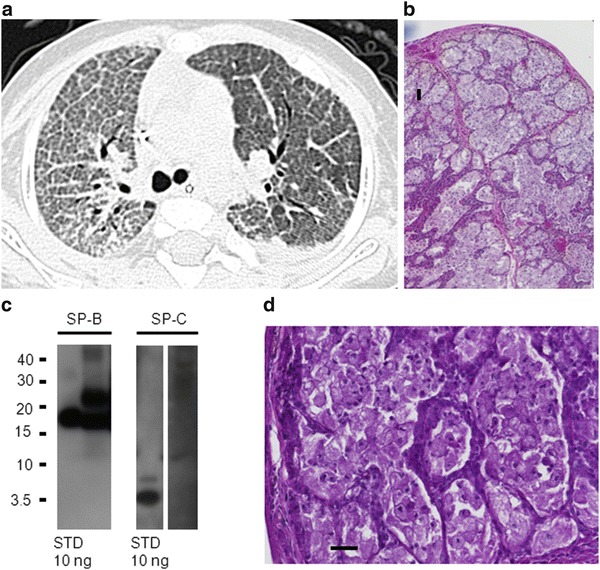
Patient 1 at the age of 7 months: (a) Upper left: CT scan showing crazy-paving pattern with pronounced inter- and intralobar septi, particular on the right side; left side after therapeutic lavage. (b) Upper right and (d) lower right: Lung biopsy (hematoxylin and eosin stain, 40-fold) showing alveolar filling with fine granular material. PAS-positive material (100 fold) mainly foamy macrophages and some extracellular surfactant material. (c) Lower left: Western blotting of bronchoalveolar lavage for surfactant protein B (SP-B) and SP-C. 5 μg of total protein of lavage fluid per lane was added and the respective standards (STD). After SDS-PAGE and transfer, the membranes were probed with antibodies against SP-B and SP-C. Molecular weights (kDa) are indicated on the left side. All bands were analyzed under nonreducing conditions. SP-B was detected as dimers (typical bands at 16 kDa; compare to standard STD of 10 ng applied to lane 1) and some higher molecular forms (bands at 24 kDa); such forms are of interest, as often seen in patients with alveolar proteinosis. No monomers or degradation products were detected. SP-C was absent at molecular weight of about 4 kDa (usually SP-C is present in amounts at least about 50% of SP-B)
Due to the combination of severe respiratory disease with hepatosplenomegaly, metabolic workup was initiated showing elevated plasma oxysterols (150 ng/ml, measured by GC/MS) and chitotriosidase activity (415 nmol/h/ml), suspicious for Niemann-Pick type C disease. The diagnosis of Niemann-Pick type C-2 was confirmed by a genetic analysis of the NPC2 gene revealing the homozygous mutation c.352G>T (p.E118X). This mutation leads to a premature stop codon and has been associated with a severe phenotype of Niemann-Pick type C-2 before (Millat et al. 2001; Schofer et al. 1998).
Considering bone marrow transplant as therapeutic option (Breen et al. 2013), extensive neurological workup was performed after diagnosis at 8 months of age. High-resolution 3T MRI of the brain revealed hypomyelinization and global brain atrophy with enlarged inner and outer cerebrospinal fluid space (Fig. 2a–c). Electroencephalography detected multiregional epileptiform discharges regardless of high dosages of benzodiazepines reflecting increased cortical excitability (Fig. 2d). Evaluation of development revealed general psychomotor retardation with muscular hypotonia (as far as possible to judge due to sedatives). Gross motor movement was unfocused without active grasping for objects, hand-knee contact, or changing of objects between hands. Major developmental milestones as sitting were not reached. Spontaneous ocular fixation and eye contact were possible though without appropriate endurance. Mild horizontal saccades, end-position nystagmus, and intermittent irregular myocloni were present. Considering these results, decision was made together with the parents for palliative care management. The patient died at 11 months of age after rapid respiratory deterioration.
Fig. 2.
