

Abstract
Background: Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is included in many newborn screening programmes worldwide. In addition to the prevalent mutation c.985A>G in the ACADM gene, potentially mild mutations like c.199T>C are frequently found in screening cohorts. There is ongoing discussion whether this mutation is associated with a clinical phenotype.
Methods: In 37 MCADD patients detected by newborn screening, biochemical phenotype (octanoylcarnitine (C8), ratios of C8 to acetylcarnitine (C2), decanoylcarnitine (C10) and dodecanoylcarnitine (C12) at screening and confirmation) and clinical phenotype (inpatient emergency treatment, metabolic decompensations, clinical assessments, psychometric tests) were assessed in relation to genotype.
Results: 16 patients were homozygous for c.985A>G (group 1), 11 compound heterozygous for c.199T>C and c.985A>G/another mutation (group 2) and 7 compound heterozygous for c.985A>G and mutations other than c.199T>C (group 3) and 3 carried neither c.985A>G nor c.199T>C but other known homozygous mutations (group 4). At screening C8/C2 and C8/C10, at confirmation C8/C2, C8/C10 and C8/C12 differed significantly between patients compound heterozygous for c.199T>C (group 2) and other genotypes. C8, C10 and C8/C2 at screening were strongly associated with time of sampling in groups 1 + 3 + 4, but not in group 2. Clinical phenotype did not differ between genotypes. Two patients compound heterozygous for c.199T>C and a severe mutation showed neonatal decompensation with hypoglycaemia.
Conclusion: Biochemical phenotype differs between MCADD patients compound heterozygous for c.199T>C with a severe mutation and other genotypes. In patients detected by newborn screening, clinical phenotype does not differ between genotypes following uniform treatment recommendations. Neonatal decompensation can also occur in patients with the presumably mild mutation c.199T>C prior to diagnosis.
Introduction
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is the most common inborn error of fatty acid oxidation, which affects about 1:20,000 to 1:8,000 newborns (Andresen et al. 2001; Grosse et al. 2006; Saudubray et al. 2012). It is included in many newborn screening programmes worldwide (National Newborn Screening and Genetics Resource Center, Austin, Texas; Bundesausschuss 2005; Wilcken et al. 2007). Outcome is predominantly excellent following presymptomatic diagnosis, if prolonged fasting is avoided and adequate emergency management is performed during episodes of metabolic stress (Wilcken et al. 2007; Lindner et al. 2011). In cohorts diagnosed symptomatically, about 20% of patients died from their first metabolic decompensation, 20% showed severe neurological sequelae (Derks et al. 2006). In these cohorts, the c.985A>G mutation in the ACADM gene accounted for about 90% of disease-causing alleles (Gregersen et al. 1991; Yokota et al. 1991; Derks et al. 2006). In addition to this most common mutation in Europe, in screening cohorts other mutations, especially c.199T>C, are frequently found (Ziadeh et al. 1995; Andresen et al. 2001; Maier et al. 2005; Waddell et al. 2006; Hsu et al. 2008). There is ongoing discussion, whether patients with this potentially mild mutation would ever show any clinical phenotype (Andresen et al. 2001) or might not require treatment at all (Sturm et al. 2012). However, findings on associations between genotype and biochemical phenotype in MCADD are divergent (Andresen et al. 1997; Maier et al. 2005; Rhead 2006; Sturm et al. 2012). A straightforward correlation between clinical phenotype and genotype could not be demonstrated so far (Wilcken et al. 1994; Andresen et al. 1997).
Methods
Patients
From 1999 until 2012, MCADD was detected in 119 newborns at the Newborn Screening Center in Heidelberg. According to our current protocol, MCADD recall is performed in all cases with C8 > 1 μmol/L and cases with C8 > 0.28 μmol/L if C8/C10 is > P 99, C8/C2 > P 99.5 and C8/12 > P 99.5. Minimal criteria for confirmation of MCADD are a characteristic acylcarnitine profile in dried blood and presence of hexanoylglycine in urine or an informative genotype or reduced enzyme activity. In this study we report follow-up data based on the biochemical and clinical phenotypes of 37 patients with MCADD who took part in a study on long-term outcome of patients with inborn errors of metabolism detected by newborn screening (Lindner et al. 2011) and in whom information on genotype (both alleles) was available.
The recommended time for newborn screening blood sampling in Germany was day 3–5 before 2002 and 36–72 h thereafter (Harms et al. 2002).
After confirmation of MCADD, all families received counselling by a metabolic specialist. Recommendations included regular feeds, avoidance of prolonged fasting and immediate contact to the metabolic centre in case of intercurrent infections to decide on further management. All children received a personalised emergency card.
Mean age of patients (17 male, 20 female) at evaluation was 7.0 years (SD 4.2, range 0.6–14.4).
Genotype
Genotype analysis was performed at the Institute of Human Genetics, University of Heidelberg. All exons and parts of the neighbouring intron regions of the ACADM gene (NM_000016.4) were analysed by direct sequencing (Zschocke et al. 2001). Mutations are described according to the recommendations of HGVS (http://www.hgvs.org/mutnomen/). The conventional nomenclature on protein level skips the first 25 amino acids of the precursor peptide and starts numbering with codon 26. Mutation analysis in patients’ parents was not routinely performed, but compound heterozygosity was assumed in these biochemically well-characterised patients carrying two mutations.
Biochemical Parameters
Acylcarnitine profiles were measured from dried blood spots as previously described (Schulze et al. 2003). Levels of octanoylcarnitine (C8) and ratios of C8 to acetylcarnitine (C2), decanoylcarnitine (C10) and dodecanoylcarnitine (C12) at screening and confirmation were used for further evaluation.
Analysis of organic acids in urine was performed using gas chromatography/mass spectrometry (Hoffmann et al. 1989). For selected patients, e.g., with mutations not previously described, enzyme activity in lymphocytes was analysed by Prof. Wanders, AMC, Amsterdam, the Netherlands (patients 18, 29), or by Prof. Spiekerkötter, University Children’s Hospital, Düsseldorf, Germany (patients 17, 28).
