

Abstract
Objective: To study the clinical manifestations and occurrence of mtDNA depletion and deletions in paediatric patients with neuromuscular diseases and to identify novel clinical phenotypes associated with mtDNA depletion or deletions.
Methods: Muscle DNA samples from patients presenting with undefined encephalomyopathies or myopathies were analysed for mtDNA content by quantitative real-time PCR and for deletions by long-range PCR. Direct sequencing of mtDNA maintenance genes and whole-exome sequencing were used to study the genetic aetiologies of the diseases. Clinical and laboratory findings were collected.
Results: Muscle samples were obtained from 104 paediatric patients with neuromuscular diseases. mtDNA depletion was found in three patients with severe early-onset encephalomyopathy or myopathy. Two of these patients presented with novel types of mitochondrial DNA depletion syndromes associated with increased serum creatine kinase (CK) and multiorgan disease without mutations in any of the known mtDNA maintenance genes; one patient had pathologic endoplasmic reticulum (ER) membranes in muscle. The third patient with mtDNA depletion was diagnosed with merosine-deficient muscular dystrophy caused by a homozygous mutation in the LAMA2 gene. Two patients with an early-onset Kearns-Sayre/Pearson-like phenotype harboured a large-scale mtDNA deletion, minor multiple deletions and high mtDNA content.
Conclusions: Novel encephalomyopathic mtDNA depletion syndrome with structural alterations in muscle ER was identified. mtDNA depletion may also refer to secondary mitochondrial changes related to muscular dystrophy. We suggest that a large-scale mtDNA deletion, minor multiple deletions and high mtDNA content associated with Kearns-Sayre/Pearson syndromes may be secondary changes caused by mutations in an unknown nuclear gene.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_438) contains supplementary material, which is available to authorized users.
Introduction
Mitochondrial DNA depletion syndromes (MDDS) are a clinically and genetically heterogeneous group of typically recessively inherited diseases with early or juvenile onset that are characterized by a severe reduction of mtDNA content (Alberio et al. 2007). Based on affected tissues and their mtDNA content, the clinical presentations of MDDS can be classified into three different forms: encephalomyopathic, myopathic and hepatocerebral. For clinical purposes, mtDNA depletion has been defined as mtDNA content of <0.30 relative to age-matched controls (Rahman and Poulton 2009). However, measuring intracellular mtDNA content is technically challenging and the amount of mtDNA is age- and tissue-related (Dimmock et al. 2010; Morten et al. 2007). Numerous pathogenic mutations have been found in the 12 nuclear genes responsible for encoding proteins vital to mtDNA maintenance (El-Hattab and Scaglia 2013; Suomalainen and Isohanni 2010).
Mitochondrial DNA (mtDNA) deletions are qualitative mitochondrial genome defects that come from the loss of mtDNA molecule fragments (Krishnan et al. 2008). They are most likely to be caused by defects that occur during the repair or replication of mtDNA (Krishnan et al. 2008; Yu-Wai-Man and Chinnery 2012). mtDNA deletions are associated with several clinical syndromes, but they also increase with age (Krishnan et al. 2007). Mutations in nuclear genes involved in mtDNA replication and maintenance, e.g., POLG1 (NM_001126131.1) encoding the catalytic subunit of mitochondrial DNA polymerase γ, can cause accumulation of mtDNA point mutations and deletions throughout life (Chan and Copeland 2009). The most common single large-scale mtDNA deletion is 4,977 base pairs (bp) spanning between the genes for cytochrome B (CytB) and cytochrome c oxidase subunit II (COXII) (Krishnan et al. 2007; Remes et al. 2005).
The aim of this study was to investigate the occurrence of mtDNA depletions and deletions in muscle biopsy samples of paediatric patients with undefined encephalomyopathy or myopathy, in order to estimate the role of mtDNA rearrangements in pathogenesis of these diseases. Further objective was to identify novel clinical phenotypes associated with mtDNA depletion or deletions.