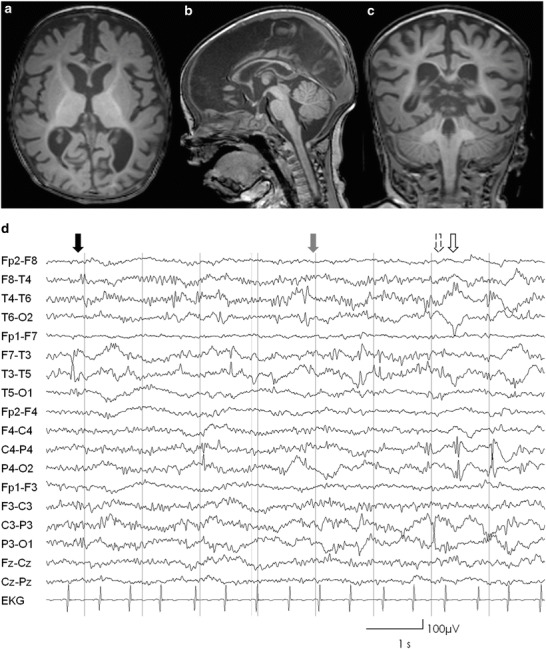
(a–c) Axial sagittal and coronal T1-weighted cranial MRI slices of patient 1, showing delayed myelinization sparing the frontal and posterior subcortical white matter and global brain atrophy; (d) EEG performed with current application of high doses of midazolam showing multiregional epileptiform discharges temporal left (black arrow), right (gray arrow), parietal left (lined arrow), and right (bare arrow). In addition, excessive beta-activity reflecting benzodiazepine effect is evident
Patient 2
The second patient was born as the third child from Tunisian consanguineous parents (first cousins). The boy was born at term with a weight of 3,320 g; Apgar scores were 8 and 9 at 5 and 10 min. At the fourth day of life, the neonate presented with hyperbilirubinemia: total bilirubin 135 μmol/L (nv 1.7–17 μmol/L), conjugated bilirubin 89 μmol/L (nv 0–3.4 μmol/L), impaired liver function (albumin 2.8%, INR 1.47), and severe splenomegaly. Initial chest X-ray and neurological examination were normal. Also blood and urine metabolic screenings were normal. After 20 days, he showed persistent hyperbilirubinemia, total bilirubin 104.7 μmol/L (nv 1.7–17 μmol/L), conjugated bilirubin 72 μmol/L (nv 0–3.4 μmol/L), and splenomegaly. Lysosomal investigations including urinary mucopolysaccharides and oligosaccharides were normal. Due to the persistent hyperbilirubinemia, oxysterol analysis was performed in EDTA plasma, showing abnormal values (515 ng/ml, measured by LC-MS/MS). The test led to the suspicion of Niemann-Pick type C. Since pulmonary involvement is a common feature of NPC2 patients, molecular analysis of the NPC2 gene was performed, and the previously described mutation c.436C>T (p.Q146X) was found in a homozygous state (Millat et al. 2005).
Filipin staining of cultured fibroblasts showed a massive intracellular accumulation of unesterified cholesterol (Fig. 3a). The chitotriosidase activity was elevated to 169.3 nmol/h/ml. A therapy with miglustat was started at the dose of 50 mg once a day at 4 weeks of life.
Fig. 3.
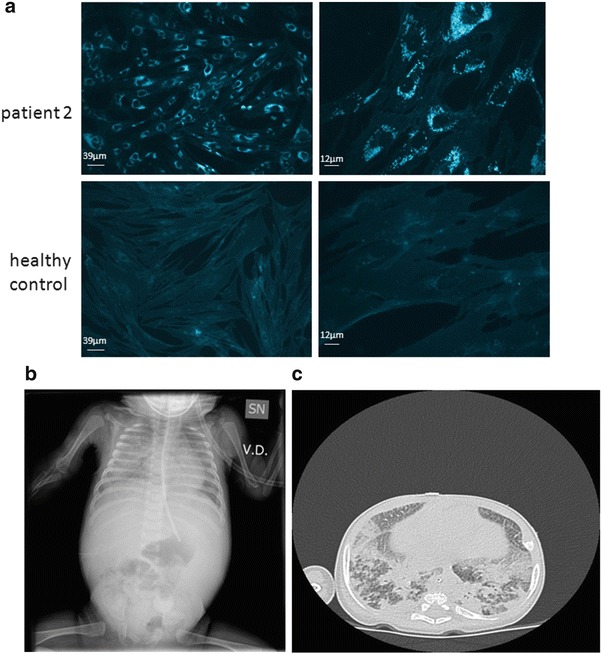
(a) Filipin staining of cultured fibroblasts in patient 2, showing a massive intracellular accumulation of unesterified cholesterol. (b, c) Chest X-ray and high-resolution CT of patient 2, showing diffuse hypo-diaphaneity with air bronchogram in the whole right pulmonary field and in the left inferior field (b) and smooth septal thickening and ground glass opacities in intermixed pattern (c)
A few days after discharge, the patient developed bronchiolitis. The chest X-ray showed bilateral micronodular infiltrates in both lungs, more pronounced in the right field. Due to a mean poor weight gain, miglustat was stopped at 2 and a half months of age and never recommenced. Ophthalmologic evaluation showed a regular fundus. The baby presented generalized hypotonia, with reduced spontaneous motility and poor visual contact. The baby was discharged with antibiotics and salbutamol aerosol.