Clinical Parameters
Standardised clinical status examination investigated 32 clinically relevant signs related to the central nervous system, peripheral nervous system, muscle, heart, eye, liver, skin, kidney, haematopoiesis and growth. A critical subset of signs relevant to MCADD was defined (available upon request). Most recent clinical assessments were evaluated, and assessments with at least one disease relevant finding were classified as abnormal. Intellectual development was evaluated by standardised psychometric instruments appropriate for age: 1.5 years Denver test or Bayley Scales of Infant Development (BSID-II), 3.5 years K-ABC or WPPSI-III and >5 years SON-R 2.5–7 or WISC-IV. IQ ≥ 85 was considered normal; results <85 and Denver results not appropriate for age were scored subnormal.
Statistical Analysis
Differences of acylcarnitine markers between groups were tested for significance by randomised one-way ANOVA (Edgington 1995; Howell 2001; Smucker et al. 2007). Cohen’s was computed as measure for the size of the differences. For group comparison of “presence of hexanoylglycine” and clinical parameters, Fisher’s exact test was used (IBM SPSS Statistics 20.0).
For analysis of acylcarnitines in relation to time of sampling, curve fitting and nonlinear regression were performed using R (Spiess 2012; Elzhov et al. 2013). Akaike weights were used as criterion for model selection (Spiess and Neumeyer 2010). Adjusted R2adj was used as measure of strength of association between time and acylcarnitine marker (strong association assumed if ). The Anderson-Darling test was computed using R (Scholz 2011).
Due to the explorative nature of the analysis, no adjustment of alpha error was performed. P-values ≤ 0.05 were considered statistically significant, values >0.05 and ≤0.1 reported as trends.
Results
Genotypes
Sixteen patients were homozygous for c.985A>G (genotype group 1), eleven patients (group 2) compound heterozygous for c.199T>C in combination with c.985A>G (n = 8) or another mutation (n = 3) and seven patients compound heterozygous for c.985A>G and mutations other than c.199T>C (group 3) and three patients carried neither c.985A>G nor c.199T>C but other known homozygous mutations (group 4). Thus, c.985A>G was the most frequent disease-causing allele accounting for 63.5%, followed by c.199T>C with 14.9% of all alleles.
Information on individual patients and assumed severity of mutations other than c.985A>G or c.199T>C according to literature (Andresen et al. 2001; Waddell et al. 2006; ter Veld et al. 2009; Smith et al. 2010; Yusupov et al. 2010; Sturm et al. 2012) is given in Table 1.
Table 1.
Genotypes, biochemical and clinical phenotypes
| Pat. No. | Mutation 1 | Mutation severity | Mutation 2 | Mutation severity | Genotype group | C8 NBS | C8 confirmation | HG at confirmation (0 = not detectable 1 = detectable) | Number of symptomatic episodes | If symptomatic episode: hypoglycaemia (1 = yes, 0 = no) | If symptomatic episode: impaired consciousness (1 = yes, 0 = no) | Clinical status (1 = abnormal, 0 = normal) | IQ | Age at evaluation (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.985A>G | S | c.985A>G | S | 1 | 1.73 | 1.91 | 0 | 0 | 0 | 128 | 14.4 | ||
| 2 | c.985A>G | S | c.985A>G | S | 1 | 7.14 | 1.3 | 1 | 1a | 1 | 1 | 0 | 109 | 13.7 |
| 3 | c.985A>G | S | c.985A>G | S | 1 | 1.05 | 5.33 | 0 | 0 | 0 | 102 | 13.7 | ||
| 4 | c.985A>G | S | c.985A>G | S | 1 | 7.58 | 10.1 | 1 | 1 | 0 | 1 | 0 | 79 | 12.3 |
| 5 | c.985A>G | S | c.985A>G | S | 1 | 6.49 | mv | mv | 1a | mv | 1 | 0 | 105 | 12.3 |
| 6 | c.985A>G | S | c.985A>G | S | 1 | 22.46 | 2.31 | mv | 0 | nd | nd | 11.3 | ||
| 7 | c.985A>G | S | c.985A>G | S | 1 | 23.19 | 5.5 | mv | 0 | 0 | 106 | 11.2 | ||
| 8 | c.985A>G | S | c.985A>G | S | 1 | 3.05 | 4.03 | mv | 0 | 0 | 104 | 9.9 | ||
| 9 | c.985A>G | S | c.985A>G | S | 1 | 4.69 | 1.73 | 1 | 0 | 0 | 104 | 6.4 | ||
| 10 | c.985A>G | S | c.985A>G | S | 1 | 1.87 | 3.1 | 1 | 0 | nd | 76 | 6.2 | ||
| 11 | c.985A>G | S | c.985A>G | S | 1 | 4.9 | 3.39 | 1 | 2 | mv | 1 | 0 | 119 | 6.8 |
| 12 | c.985A>G | S | c.985A>G | S | 1 | 6.43 | 3.96 | 1 | 0 | 0 | 118 | 5.9 | ||
| 13 | c.985A>G | S | c.985A>G | S | 1 | 3.92 | 3.06 | 1 | 0 | 0 | 120 | 5.3 | ||
| 14 | c.985A>G | S | c.985A>G | S | 1 | 6.54 | 1.59 | 1 | 0 | 0 | AA | 2.4 | ||
| 15 | c.985A>G | S | c.985A>G | S | 1 | 40.22 | 0.97 | 1 | 1a | 1 | 0 | 0 | AA | 1.8 |
| 16 | c.985A>G | S | c.985A>G | S | 1 | 7.89 | 1.46 | 1 | 0 | 0 | nd | 1.2 | ||
| 17 | c.799G>A | Sb | c.199T>C | M | 2 | 2.15 | 6.51c | 1 | 1a | 1 | 1 | 0 | 93 | 4.3 |
| 18 | c.403_405delATT | Sd | c.199T>C | M | 2 | 2.51 | 0.39 | 1 | 0 | 0 | 114 | 9.9 | ||
| 19 | c.985A>G | S | c.199T>C | M | 2 | 1.7 | 1.34 | 0 | 0 | 0 | 108 | 14.1 | ||