Subjects and Methods
Patients and Controls
Skeletal muscle biopsy samples were taken as a part of the diagnostic protocol and collected from patients under 18 years with undefined encephalomyopathy or myopathy, who were examined at the Department of Paediatrics of Oulu University Hospital between 1990 and 2012 by the protocol including both clinical assessments and histological, biochemical and/or molecular genetic analyses (Uusimaa et al. 2000). All 104 patients were screened for the eight common POLG1 mutations (p.T251I, p.A467T, p.N468N, p.G517V, p.P587L, p.R722H, p.W748S and p.Y955C) and the common MELAS m.3423A>G, MERRF m.8834A>G and NARP m.8993T>G mutations in mtDNA. The control muscle samples were collected by the Department of Paediatrics at Oulu University Hospital between the years 2008 and 2012. The samples were taken from patients with no signs of mitochondrial or other neurological diseases during surgical treatments for orthopaedic conditions where mitochondrial function has not been shown to play a major role. Muscle biopsies of 0.25–0.5 cm3 in diameter were performed during an operation requiring an incision through muscle tissue. The biopsies were only done if unnecessary trauma could be avoided. The collection of muscle tissue samples did not interfere with the result of the operation or the recovery of the patient.
The study protocol has been approved by the Ethics Committee of the Faculty of Medicine at the University of Oulu and the Ethics Committee of the Northern Ostrobothnia Hospital District and it is in compliance with the Helsinki Declaration. Written informed consent was given by the guardians of the subjects prior to the study.
DNA Extraction from Muscle Biopsy Samples and Fibroblasts
Total genomic DNA was extracted from skeletal muscle samples using standard phenol-chloroform-isoamyl alcohol extraction (PCIAA) and ethanol precipitation or by using a commercially available Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). All extracted DNA samples were aliquoted into small fractions and stored in −80°C. DNA for whole-exome sequencing was extracted from patient fibroblasts by using a commercially available QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and ethanol precipitation. Fibroblasts were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) including 5 mM glucose, sodium bicarbonate and pyridoxine and supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 IU/mL penicillin and 100 μg/mL streptomycin.
Real-Time Quantitative PCR of Muscle DNA Samples
Real-time quantitative PCR (qRT-PCR) was used to determine the amount of mtDNA relative to nuclear DNA in both patient and control muscle samples and was shown as an mtDNA/nDNA ratio. mtDNA was amplified using PCR primers targeted at the mitochondrial NADH dehydrogenase 1 (ND1) gene as described by He et al. (He et al. 2002). The values were normalized using the nuclear-encoded brain natriuretic peptide (BNP) gene as a single-copy nuclear gene. Amplification products were detected by sequence-specific 6FAM/TAMRA-labelled fluorogenic probes (Sigma-Genosys, Suffolk, UK). The sequences of the primers and probes and description of reaction conditions are available on request. The PCR programme was performed and amplification products were detected by an iCycler thermal cycler and an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA). The mtDNA/nDNA ratio was calculated with the ΔCT method described by Pfaffl (Pfaffl 2001). mtDNA depletion was defined as an mtDNA ratio of <0.30 relative to median of age-matched controls (Rahman and Poulton 2009). Unfortunately, we were not able to obtain controls below 5 years of age. Thus, patients below 5 years were compared to median of 5-year-old controls.
Detection of Mitochondrial DNA Deletions in Muscle DNA Samples
Deletions of mitochondrial DNA were detected by long-range PCR (XL-PCR) amplification of mtDNA using Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA). The amplification reaction was carried out with light-strand primer starting from the nucleotide 10 (L10) and heavy-strand primer starting from the nucleotide 16496 (H16496). The sequences of the primers and probes and description of reaction conditions are available on request. The PCR products, along with a GeneRuler λ mix ladder and 1 kb ladder (New England Biolabs, Ipswich, MA, USA), were electrophoresed on 0.7% agarose gel stained with a SYBR Safe DNA gel stain (Invitrogen, Eugene, OR, USA).
Molecular Genetic Studies on Patients with Mitochondrial DNA Depletion or Deletions
Based on the clinical phenotype and muscle histology findings, one patient (Patient 3) was analysed for laminin subunit alpha 2 (LAMA2) gene. All other patients with mtDNA depletion or deletions (Patients 1–2 and 4–5) were analysed for all the exons and intron-exon boundaries of the POLG1, C10orf2 and thymidine kinase 2 (TK2) genes and the entire mtDNA coding region by direct sequencing. In addition, two patients (Patients 4–5) with a large single mtDNA deletion associated with Kearns-Sayre/Pearson-like phenotypes were analysed also for ADP/ATP translocase 1 (ANT1) gene.