He was readmitted about 1 month later with respiratory distress that needed high oxygen supply. Chest X-ray images were dramatically altered with diffuse hypo-diaphaneity and air bronchogram in the whole right pulmonary field and in the left inferior field. High-resolution CT showed smooth septal thickening and ground glass opacities in intermixed pattern (Fig. 3b, c). Cholestatic disease was stable. Although intensive respiratory therapy was initiated, the patient gradually deteriorated and died 1 month after admission, at the age of four months and a half.
Patient 3
The third patient was already described in detail by Griese et al. (2010). The baby girl was born at term after an uneventful pregnancy as the second child. She presented with progressive tachypnea, failure to thrive, and poor feeding since the second month of life. The X-ray showed bilateral micronodular infiltrates especially in the lower left lung and apical right lobe. Hepatosplenomegaly was present with the liver enlarged to 4 cm and the spleen enlarged to 5 cm below the costal margin. Neurological development and blood and urine metabolic screening were normal. The suspicion for NPC was confirmed by a genetic analysis which revealed a homozygous deletion (c.408_409delAA) in NPC2, leading to a frame shift and an elongated protein with additional 80 amino acids (Griese et al. 2010). Treatment of the pulmonary alveolar proteinosis with GM-CSF and half lung lavages did not improve the patient’s general condition. She died after the fifth lavage after developing a pneumothorax (Griese et al. 2010). The oxysterol amount in a preserved serum sample of this patient was highly elevated to 226 ng/ml.
Discussion
The heterogeneous phenotype and the variable progression of Niemann-Pick type C disease often lead to a delay of diagnosis. Available screening methods are limited to filipin staining of unesterified cholesterol in cultured fibroblasts, measurement of chitotriosidase activity, and genetic analysis of the NPC1 and NPC2 genes. Filipin staining of fibroblasts is currently the most specific diagnostic method for NPC (Wraith et al. 2009; Patterson et al. 2012), showing the classical storage pattern, characterized by massive accumulation of unesterified cholesterol in 85% of NPC patients. However, 15% of patients present the so-called variant biochemical phenotype, characterized by mild to moderate intracellular cholesterol accumulation. In these cases the diagnosis of NPC might still be uncertain (Vanier et al. 1991; Wraith et al. 2009; Patterson et al. 2012). Since filipin staining requires living cells, a skin biopsy is needed, and the procedure is time-consuming. Chitotriosidase activity may be a useful indication for NPC, but is not sensitive or specific for NPC (Ries et al. 2006) and often normal in adults. In order to speed up the diagnosis, Porter et al. (2010) published a sensitive and specific biomarker by measuring plasma oxysterols in NPC1 patients. The elevation of several oxysterols in plasma of NPC patients was used to distinguish NPC1 patients from controls.