| 20 | c.985A>G | S | c.199T>C | M | 2 | 2.2 | 0.5 | 1 | 0 | 0 | 127 | 8.4 | ||
| 21 | c.985A>G | S | c.199T>C | M | 2 | 2.21 | 1.13 | 1 | 1a | 1 | 0 | 0 | 100 | 8.4 |
| 22 | c.985A>G | S | c.199T>C | M | 2 | 3.1 | 0.53 | 0 | 0 | 0 | 92 | 7.8 | ||
| 23 | c.985A>G | S | c.199T>C | M | 2 | 1.36 | 0.35 | 1 | 0 | 0 | 122 | 7.7 | ||
| 24 | c.985A>G | S | c.199T>C | M | 2 | 0.97 | 1.94 | mv | 0 | 0 | 122 | 7.8 | ||
| 25 | c.985A>G | S | c.199T>C | M | 2 | 1.17 | 0.37 | 1 | 0 | 0 | AA | 1.9 | ||
| 26 | c.985A>G | S | c.199T>C | M | 2 | 1.94 | 0.28 | 1 | 0 | 0 | AA | 1.5 | ||
| 27 | c.1140_1141insG | Ue/S | c.199T>C | M | 2 | 0.78 | 0.48 | mv | 0 | 0 | AA | 7.8 | ||
| 28 | c.985A>G | S | c.424_426delAAG | Sf | 3 | 6.17 | 4.6 | mv | 0 | 0 | nd | 4.3 | ||
| 29 | c.985A>G | S | c.721G>A | Ug | 3 | 14.38 | 5.72 | 1 | 0 | 0 | AA | 3.2 | ||
| 30 | c.985A>G | S | c.799G>A | Sb | 3 | 6.47 | 1.52 | 1 | 0 | 0 | nd | 1.6 | ||
| 31 | c.985A>G | S | c.387+1G>A | Sh | 3 | 4.97 | 1.06 | 1 | 0 | 1 | NAA | 3.9 | ||
| 32 | c.985A>G | S | c.362C>T | Si | 3 | 2.15 | 3.86 | 1 | 0 | nd | 107 | 7.1 | ||
| 33 | c.985A>G | S | c.244_245insT | Si | 3 | 10.82 | 3.88 | 1 | 0 | 0 | nd | 1.7 | ||
| 34 | c.985A>G | S | c.244_245insT | Si | 3 | 0.57 | 1.67 | 1 | 0 | 0 | nd | 0.6 | ||
| 35 | c.583G>A | Sj | c.583G>A | Sj | 4 | 9.27 | 3.82 | 1 | 0 | 0 | AA | 2.7 | ||
| 36 | c.799G>A | Sb | c.799G>A | Sb | 4 | 4.17 | 2.67 | 1 | 0 | 0 | 107 | 11.0 | ||
| 37 | c.799G>A | Sb | c.799G>A | Sb | 4 | 6.01 | 1.24 | mv | 0 | 0 | 90 | 7.7 |
aDecompensation in neonatal period
bSmith et al. (2010)
cSample was taken in period of decompensation
dSturm et al. (2012) (Nomenclature c.397_399delATT, enzyme activity 3%)
eSequence variant of unknown clinical significance; theoretically causing frameshift and premature stop codon, therefore presumably severe mutation; enzyme activity in this patient 13.9% according to ter Veld et al. (2009)
fEnzyme activity in lymphocytes 0% (performed at the University Children’s Hospital, Duesseldorf)
gMutation not previously described
hYusupov et al. (2010)
iAndresen et al. (2001)
jWaddell et al. (2006)
AA result of Denver test appropriate for age, C8 octanoylcarnitine, HG hexanoylglycine in urine, mv missing value, NAA result of Denver test not appropriate for age, NBS newborn screening, nd not done, S severe (assumed mutation severity), M mild (assumed mutation severity), U mutation severity unknown
In one patient of German origin, a previously unreported mutation (HGMD 2013) was found. Patient 29 carried c.985A>G and c.721G>A/p.(Gly241Ser) (conventional nomenclature: Gly216Ser). A pathogenetic relevance of c.721G>A seems likely, as glycine at position 241 lies in a conserved region. Enzyme activity in lymphocytes (AMC Amsterdam) was <0.06 nmol/min mg protein (norm 0.43–1.63). The patient never showed a symptomatic episode, but was so far admitted five times for emergency treatment during intercurrent infections.
Patient 18 carried c.403_405delATT/p.(Ile135del) (conventional nomenclature: Ile110del), causing loss of isoleucine at position 135, together with c.199T>C. Enzyme activity in lymphocytes (AMC Amsterdam) was 0.18 nmol/min mg protein (norm 0.43–1.63). The patient never showed a symptomatic episode and never required inpatient emergency treatment. Sturm et al. described this mutation under a different nomenclature as c.397_399delATT in a homozygous patient associated with residual enzyme activity of 3% (Sturm et al. 2012).
Patient 27 carried c.199T>C and c.1140_1141insG, the latter being a sequence variant of unknown clinical significance. Theoretically it causes frame shift and a premature stop codon and is therefore a presumably severe mutation. Enzyme activity in this patient was 13.9% according to ter Veld et al. (2009). This patient never showed a symptomatic episode.
Of the patients in genotype group 4 (neither c.985A>G nor c.199T>C), two were homozygous for c.799G>A/p.(Gly267Arg) (conventional nomenclature: Gly242Arg), and one was homozygous for c.583G>A/p.(Gly195Arg) (conventional nomenclature: Gly170Arg). All patients were of Turkish origin.
Biochemical Parameters
Levels of C8, C8/C2, C8/C10 and C8/C12 at screening and confirmation in genotype groups 1–4 are shown in Table 2. At screening C8/C2 (F(3,34) = 3.26, p = 0.049, d = 0.44) and C8/C10 (F(3,34) = 37.28, p < 0.001, d = 1.58) differed significantly between genotype groups. For both ratios, post hoc comparisons were significant between groups 1 and 2, 2 and 3, and 2 and 4 (all p-values ≤ 0.03). There was a trend towards a difference in C8 levels (F(3,33) = 2.24, p = 0.096, d = 0.37) and C8/C12 (F(3,33) = 2.49, p = 0.087, d = 0.39) between genotype groups.
Table 2.