Whole-exome sequencing was performed in two patients with novel MDDS phenotypes with unknown genetic aetiology (Patients 1–2) and patients with single large-scale mtDNA deletions and minor multiple deletions (Patients 4–5). For whole-exome sequencing, total genomic DNA was extracted from skin fibroblasts. DNA samples of Patients 1–2 were analysed by the commercially available Agilent SureSelect in-solution target enrichment system (Agilent SureSelect Human All Exon V5, Agilent Technologies, Santa Clara, CA, USA) with mean sequencing coverage of 30× using the Illumina HiSeq sequencing platform at the FIMM Technology Center, Helsinki, Finland, as described by Sulonen et al. (Sulonen et al. 2011). DNA samples of Patients 4–5 were analysed using Agilent SureSelect Human All Exon Kit V1 (Agilent Technologies) and sequenced on the Illumina GAIIx sequencing platform at the McGill University, Montreal, Canada, and Genome Quebec Innovation Center, Montreal, Canada. Bioinformatics of the whole-exome sequencing data (variant filtering and interpretation) on these four patients (Patients 1–2 and 4–5) was performed by Dr. Javad Nadaf, Dr. Somayyeh Fahiminiya and Prof. Jacek Majewski at the Department of Human Genetics, McGill University, and Genome Quebec Innovation Center.
Results
Characteristics of Patient and Control Cohorts
Muscle samples were obtained from 104 paediatric patients with encephalomyopathy or myopathy. The median age was 4.1 years (range of 1 month to 17 years). Control muscle samples were obtained from 14 subjects under 18 years with no suspicion of mitochondrial or other neurological diseases. The median age of controls was 12 years (range of 5–16 years). Muscle mtDNA content of the controls is presented in Fig. 1a.
Fig. 1.
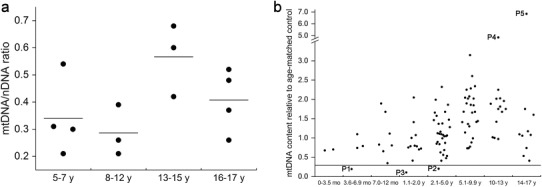
mtDNA content determined using quantitative real-time PCR. (a) Muscle mitochondrial DNA content of the control samples. The results are presented as mitochondrial DNA/nuclear DNA ratio. Each dot represents one sample. (b) Muscle mitochondrial DNA content of paediatric patients with encephalomyopathies and myopathies (N = 104) compared to median muscle mtDNA content of the age-matched control samples. mtDNA depletion was determined as mtDNA content <0.30 of age-matched controls. The cutoff point at 0.30 is marked with a horizontal line. Three patients with mtDNA depletion were identified (Patients 1–3). Two samples present with marked increase in mtDNA content relative to median of age-matched controls: a single sample in the age group 10–13 years (Patient 4) presents with a 4.9-fold increase and another sample in the age group 14–17 years (Patient 5) show a 6.9-fold increase in mtDNA content relative to age-matched controls. mo months, y years
Molecular Studies and Clinical Features of the Patients
Among the 104 paediatric patients with neuromuscular disorders, we identified three patients (Patients 1–3 in Table 1) with mtDNA depletion (Fig. 1b). Two of them presented with severe multiorgan phenotype of MDDS and increased serum creatine kinase (CK) levels. Muscle histology of Patient 2 disclosed myopathic muscle tissue with rounded fibres and endomysial collagen and electron micrograph (EM) showed disordered myofibrillar structure, pathologic ER membranes and accumulation of glycogen and extracellular collagen fibres (Fig. 2a, b). Molecular genetic studies including whole-exome sequencing did not reveal any mtDNA point mutations or mutations in any of the known mtDNA maintenance genes. Patient 3 presenting with severe myopathy and mtDNA depletion was diagnosed with merosine-deficient muscular dystrophy (MDC1A) based on histological and clinical features. This patient was found to harbour a homozygous p.G1591X mutation creating a stop codon in the LAMA2 gene.
Table 1.