Our data show that the oxidized cholesterol derivate cholestane-3β,5α,6β-triol may also be used as appropriate biomarker for identification of NPC2 patients (Fig. 4). Patients 1 and 2 presented with an elevated plasma cholestane-3β,5α,6β-triol concentration. The diagnosis of NPC2 was then confirmed by a genetic analysis. The third patient was already diagnosed with NPC2 but also showed elevated amounts of cholestane-3β,5α,6β-triol. The cholestane-3β,5α,6β-triol levels in plasma of the NPC2 patients show a clear discrimination from 50 plasma samples that have been in normal range. In contrast, the NPC2 patients could not be discriminated from 20 genetically confirmed NPC1 patients by measuring plasma cholestane-3β,5a,6β-triol (see Fig. 4). These data demonstrate the eligibility of cholestane-3β,5α,6β-triol as biomarker not only for NPC1 but also for NPC2. The specificity of oxysterols as biomarker for other lysosomal diseases with a similar clinical phenotype has already been investigated by Porter et al. (2010) and Lin et al. (2014). Porter et al. (2010) showed that cholestane-3β,5α,6β-triol and 7-ketocholesterol can be used to distinguish between patients suffering from NPC and patients suffering from other lysosomal diseases (infantile neuronal ceroid lipofuscinosis (INCL), GM-1 gangliosidosis, GM-2 gangliosidosis, and Gaucher disease (GD)), where the plasma oxysterols showed normal levels.
Fig. 4.
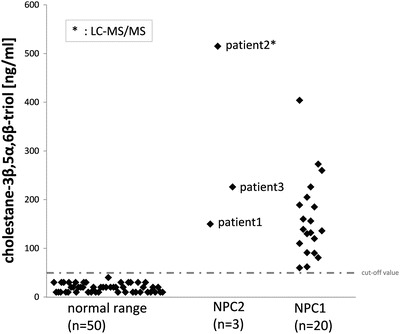
Cholestane-3β,5α,6β-triol levels in plasma of patients with genetically confirmed NPC2 (n = 3) showing a clear discrimination from 50 plasma samples in normal range. The patients with confirmed NPC2 mutations show similar cholestane-3β,5α,6β-triol levels as NPC1 (n = 20) patients. The dotted line indicates the cutoff value for the GC/MS method (50 ng/ml)
Lin et al. (2014) additionally demonstrated that 7-ketocholesterol was not elevated in glycogen storage disorder type II (GSDII), Krabbe disease (KD), metachromatic leukodystrophy (MLD), and mucopolysaccharidosis type II (MPSII). A limitation in specificity of oxysterols as biomarkers for NPC becomes evident in the elevation of 7-ketocholesterol in the plasma of patients with a defect in the acid sphingomyelinase (Niemann-Pick A/B). This might also apply for plasma cholestane-3β,5α,6β-triol. It has been shown for at least one NPB patient (SSIEM 2014 Annual Symposium: Abstracts suppl: S150, 2014). In their publication, Lin et al. (2014) stated that cholestane-3β,5α,6β-triol was not detectable by ESI-MS due to the relatively low ionization efficiency.
NPC2 patients usually present with early pulmonary involvement and progression to severe respiratory disease. Although minimizing diagnostic delay is an important factor for all NPC patients, it is particularly critical for NPC2 patients with severe lung problems, in whom immediate causative treatment is of great importance. Indeed, in one NPC2 patient, early bone marrow transplantation improved the respiratory illness and the general developmental outcome (Bonney et al. 2010; Breen et al. 2013).
In patient 1, metabolic workup was already done at 3 month of age, showing mildly elevated chitotriosidase activity up to 117 nmol/ml/h (normal <100 nmol/ml/h) suggesting a lysosomal storage disease. A subsequent investigation for lysosomal diseases, such as Gaucher and Niemann-Pick A/B, was done by enzymatic analyses, showing negative results. Further testing for NPC was not performed. If the oxysterols would have been tested at the initial clinic suspicion, the diagnosis would have been confirmed earlier, and further diagnostics would have been spared (multiple genetic analysis, whole exome sequencing, lung biopsy).
In patient 2 the prolonged neonatal cholestatic jaundice together with a splenomegaly suggested an early neonatal lysosomal disease. In the metabolic investigation protocol, oxysterol analysis was recently included as it allows a rapid screening of NPC. The diagnosis in patient 2 was puzzling for the lack of inflammatory lung disease. Nevertheless, oxysterol analysis established the correct diagnosis.
Other patients with the same mutation as patients 1 and 2 have been described before. One patient carrying the homozygous p.E118X mutation also came from Germany and showed a similar clinical phenotype (Schofer et al. 1998; Millat et al. 2001). An Algerian patient described by Verot et al. (2007) with the same mutation as patient 2 did not show a pronounced pulmonary involvement.