Biochemical parameters for the four genotype groups in the first newborn screening sample (upper lines) and at confirmation (lower lines)
| Genotype group | C8 | C8/C2 | C8/C10 | C8/C12 | |
|---|---|---|---|---|---|
| Group 1 | |||||
| c.985A>G/c.985A>G | Mean | 9.32 | 0.42 | 10.99 | 95.55 |
| SD | 10.47 | 0.41 | 2.43 | 114.74 | |
| Range | 1.05; 40.22 | 0.08; 1.83 | 6.41; 15.71 | 9.55; 449.20 | |
| Mean | 3.23 | 0.45 | 11.37 | 44.39 | |
| SD | 2.44 | 0.39 | 2.95 | 28.52 | |
| Range | 0.97; 10.10 | 0.08; 1.29 | 6.30; 18.25 | 5.80; 82.00 | |
| Group 2 | |||||
| c.199T>C/c.985A>G or another mutation | Mean | 1.83 | 0.07 | 2.67 | 13.21 |
| SD | 0.71 | 0.03 | 0.44 | 7.20 | |
| Range | 0.78; 3.10 | 0.03; 0.14 | 1.87; 3.33 | 5.86; 31.38 | |
| Group 3 | |||||
| c.985A>G/other than c.199T>C | Mean | 1.26 | 0.06 | 2.14 | 8.84 |
| SD | 2.14 | 0.05 | 0.65 | 4.11 | |
| Range | 0.28; 6.51 | 0.01; 0.20 | 1.55; 4.12 | 4.41; 15.50 | |
| Mean | 6.50 | 0.27 | 10.97 | 46.43 | |
| SD | 4.78 | 0.16 | 2.99 | 22.53 | |
| Range | 0.57; 14.38 | 0.10; 0.55 | 7.68; 15.06 | 14.25; 79.99 | |
| Mean | 2.95 | 0.23 | 10.35 | 42.49 | |
| SD | 1.82 | 0.12 | 2.59 | 19.31 | |
| Range | 1.06; 5.72 | 0.07; 0.36 | 7.24; 13.62 | 21.20; 71.50 | |
| Group 4 | |||||
| Neither c.985A>G nor c.199T>C | Mean | 6.48 | 0.29 | 8.49 | 46.90 |
| SD | 2.58 | 0.26 | 1.98 | 28.33 | |
| Range | 4.17; 9.27 | 0.03; 0.55 | 6.20; 9.70 | 14.38; 66.21 | |
| Mean | 3.25 | 0.20 | 9.29 | 28.49 | |
| SD | 0.81 | 0.21 | 0.86 | 8.82 | |
| Range | 1.24; 3.82 | 0.04; 0.35 | 8.68; 9.89 | 22.25; 34.73 | |
At confirmation C8/C2 (F(3,27) = 3.42, p = 0.033, d = 0.51), C8/C10 (F(3,28) = 29.80, p < 0.001, d = 1.49) and C8/C12 (F(3,26) = 4.89, p = 0.008, d = 0.66) differed significantly between genotype groups. Post hoc comparisons were significant between groups 1 and 2 and 2 and 3 for all ratios and between 2 and 4 only for C8/C10 and C8/C12 (all p-values ≤ 0.022).
Comparing only patients from genotype group 1 and 2, all acylcarnitine parameters evaluated were significantly higher (all p-values ≤ 0.018) in group 1 at screening and confirmation (compare Table 2). The best discriminative parameter between these two groups was C8/C10, both at screening and confirmation, as there was no overlap between the two groups.
Presence of hexanoylglycine in urine did not differ between genotype groups (Fisher’s exact test, p = 0.741).
Time of Sampling
Median age at blood sampling for screening was 3.0 days (mean 3.5, SD 2.2, range 1.5–14.1) and at confirmation 10.5 days (mean 12.9, SD 10.3, range 4.0–56.0). There was no significant difference between genotype groups regarding the distribution of time of blood sampling for screening (Anderson-Darling test adjusted for ties: AD (N = 37) = −0.93, p = 0.693) and confirmation (AD (N = 36) = 0.80, p = 0.182). Regression equations and models for curve fitting are shown for parameters at screening for genotype groups 1 + 3 + 4 vs. group 2 in Fig. 1. At screening, in genotype groups 1 + 3 + 4, C8, C8/C2 and C10 were strongly associated with age at sampling (C8, ; C8/C2, ; C10, ). In genotype group 2, there was no association between time of sampling and any of the parameters assessed (maximum ). At confirmation none of the parameters showed an association with time of sampling in any genotype group (maximum ).
Fig. 1.