Clinical features of paediatric patients with neuromuscular diseases associated with mtDNA depletion (Patients 1–3)
| Patient | Age at onset | Age at biopsy | Deletion status | mtDNA/nDNA ratio (± S.D.) | mtDNA relative to control | Enc | Myo | Hep | FTT | Epi | Ata | Others | Laboratory findings | OXPHOS activity | Muscle histology | Genetic aetiology |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | Newborn | 5 mo | No | 0.06 ± 0.05 | 0.19 | + | − | − | + | + | − | Spasticity, microcephaly | L: + | N | Atr, F | Unknown |
| EEG, burst suppression; MRI, white matter necrosis, basal ganglia and thalamic lesions | CFSL: + | |||||||||||||||
| P: + | ||||||||||||||||
| CK: + | ||||||||||||||||
| UAA: + | ||||||||||||||||
| P2 | Newborn | 3 y 5 mo | No | 0.06 ± 0.02 | 0.2 | + | + | − | + | − | − | Optic atrophy, pigmentary retinopathy, nystagmus, SNHI, death at age 4 y 8 mo | L: N | N.A. | Gly, ER, Fib, | Unknown |
| P: N | ||||||||||||||||
| CK: + | ||||||||||||||||
| P3 | 9 mo | 1 y 10 mo | No | 0.03 ± 0.01 | 0.1 | − | + | − | − | − | − | Merosine-deficient muscular dystrophy (MDC1A), severe muscular hypotonia | CK: + | N.A. | Atr, F, Fib | LAMA2 p.G1591X +/+ |
| ENMG: myopathy | ||||||||||||||||
| Brain MRI: leukodystrophy |
Abbreviations for clinical features: Enc encephalopathy, Myo myopathy, Hep hepatopathy, FTT failure to thrive, Epi epilepsy, Ata ataxia, EEG electroencephalography, MRI magnetic resonance imaging, SNHI sensorineural hearing impairment
Abbreviations for laboratory findings and OXPHOS activity: CK creatine kinase, L blood lactate, CSFL cerebrospinal fluid lactate, P blood pyruvate, N normal, + elevated, − decreased, UAA + in patient P1 generalized aminoaciduria, OXPHOS respiratory chain, N.A. not available
Abbreviations for histology: N normal, Atr atrophy, F fat excess, Gly increased glycogen, ER pathologic endoplasmic reticulum membranes, Fib accumulation of extracellular collagen fibres, N.A. not available
Abbreviations and footnotes: y years, mo months
Fig. 2.
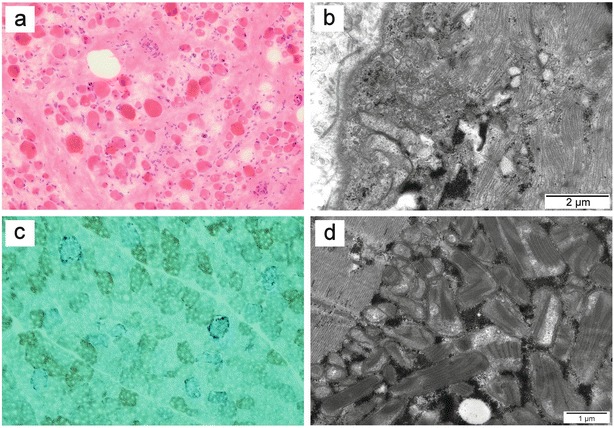
Histological findings in skeletal muscle biopsies associated with mtDNA depletion or deletions. (a) Muscle histology of Patient 2 disclosed myopathic muscle tissue with rounded fibres and endomysial collagen. HE staining. (b) Electron micrograph of the muscle of Patient 2 showing disordered myofibrillar structure, pathologic endoplasmic reticulum membranes, accumulation of glycogen and extracellular collagen fibres. (c) Muscle histology of Patient 5 demonstrated COX-negative fibres. SDH-COX staining. (d) Electron micrograph of the muscle of Patient 5 displayed accumulation of mitochondria with pathologic internal structure and abnormal cristae
Two patients (Patients 4 and 5; Table 2) harboured a major large-scale 5 kb deletion and minor multiple deletions (Fig. 3a, b). In addition, qRT-PCR of these two patients showed high mtDNA content (4.9- and 6.9-fold increases, respectively) relative to the median of age-matched controls (Fig. 1a, b). The clinical features of both patients included early-onset external ophthalmoplegia, sensorineural hearing impairment, progressive tremor, severe ataxia, migraine and dysarthria. Furthermore, Patient 5 presented with transient anaemia and granulocytopaenia, pigmentary retinopathy and a trifascicular cardiac conduction block requiring a pacemaker in connection to Kearns-Sayre and Pearson syndromes. Muscle histology of both patients disclosed ragged-red fibres and COX-negative fibres as demonstrated in Fig. 2c. In addition, EM of muscle biopsy of Patient 5 revealed accumulation of mitochondria with pathologic internal structure and abnormal cristae (Fig. 2d). Respiratory chain activity measurement showed decreased activity of complexes I and I + III in muscle of Patient 5. Molecular genetic studies including whole-exome sequencing did not disclose any mtDNA point mutations or mutations in any of the known mtDNA maintenance genes.