In all three patients, NPC2 mutations were deleterious, leading to death in the first year of life, mainly due to respiratory manifestations. Usually, NPC2 patients present with a broad spectrum of clinical severity including progressive neurological dysfunction (Millat et al. 2001; Verot et al. 2007; Alavi et al. 2013; Klünemann 2002). However, in these cases, initial neurological symptoms were unspecific and did not lead to NPC diagnosis. Since systemic symptoms precede neurological signs, it is likely that these patients did not live long enough to develop characteristic neurological symptoms. These findings were also mentioned by Millat et al. (2001), who described six NPC2 patients having a short lifespan, 4 of those did not show any neurological symptoms and died of respiratory failure in the first year of life. Two other patients were described who survived until 19 month and 4 years, respectively. These two patients developed neurological disease (Millat et al. 2001).
In conclusion, the measurement of oxysterols should be well kept in mind in the differential diagnosis of lysosomal diseases. Our data confirm that the elevation of oxysterols in plasma presents a valuable tool and may speed up the diagnosis of NPC1 and NPC2. Early detection of the NPC2 diagnosis is a significant step toward slowing of disease progression and allows genetic counseling for the family.
Compliance with Ethics Guidelines
Conflict of Interest
The work of Thorsten Marquardt and Janine Reunert is part of an investigator-initiated study, funded by a grant from Actelion Pharmaceuticals Ltd.
The work of Matthias Griese was supported by eRARE-2009 (EUPAPNet), DFG-970/8-1, and FP7-chILD-EU. The funding sources had no involvement in the collection, analysis, and interpretation of data or in the writing of the report.
Janine Reunert has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Giulia Polo has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Frank Kannenberg has received speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Eugen Mengel has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Andrea Dardis has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Alessandro Burlina has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Bruno Bembi has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Alberto Burlina has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Thorsten Marquardt has received travel reimbursements and speaker honorarium from Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
Amelie Lotz-Havla, Manfred Fobker, Ania Muntau, Philipp Schnabel, Olaf Sommerburg, Ingo Borggraefe, Matthias Mall, and Giovanni Ciana declare that they have no conflict of interest. None of the authors has nonfinancial interests that may be relevant to the submitted work.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
Janine Reunert: acquisition and analysis of data, drafting, and revising the manuscript.
Amelie Lotz-Havla and Andrea Dardis: acquisition and interpretation of data, involved in drafting and revising the manuscript.
Giulia Polo, Matthias Griese, Frank Kannenberg, Eugen Mengel, Manfred Fobker, Ania Muntau, Ingo Borggraefe, Philipp Schnabel, Olaf Sommerburg, Alessandro Burlina, Matthias Mall, Giovanni Ciana, and Bruno Bembi: acquisition and interpretation of data, revising the manuscript.
Alberto Burlina and Thorsten Marquardt: supervising and design of the study, acquisition and interpretation of data, revising the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
T. Marquardt, Email: marquat@uni-muenster.de
Collaborators: Johannes Zschocke
References
- Alavi A, Nafissi S, Shamshiri H, Nejad MM, Elahi E. Identification of mutation in NPC2 by exome sequencing results in diagnosis of Niemann-Pick disease type C. Mol Genet Metab. 2013;110:139–144. doi: 10.1016/j.ymgme.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Blanchette-Mackie EJ, Dwyer NK, Amende LM, et al. Type-C Niemann-Pick disease: low density lipoprotein uptake is associated with premature cholesterol accumulation in the Golgi complex and excessive cholesterol storage in lysosomes. Proc Natl Acad Sci U S A. 1988;85:8022–8026. doi: 10.1073/pnas.85.21.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boenzi S, Deodato F, Taurisano R, et al. A new simple and rapid LC–ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann–Pick type C disease. Clin Chim Acta. 2014;437:93–100. doi: 10.1016/j.cca.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Bonney DK, O’Meara A, Shabani A, et al. Successful allogeneic bone marrow transplant for Niemann–Pick disease type C2 is likely to be associated with a severe “graft versus substrate” effect. J Inherit Metab Dis. 2010;33:171–173. doi: 10.1007/s10545-010-9060-3. [DOI] [PubMed] [Google Scholar]