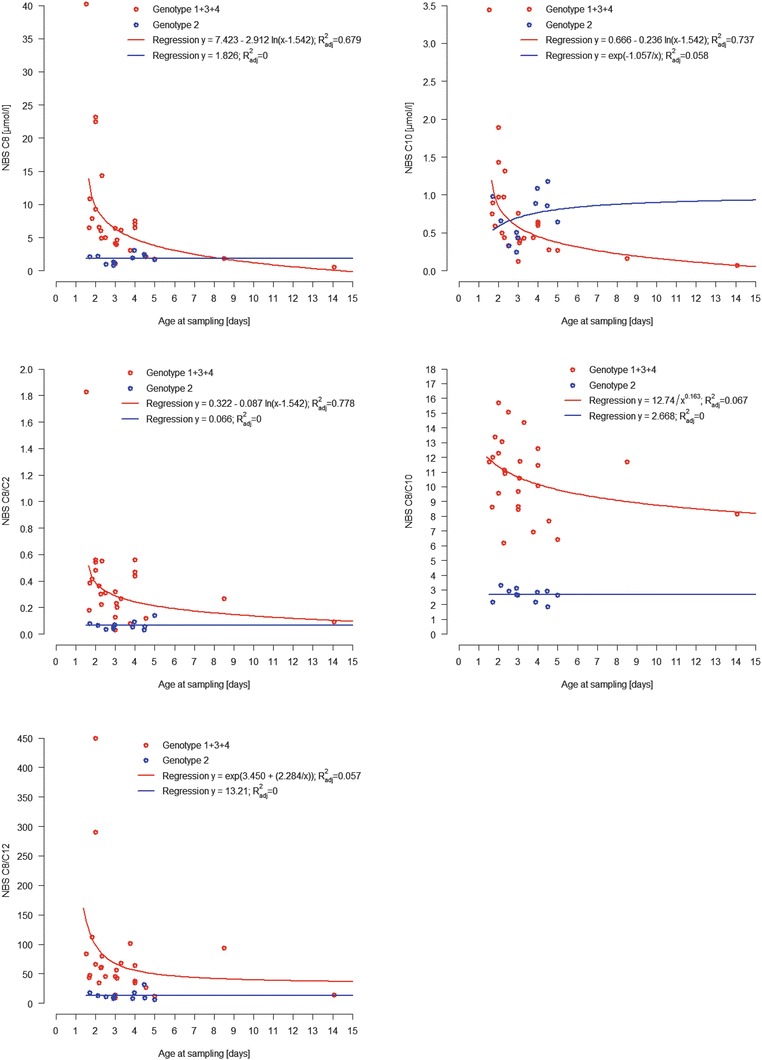
Biochemical parameters at first screening in relation to age at sampling in genotype groups 1 + 3 + 4 and 2. Regression equations and regression models for nonlinear curve fitting are shown for C8 (logarithmic and linear model), C10 (logarithmic and S-model), C8/C2 (logarithmic and linear model), C8/C10 (hyperbolic and linear model) and C8/C12 (S-model and linear model)
Clinical Outcome
Symptomatic episodes, defined as episodes with hypoglycaemia and/or reduced consciousness, occurred in 7 of 37 patients (18.9%; details in Table 1). None of these episodes resulted in death. Four patients showed an episode with hypoglycaemia (Patients 2, 15, 17, 21), all in the neonatal period before screening results were available. Two of these patients were homozygous for c.985A>G (patients 2 and 15), one compound heterozygous for c.799G>A and c.199T>C (patient 17) and one for c.985A>G and c.199T>C (patient 21). Patient 17 was admitted to hospital on day 4 of life because of pronounced weight loss and tachypnea and showed metabolic acidosis, hypoglycaemia (blood glucose 2 mmol/L) and ketonuria (ketonstix +++). Residual enzyme activity in lymphocytes was 19% (University Children’s Hospital, Düsseldorf). Patient 21 showed postnatal hypoglycaemia (blood glucose 1.2 mmol/L) and was treated on the neonatal intensive care unit with oral feeds of carbohydrates on the first day of life. Patients 2 and 17 also showed reduced consciousness in these episodes.
Three patients, all c.985A>G homozygous, had episodes with reduced consciousness without hypoglycaemia or without information on blood glucose level. Patient 11 showed two episodes (aged 20 and 21 months) with reduced consciousness after 12 h overnight fast. Blood glucose was not measured, but after application of glucose polymer by the parents, the patient was reported to have recovered quickly and to be vigilant again. Patient 4 was admitted to intensive care aged 5 weeks with severe dehydration and reduced consciousness, but normal blood glucose. Patient 5 showed neonatal decompensation with coma and need for resuscitation. This event took place at an external hospital, and unfortunately we do not have information on blood glucose levels from this episode.
The number of symptomatic episodes (p = 0.675) and hospital admissions for emergency treatment (p = 0.392) did not differ between genotype groups. Except for delayed speech development in patient 31, there were no relevant pathological findings in clinical status in any of the patients. Results of developmental tests were normal in 90.3% of patients (28 of 31), which is not different from a standard cohort (about 16% IQ < 85). Percentage of subnormal developmental test results or abnormal clinical findings did not differ between genotype groups. Patient 5 who had experienced neonatal decompensation with coma and need for resuscitation showed normal clinical status and intellectual development (IQ 105).
Discussion
MCADD is widely accepted as suitable condition for newborn screening, as death or neurological sequelae can mostly be prevented by prophylactic measures (Nennstiel-Ratzel et al. 2005; Wilcken et al. 2007; Lindner et al. 2011). This finding could be replicated in our follow-up study. As for many other conditions, also in MCADD the number of patients identified by screening is higher than the number of patients identified clinically (Derks et al. 2005; Wilcken et al. 2007; Wilcken et al. 2009). The genotypic spectrum in screened cohorts includes presumably mild mutations like c.199T>C (Andresen et al. 2001; Maier et al. 2005), and there is ongoing discussion about genotype-phenotype correlation. In our study we assessed biochemical and clinical phenotype in relation to genotype in patients detected by newborn screening.
Biochemical Phenotype
We found significant differences and trends between genotype groups in several acylcarnitine markers at screening and confirmation. Patients compound heterozygous for c.199T>C and c.985A>G or another mutation showed lower acylcarnitine markers than the other genotype groups, which is in line with previous reports (Maier et al. 2005; Waddell et al. 2006). In contrast to results from an Australian cohort (Waddell et al. 2006), we found no difference between patients homozygous for c.985A>G and compound heterozygous for c.985A>G in combination with mutations other than c.199T>C. This is in accordance with the report by Maier and colleagues (2005). Other authors (Sturm et al. 2012) reported a lack of association between genotype and octanoylcarnitine levels at screening due to an overlap between patients homozygous for c.985A>G and compound heterozygous for c.985A>G and c.199T>C. However, this study did not consider ratios between acylcarnitines and did not apply statistical methods to compare biochemical parameters between genotypes.
In our study, patients carrying neither c.985A>G nor c.199T>C but other known homozygous mutations did not differ biochemically from patients carrying c.985A>G (homozygous or in combination with mutations other than c.199T>C), but from patients carrying c.199T>C compound heterozygous with c.985A>G or another mutation. Two of these three patients were homozygous for c.799G>A, which has been described as severe (Smith et al. 2010; Sturm et al. 2012) and has been found in symptomatic patients (Yokota et al. 1991; Andresen et al. 1997). In contrast to only mild elevations of acylcarnitine markers found in three patients homozygous for c.799G>A reported by Maier et al. (2005), which lay in the range of patients carrying c.199T>C and another mutation, C8 levels in our patients homozygous for c.799G>A lay above the range of patients carrying c.199T>C and another mutation.
In our patient collective, we found C8/C10 to best discriminate between patients homozygous for c.985A>G and compound heterozygous for c.199T>C and c.985A>G or another mutation, which is in line with other reports (Maier et al. 2009; Smith et al. 2010).