Table 2.
Clinical features of patients with Kearns-Sayre/Pearson syndrome associated with a single large-scale deletion and minor multiple mtDNA deletions and high mtDNA content (Patients 4–5)
| Patient | Aget at onset | Age at biopsy | Deletion status | mtDNA/nDNA ratio (± S.D.) | mtDNA relative to control | CPEO | Pt | Mig | SNHI | Myo | Ata | Others | Laboratory findings | Muscle histology | Genetic aetiology |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P4 | 5 y | 12 y | Single 11 kb band + M | 1.89 ± 0.25 | 4.9 | + | − | + | + | − | + | Progressive tremor | L: + | C−, RRF, F, Fib | Unknown |
| Brain CT: subcortical and cerebellar hypodensity | CFSL: + | ||||||||||||||
| Brain MRI: T2-weighted lesions in globus pallidus and thalamic and frontal subcortical regions | P: N | ||||||||||||||
| CK: N | |||||||||||||||
| P5 | 1 y | 16 y | Single 11 kb band + M | 2.81 ± 0.89 | 6.9 | + | + | + | + | + | + | Transient anaemia and granulocytopaenia, pigmentary retinopathy, growth retardation, progressive cognitive impairment, trifascicular conduction block | LE: + | C−, RRF, Mito, Cr | Unknown |
| Ultrasound: echogenic liver and kidneys | OXPHOS: CI + III and CIII− | ||||||||||||||
| Brain MRI: symmetric T2-weighted lesions in inferior colliculi and right thalamic and frontal subcortical regions |
Abbreviations for mtDNA deletion status: M multiple deletions
Abbreviations for clinical features: CPEO chronic external ophthalmoplegia, Pt bilateral ptosis, Mig migraine, SNHI sensorineural hearing impairment, Myo myopathy, Ata ataxia, CT computed tomography, MRI magnetic resonance imaging
Abbreviations for laboratory findings: L blood lactate, CSFL cerebrospinal fluid lactate, P blood pyruvate, CK creatine kinase, LE liver enzymes, OXPHOS C respiratory chain complex activity, N normal, + elevated, − decreased, N.A. not available
Abbreviations for histology: C− COX-negative fibres, RRF ragged-red fibres, F fat excess, Fib fibrosis, Mito pathologic inner mitochondrial structures, Cr abnormal cristae in mitochondria, N.A. not available
Abbreviations and footnotes: y years, mo months
Fig. 3.
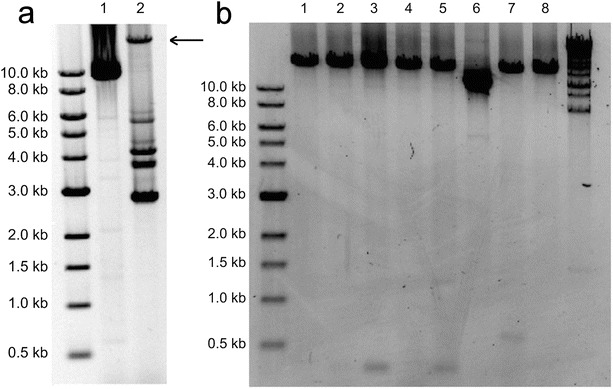
Long-range PCR of muscle DNA. (a) Lane 1, patient 4 presenting with Kearns-Sayre/Pearson-like syndrome; lane 2, a positive control for mtDNA deletions. Intact 16.6 kb mtDNA is marked with a black arrow. (b) Lanes 1 and 8, 5-year-old healthy control; lanes 2–5, patient samples with unspecific deletion bands; lane 6, Patient 5 presenting with a single large-scale deletion with no intact 16.6 kb mtDNA band. In addition, smaller deletion bands are visible signifying multiple mtDNA deletions; lane 7, a patient sample with unspecific 0.5 kb band
Discussion
We investigated mtDNA depletion and deletion in relation to paediatric neuromuscular diseases. As it is not ethically justifiable to collect muscle biopsies from healthy children with a sole intention to obtain control samples, the control samples had to be collected from patients who underwent surgical operation. This obviously had an effect on the selection process. Thus, we were unfortunately lacking control muscle samples from healthy children younger than 5 years old, but as presented by Dimmock et al. (Dimmock et al. 2010), muscle mtDNA content is relatively stable between 0 and 6 years of age. Therefore, mtDNA ratio of patients under 5 years was determined relative to median of 5-year-old controls. In all, we identified 4 patients with a typical presentation of a mitochondrial disorder in association with mtDNA depletion or deletions. Furthermore, mtDNA depletion was found as a secondary finding in hereditary muscular dystrophy.