- Breen C, Wynn RF, O’Meara A, et al. Developmental outcome post allogenic bone marrow transplant for Niemann Pick Type C2. Mol Genet Metab. 2013;108:82–84. doi: 10.1016/j.ymgme.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Griese M, Brasch F, Aldana V, et al. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin Genet. 2010;77:119–130. doi: 10.1111/j.1399-0004.2009.01325.x. [DOI] [PubMed] [Google Scholar]
- Higgins ME, Davies JP, Chen FW, Ioannou YA. Niemann–Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab. 1999;68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93:1288–1292. doi: 10.1172/JCI117084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Sidhu R, Porter FD, et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res. 2011;52:1435–1445. doi: 10.1194/jlr.D015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klansek JJ, Yancey P, Clair RWS, et al. Cholesterol quantitation by GLC: artifactual formation of short-chain steryl esters. J Lipid Res. 1995;36:2261–2266. [PubMed] [Google Scholar]
- Klünemann HH, Elleder M, Kaminski WE, et al. Frontal lobe atrophy due to a mutation in the cholesterol binding protein HE1/NPC2. Ann Neurol. 2002;52:743–749. doi: 10.1002/ana.10366. [DOI] [PubMed] [Google Scholar]
- Lin N, Zhang H, Qiu W, et al. Determination of 7-ketocholesterol in plasma by LC-MS for rapid diagnosis of acid SMase-deficient Niemann-Pick disease. J Lipid Res. 2014;55:338–343. doi: 10.1194/jlr.D044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel E, Klunemann H-H, Lourenco CM, et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis. 2013;8:166. doi: 10.1186/1750-1172-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millat G, Chikh K, Naureckiene S, et al. Niemann-Pick disease type C: spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am J Hum Genet. 2001;69:1013–1021. doi: 10.1086/324068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millat G, Baïlo N, Molinero S, et al. Niemann–Pick C disease: use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol Genet Metab. 2005;86:220–232. doi: 10.1016/j.ymgme.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Okamura N, Kiuchi S, Tamba M, et al. A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim Biophys Acta BBA Mol Cell Biol Lipids. 1999;1438:377–387. doi: 10.1016/S1388-1981(99)00070-0. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Hendriksz CJ, Walterfang M, et al. Recommendations for the diagnosis and management of Niemann–Pick disease type C: an update. Mol Genet Metab. 2012;106:330–344. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Porter FD, Scherrer DE, Lanier MH et al (2010) Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann–Pick C1 disease. Sci Transl Med 2:56ra81. doi:10.1126/scitranslmed.3001417 [DOI] [PMC free article] [PubMed]
- Reddy JV, Ganley IG, Pfeffer SR. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PloS One. 2006;1:e19. doi: 10.1371/journal.pone.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries M, Schaefer E, Lührs T, et al. Critical assessment of chitotriosidase analysis in the rational laboratory diagnosis of children with Gaucher disease and Niemann–Pick disease type A/B and C. J Inherit Metab Dis. 2006;29:647–652. doi: 10.1007/s10545-006-0363-3. [DOI] [PubMed] [Google Scholar]
- Schofer O, Mischo B, Püschel W, et al. Early-lethal pulmonary form of Niemann-Pick type C disease belonging to a second, rare genetic complementation group. Eur J Pediatr. 1998;157:45–49. doi: 10.1007/s004310050764. [DOI] [PubMed] [Google Scholar]
- SSIEM (2014) Annual symposium: abstracts. J Inherit Metab Dis 37:27–185. doi:10.1007/s10545-014-9740-5 [DOI] [PubMed]
- Storch J, Xu Z. Niemann–Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim Biophys Acta BBA Mol Cell Biol Lipids. 2009;1791:671–678. doi: 10.1016/j.bbalip.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT, Rodriguez-Lafrasse C, Rousson R, et al. Type C Niemann–Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim Biophys Acta. 1991;1096:328–337. doi: 10.1016/0925-4439(91)90069-L. [DOI] [PubMed] [Google Scholar]
- Verot L, Chikh K, Freydière E, et al. Niemann–Pick C disease: functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin Genet. 2007;71:320–330. doi: 10.1111/j.1399-0004.2007.00782.x. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Baumgartner MR, Bembi B, et al. Recommendations on the diagnosis and management of Niemann–Pick disease type C. Mol Genet Metab. 2009;98:152–165. doi: 10.1016/j.ymgme.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Zampieri S, Mellon SH, Butters TD, et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J Cell Mol Med. 2009;13:3786–3796. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]