Time of Sampling
It has been reported that time of sampling affects octanoylcarnitine levels in MCADD (Rhead 2006; Maier et al. 2009), but not in unaffected newborns (Khalid et al. 2010). As age at sampling did not differ between genotype groups, our collective was eligible for assessment of both the association between biochemical parameters and genotype and biochemical parameters and time of sampling. Only octanoylcarnitine, C8/C2 and C10 at screening showed a strong association with age at sampling in patients with genotypes not including c.199T>C, with both levels decreasing with time. In patients compound heterozygous for c.199T>C and c.985A>G or another mutation, no association was found between time of sampling and any of the parameters at screening. At confirmation, none of the parameters showed an association with time in any of the genotype groups. It is important to be aware of this age dependency in interpretation of screening results, especially if the first sample has been drawn later than the recommended age of 36–72 h.
Clinical Phenotype
Previous studies have come to the conclusion that in MCADD, correlation between genotype and clinical phenotype is not straightforward (Wilcken et al. 1994; Heptinstall et al. 1995; Andresen et al. 1997) and that clinical variation may result from differences in metabolic stress experienced rather than differences in genotype (Andresen et al. 1997). In our study neonatal episodes of hypoglycaemia were also documented for two patients carrying the presumably mild mutation c.199T>C. So far it had been stated that patients compound heterozygous for c.199T>C and another mutation had never been found to be symptomatic (Andresen et al. 2001; Maier et al. 2005; Waddell et al. 2006; Smith et al. 2010; Sturm et al. 2012). O’Reilly reported that patients carrying c.199T>C compound heterozygous with c.985A>G had shown significant lethargy without hypoglycaemia in an episode of vomiting, requiring hospital admission (O’Reilly et al. 2004). Both our patients carried a known disease-causing mutation on the second allele (patient 17 c.199T>C and c.799G>A; patient 21 c.199T>C and c.985A>G). The mutation c.799G>A has been found in patients detected asymptomatically by newborn screening (Zschocke et al. 2001), as well as in symptomatic patients (Yokota et al. 1991; Andresen et al. 1997). This mutation has been classified as severe (Smith et al. 2010; Sturm et al. 2012). Residual enzyme activity of 19% in our patient 17 (c.199T>C and c.799G>A) was in the range of disease-causing mutations.
Sturm et al. (2012) found residual enzyme activities in patients compound heterozygous for c.199T>C and a severe mutation between 28% and 49% and discussed that c.199T>C may be an innocuous polymorphism without clinical relevance. As a consequence of hypoglycaemia in our two patients carrying c.199T>C and a severe mutation, it should be considered that also patients with presumably mild mutations might become symptomatic under metabolic stress. Thus, it does not seem safe to predict a completely asymptomatic clinical phenotype from the milder biochemical phenotype. Neonatal hypoglycaemia or death has been reported in patients with MCADD diagnosed symptomatically (Wilcken et al. 1994; Derks et al. 2006; Wilcken et al. 2007). However, it could be discussed that neonatal hypoglycaemia in our patients might have been unrelated to MCADD. Neonatal hypoglycaemia affects 5–15% of otherwise healthy newborns (Harris et al. 2013). Patient 17 showed ketotic hypoglycaemia, which is not typical for MCADD, but has been reported in a relevant number of MCADD patients before (Iafolla et al. 1994). This finding can be explained by some residual MCAD function. Hsu also reported a patient with MCADD (c.985A>G and c.127G>A) with marked ketonuria during an intercurrent infection (Hsu et al. 2008).
In our study, reduced consciousness was documented in one patient homozygous for c.985A>G without hypoglycaemia. Reduced vigilance might be explained by severe dehydration in this patient. However, some authors postulate that in MCADD episodes of reduced consciousness can occur without hypoglycaemia due to accumulation of free fatty acids and their carnitine and CoA esters (Mayell et al. 2007; Saudubray et al. 2012).
So far, all our patients with MCADD detected by newborn screening receive uniform recommendations regarding feeding intervals and emergency management. Under this prophylactic treatment, no difference was found between different genotype groups concerning number of metabolic decompensations, inpatient emergency treatment and the overall excellent clinical outcome. Concerning clinical evaluation, the finding of delayed speech development in one patient – who had never experienced a metabolic decompensation – may well be unrelated to MCADD, as it is common in the general population (Gottschling et al. 2012). The percentage of MCADD patients with subnormal psychometric test results (10%) was not higher than expected in the general population.
Conclusion
Patients with MCADD homozygous for c.985A>G did not differ biochemically from patients compound heterozygous for c.985A>G and mutations other than c.199T>C. Patients carrying c.199T>C in combination with c.985A>G or another mutation showed significantly lower acylcarnitine markers compared to other genotypes and thus a “milder” biochemical phenotype. Evaluation of clinical phenotype of our patients showed – to our knowledge for the first time – that neonatal decompensation with hypoglycaemia can also occur in patients carrying c.199T>C and a severe mutation prior to diagnosis. Therefore, it does not seem safe to predict an asymptomatic clinical phenotype from the milder biochemical phenotype. This leads to the conclusion that also in patients carrying c.199T>C compound heterozygous with a severe mutation, there may be the risk of MCADD-associated hypoglycaemia under severe metabolic stress. Given uniform treatment recommendations, no differences were found in clinical phenotype between different genotypes.
Acknowledgements
This study was made possible by the continuous and generous support of the Dietmar Hopp Foundation, St. Leon-Rot.
The authors thank all patients and their families for their participation and trust.
Many thanks to all the colleagues who provided information on their patients: U. Wendel, E. Thimm (Düsseldorf), E. Mengel (Mainz), F.K. Trefz (Reutlingen) and M. Baumgartner (Zürich).
Enzyme activity in patients 18 and 29 was analysed by Prof. Wanders, AMC, Amsterdam, the Netherlands, and in patients 17 and 28 by Prof. Spiekerkötter at the University Children’s Hospital, Düsseldorf, Germany.
Take-Home Message
Biochemical but not clinical phenotype differs between MCADD patients with different genotypes detected by newborn screening following uniform treatment recommendations. Neonatal decompensation can also occur in patients carrying the presumably mild mutation c.199T>C compound heterozygous with a severe mutation prior to diagnosis.