Two patients with mtDNA depletion (Patients 1–2) presented with novel types of mitochondrial DNA depletion syndromes manifesting with multiorgan disease. The clinical features of these two patients with the severe MDDS associated with increased CK levels suggested mutations in the TK2 gene (Lesko et al. 2010), but sequencing of TK2 gene disclosed no mutations. Patient 1 manifested with a severe progressive metabolic encephalomyopathy and the brain MRI of Patient 1 disclosed white matter degeneration as well as basal ganglia and thalamic lesions resembling Leigh syndrome. Similar encephalopathic phenotypes have been described in MDDS caused by several mtDNA maintenance genes including in SUCLA2 and SUCLG1 (Ostergaard et al. 2007a, b; Carrozzo et al. 2007; Elpeleg et al. 2005), DGUOK (Mandel et al. 2001; Dimmock et al. 2008) and RRM2B (Bornstein et al. 2008; Acham-Roschitz et al. 2009; Kollberg et al. 2009). Patient 1 was also found with generalized aminoaciduria that has previously been described in MDDS patients (Uusimaa et al. 2014). Patient 2 presented with a novel early-onset and fatal MDDS phenotype including encephalopathy, optic atrophy, pigmentary retinopathy, sensorineural hearing impairment and pathologic ER membranes in histologically disordered muscle with accumulation of glycogen and extracellular collagen fibres (Fig. 2b). A patient with mtDNA depletion and similar clinical features has previously been described to harbour a missense mutation in MFN2 gene encoding mitofusin 2, a protein essential in mitochondrial network dynamics (Renaldo et al. 2012). As ER and mitochondria are functionally related (Kornmann 2013), the muscle histology of Patient 2 suggests involvement of ER in the pathogenesis of the disease.
Two patients (Patients 4 and 5) harboured a large-scale mtDNA deletion in addition to minor deletions in muscle DNA (Fig. 3). Although XL-PCR is not a quantitative method for mtDNA deletion analysis, we were not able to detect intact 16.6 kb mtDNA in an XL-PCR assay of these patients suggesting a very high heteroplasmy rate of the mutated mtDNA molecule. Most interestingly, we found markedly increased muscle mtDNA content with the qRT-PCR analysis in both cases referring to overamplification of mtDNA as a compensation mechanism for large-scale mtDNA deletion. These patients developed ophthalmoplegia at a young age along with other neurological symptoms including sensorineural hearing loss, muscle hypotonia and ataxia. Patient 5 also presented with pigmentary retinopathy and cardiac conduction block associated with KSS (#OMIM 530000), as well as transient anaemia and granulocytopaenia suggesting Pearson syndrome (OMIM #557000). In the previous literature, we found only one similar case of mtDNA overamplification associated with mtDNA deletions. This case was a female patient presenting with KSS (Wong et al. 2003) including progressive external ophthalmoplegia, ptosis and pigmentary retinopathy; the atypical onset of the disease happened at 30 years of age. She harboured a 3,078 bp mtDNA deletion with a 92% proportion of mutant mtDNA and partial mtDNA duplication. Ragged-red fibres were seen in muscle histology of this patient, but respiratory chain activity was normal. Quantitative analysis showed a ninefold increase in muscle mtDNA content, thus suggesting compensatory amplification of mtDNA due to a high deletion mutant load.