Compliance with Ethics Guidelines
Conflict of Interest
Authors’ Disclosures
G. Gramer received support for travel expenses to a scientific meeting from Merck Serono.
G. Haege reports no disclosures.
J. Fang-Hoffmann reports no disclosures.
G.F. Hoffmann received lecture fees from Nutricia, Friedrichsdorf i.T.
C. R. Bartram reports no disclosures.
K. Hinderhofer reports no disclosures.
P. Burgard is member of an advisory board for Merck Serono, Darmstadt, and received lecture fees from Merck Serono, Darmstadt; Vitaflo, Bad Homburg; and Nutricia, Friedrichsdorf i.T.
M. Lindner received lecture fees from Orphan Europe, Milupa and Nutricia.
Conflict of Interest
None
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients/their parents for being included in the study. The study was approved by the University Hospital Heidelberg ethical committee (IRB code 104/2005).
Authors’ Contributions
G. Gramer: Study design; recruitment of patients; collection, evaluation and interpretation of data; drafting and writing the manuscript
G. Haege: Statistical analysis; evaluation and interpretation of data; writing and revision of the manuscript
J. Fang-Hoffmann: Data collection; revision of the manuscript
G. F. Hoffmann: Study design; evaluation and interpretation of data; revision of the manuscript
C. R. Bartram: Molecular genetic analyses; revision of the manuscript
K. Hinderhofer: Molecular genetic analyses; revision of the manuscript
P. Burgard: Study design; recruitment of patients; collection, evaluation and interpretation of data; statistical analysis; revision of the manuscript
M. Lindner: Study design; recruitment of patients; collection, evaluation and interpretation of data; revision of the manuscript
All authors approved the final version of the manuscript.
G. Gramer serves as guarantor for the article.
Funding
This study was supported by the Dietmar Hopp Foundation, St. Leon-Rot, Germany. The authors confirm independence from the sponsor; the content of the article has not been influenced by the sponsor.
G. Gramer was supported by a research scholarship (Olympia Morata Programme) of the Medical Faculty of the University of Heidelberg.
Contributor Information
Gwendolyn Gramer, Email: gwendolyn.gramer@med.uni-heidelberg.de.
Collaborators: Johannes Zschocke
References
- Andresen BS, Bross P, Udvari S, et al. The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: is there correlation between genotype and phenotype? Hum Mol Genet. 1997;6:695–707. doi: 10.1093/hmg/6.5.695. [DOI] [PubMed] [Google Scholar]
- Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001;68:1408–1418. doi: 10.1086/320602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundesausschuss GBdÄuKG (2005) Beschluss über eine Änderung der Richtlinen des Bundesausschusses der Ärzte und Krankenkassen über die Früherkennung von Krankheiten bei Kindern bis zur Vollendung des 6. Lebensjahres (Kinder-Richtlinien) zur Einführung des erweiterten Neugeborenen-Screenings, 2011. http://www.g-ba.de/informationen/beschluesse/zur-richtlinie/15/#170
- Derks TG, Duran M, Waterham HR, Reijngoud DJ, Ten Kate LP, Smit GP. The difference between observed and expected prevalence of MCAD deficiency in The Netherlands: a genetic epidemiological study. Eur J Hum Genet. 2005;13:947–952. doi: 10.1038/sj.ejhg.5201428. [DOI] [PubMed] [Google Scholar]
- Derks TG, Reijngoud DJ, Waterham HR, et al. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006;148:665–670. doi: 10.1016/j.jpeds.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Edgington ES. Randomization tests. New York: Marcel-Dekker; 1995. [Google Scholar]
- Elzhov TV, Mullen KM, Spiess A, Bolker B (2013) Minpack.lm: R interface to the Levenberg-Marquardt nonlinear least-squares algorithm found in MINPACK, plus support for bounds. http://cran.r-project.org/web/packages/minpack.lm/index.html
- Gottschling A, Franze M, Hoffmann W. Entwicklungsverzögerungen bei Kindern: Screening als Grundlage für eine gezielte Förderung. Dtsch Arztebl. 2012;3:123–125. [Google Scholar]
- Gregersen N, Blakemore AI, Winter V, et al. Specific diagnosis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in dried blood spots by a polymerase chain reaction (PCR) assay detecting a point-mutation (G985) in the MCAD gene. Clin Chim Acta. 1991;203:23–34. doi: 10.1016/0009-8981(91)90153-4. [DOI] [PubMed] [Google Scholar]
- Grosse SD, Khoury MJ, Greene CL, Crider KS, Pollitt RJ. The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: an update. Genet Med. 2006;8:205–212. doi: 10.1097/01.gim.0000204472.25153.8d. [DOI] [PubMed] [Google Scholar]
- Harms E, Roscher A, Grüters A, et al. Neue Screening-Richtlinien - Richtlinien zur Organisation und Durchführung des Neugeborenenscreenings auf angeborene Stoffwechselstörungen und Endokrinopathien in Deutschland. Monatsschr Kinderheilkd. 2002;150:1424–1440. doi: 10.1007/s00112-002-0611-z. [DOI] [Google Scholar]
- Harris DL, Weston PJ, Signal M, Chase JG, Harding JE. Dextrose gel for neonatal hypoglycaemia (the Sugar Babies Study): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382:2077–2083. doi: 10.1016/S0140-6736(13)61645-1. [DOI] [PubMed] [Google Scholar]
- Heptinstall LE, Till J, Wraith JE, Besley GT. Common MCAD mutation in a healthy parent of two affected siblings. J Inherit Metab Dis. 1995;18:638–639. doi: 10.1007/BF02436011. [DOI] [PubMed] [Google Scholar]
- HGMD THGMD (2013) The Human gene mutation database HGMD. Retrieved 13 June, 2013 from http://www.hgmd.cf.ac.uk/ac/index.php
- Hoffmann G, Aramaki S, Blum-Hoffmann E, Nyhan WL, Sweetman L. Quantitative analysis for organic acids in biological samples: batch isolation followed by gas chromatographic-mass spectrometric analysis. Clin Chem. 1989;35:587–595. [PubMed] [Google Scholar]
- Howell DC (2001) Resampling Procedures Version 1.3. http://www.uvm.edu/~dhowell/StatPages/Resampling/ResamplingPackage.zip. Accessed 9 Nov 2010
- Hsu HW, Zytkovicz TH, Comeau AM, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008;121:e1108–e1114. doi: 10.1542/peds.2007-1993. [DOI] [PubMed] [Google Scholar]
- Iafolla AK, Thompson RJ, Jr, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994;124:409–415. doi: 10.1016/S0022-3476(94)70363-9. [DOI] [PubMed] [Google Scholar]
- Khalid JM, Oerton J, Besley G, et al. Relationship of octanoylcarnitine concentrations to age at sampling in unaffected newborns screened for medium-chain acyl-CoA dehydrogenase deficiency. Clin Chem. 2010;56:1015–1021. doi: 10.1373/clinchem.2010.143891. [DOI] [PubMed] [Google Scholar]
- Lindner M, Gramer G, Haege G, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases-report of 10 years from South-West Germany. Orphanet J Rare Dis. 2011;6:44. doi: 10.1186/1750-1172-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier EM, Liebl B, Roschinger W, et al. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat. 2005;25:443–452. doi: 10.1002/humu.20163. [DOI] [PubMed] [Google Scholar]
- Maier EM, Pongratz J, Muntau AC, et al. Dissection of biochemical borderline phenotypes in carriers and genetic variants of medium-chain acyl-CoA dehydrogenase deficiency: implications for newborn screening [corrected] Clin Genet. 2009;76:179–187. doi: 10.1111/j.1399-0004.2009.01217.x. [DOI] [PubMed] [Google Scholar]
- Mayell SJ, Edwards L, Reynolds FE, Chakrapani AB. Late presentation of medium-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2007;30:104. doi: 10.1007/s10545-006-0488-4. [DOI] [PubMed] [Google Scholar]
- National Newborn Screening & Genetics Resource Center Austin Texas. National Newborn Screening & Genetics Resource Center, Austin, Texas. http://genes-r-us.uthscsa.edu/
- Nennstiel-Ratzel U, Arenz S, Maier EM, et al. Reduced incidence of severe metabolic crisis or death in children with medium chain acyl-CoA dehydrogenase deficiency homozygous for c.985A>G identified by neonatal screening. Mol Genet Metab. 2005;85:157–159. doi: 10.1016/j.ymgme.2004.12.010. [DOI] [PubMed] [Google Scholar]
- O’Reilly L, Bross P, Corydon TJ, et al. The Y42H mutation in medium-chain acyl-CoA dehydrogenase, which is prevalent in babies identified by MS/MS-based newborn screening, is temperature sensitive. Eur J Biochem. 2004;271:4053–4063. doi: 10.1111/j.1432-1033.2004.04343.x. [DOI] [PubMed] [Google Scholar]
- Rhead WJ. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: a global perspective. J Inherit Metab Dis. 2006;29:370–377. doi: 10.1007/s10545-006-0292-1. [DOI] [PubMed] [Google Scholar]
- Saudubray JM, Van den Berghe G, Walter JH, editors. Inborn metabolic diseases. Berlin: Springer; 2012. [Google Scholar]
- Scholz F (2011) adk: Anderson-Darling K-sample test and combinations of such tests. Retrieved 27 Sept 2013 from http://cran.r-project.org/web/packages/adk/index.html
- Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111:1399–1406. doi: 10.1542/peds.111.6.1399. [DOI] [PubMed] [Google Scholar]
- Smith EH, Thomas C, McHugh D, et al. Allelic diversity in MCAD deficiency: the biochemical classification of 54 variants identified during 5 years of ACADM sequencing. Mol Genet Metab. 2010;100:241–250. doi: 10.1016/j.ymgme.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Smucker MD, Allan J, Carterette B (2007) A comparison of statistical significance tests for information retrieval evaluation. Conference on information and knowledge management (CIKM). ACM Press, Lisbon
- Spiess A (2012) qpcR: modelling and analysis of real-time PCR data. http://cran.r-project.org/web/packages/qpcR/index.html.
- Spiess AN, Neumeyer N. An evaluation of R2 as an inadequate measure for nonlinear models in pharmacological and biochemical research: a Monte Carlo approach. BMC Pharmacol. 2010;10:6. doi: 10.1186/1471-2210-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm M, Herebian D, Mueller M, Laryea MD, Spiekerkoetter U. Functional effects of different medium-chain acyl-CoA dehydrogenase genotypes and identification of asymptomatic variants. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Veld F, Mueller M, Kramer S, et al. A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell L, Wiley V, Carpenter K, et al. Medium-chain acyl-CoA dehydrogenase deficiency: genotype-biochemical phenotype correlations. Mol Genet Metab. 2006;87:32–39. doi: 10.1016/j.ymgme.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Wilcken B, Hammond J, Silink M. Morbidity and mortality in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child. 1994;70:410–412. doi: 10.1136/adc.70.5.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken B, Haas M, Joy P, et al. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet. 2007;369:37–42. doi: 10.1016/S0140-6736(07)60029-4. [DOI] [PubMed] [Google Scholar]
- Wilcken B, Haas M, Joy P, et al. Expanded newborn screening: outcome in screened and unscreened patients at age 6 years. Pediatrics. 2009;124:e241–e248. doi: 10.1542/peds.2008-0586. [DOI] [PubMed] [Google Scholar]
- Yokota I, Coates PM, Hale DE, Rinaldo P, Tanaka K. Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet. 1991;49:1280–1291. [PMC free article] [PubMed] [Google Scholar]
- Yusupov R, Finegold DN, Naylor EW, Sahai I, Waisbren S, Levy HL. Sudden death in medium chain acyl-coenzyme a dehydrogenase deficiency (MCADD) despite newborn screening. Mol Genet Metab. 2010;101:33–39. doi: 10.1016/j.ymgme.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Ziadeh R, Hoffman EP, Finegold DN, et al. Medium chain acyl-CoA dehydrogenase deficiency in Pennsylvania: neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr Res. 1995;37:675–678. doi: 10.1203/00006450-199505000-00021. [DOI] [PubMed] [Google Scholar]
- Zschocke J, Schulze A, Lindner M, et al. Molecular and functional characterisation of mild MCAD deficiency. Hum Genet. 2001;108:404–408. doi: 10.1007/s004390100501. [DOI] [PubMed] [Google Scholar]