KSS is commonly associated with 4,977 bp mtDNA deletion (Remes et al. 2005). It has also been suggested that mtDNA duplications can be a typical feature of early-onset KSS (Poulton 1992; Odoardi et al. 2003). Consideration of the role of mtDNA duplications in KSS raises a question whether the high mtDNA content in qRT-PCR is actually an artefact caused by duplicated target gene in mtDNA molecules. In this work we did not analyse the DNA samples for duplications, but previous studies (Wong et al. 2003; Odoardi et al. 2003) suggest that the ND1 gene, the target gene for qRT-PCR in our study, is not situated in common locations of either mtDNA deletions or duplications. This may indicate that the high mtDNA content found in our two KSS patients actually refers to a high copy number in the muscle suggesting compensatory overamplification of mtDNA, which is also suggested in the previous study (Wong et al. 2003). In addition, muscle histology of Patient 5 showed mitochondrial inclusion bodies, which could be related to high mtDNA content observed in quantitative analysis.
In general, in 40% of cases of mtDNA-related clinical syndromes, the genetic aetiology remains unsolved even after whole-exome sequencing (Calvo et al. 2012). In this study, with whole-exome sequencing, we did not identify pathogenic mutations in any of the known mtDNA maintenance genes (El-Hattab and Scaglia 2013; Suomalainen and Isohanni 2010) to disclose the genetic aetiology of the disease in Patients 1 and 2, but further studies are ongoing to evaluate the role of the candidate genes identified by whole-exome sequencing (unpublished data provided by Dr. Javad Nadaf, Dr. Somayyeh Fahiminiya and Prof. Jacek Majewski, Department of Human Genetics, McGill University and Genome Quebec Innovation Center, Montreal, Canada) associated with these novel MDDS phenotypes. Most single mtDNA deletions are thought to be sporadic and thus not genetically transmitted (Chinnery et al. 2004), but multiple mtDNA deletions can be inherited as an autosomal dominant or recessive trait (Zeviani et al. 1990; Nishino et al. 1999). Our two patients with a KSS/Pearson-like phenotype presented with minor multiple deletions in addition to a major large-scale mtDNA deletion, which led us to suggest a genetic origin of the mtDNA arrangements leading to these clinical phenotypes. Whole-exome sequencing was performed in Patients 4–5, but no mutations in the known mtDNA maintenance genes were found. These circumstances could refer to mutations in a yet unknown nuclear gene responsible for mtDNA maintenance.
Patient 3, presenting with severe mtDNA depletion (mtDNA content only 0.10 compared to the age-matched controls) and severe myopathy with muscle fibrosis and atrophy, was diagnosed with merosine-deficient muscular dystrophy (MDC1A) based on further histological and clinical examinations. Molecular studies on this patient disclosed homozygous p.G1591X mutation in the LAMA2 gene. Secondary mtDNA rearrangements have been previously found in neuromuscular disorders that are not primary mitochondrial diseases (Katsetos et al. 2013), but so far only a few studies concerning mtDNA rearrangements in the pathogenesis of muscle dystrophies have been performed. mtDNA deletions have been described in myotonic dystrophy (Sahashi et al. 1992) and oculopharyngeal muscular dystrophy (Lezza et al. 1997; Muqit et al. 2008). Another study on congenital myotonic dystrophy associated mtDNA depletion to a minor degree with the disease, but no evidence of mtDNA rearrangements in the pathogenesis was found and therefore mild mtDNA depletion was suggested to be secondary to the disease (Poulton et al. 1995).
In conclusion, we describe two novel early-onset phenotypes associated with mtDNA depletion, one of them presenting with pathologic endoplasmic reticulum membranes. In addition, we suggest that Kearns-Sayre and Pearson syndrome can present with multiple deletions and may be caused by mutations in an unknown mtDNA maintenance gene. Furthermore, mtDNA depletion can be a secondary finding in hereditary muscular dystrophy.
Electronic Supplementary Material
Supplemental Fig. 1. Muscle mitochondrial DNA content of paediatric patients with encephalomyopathic disorder (N = 77). Patients 1–2 and 4–5 are marked in the graph. The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 3; b = 20; c = 24 (patient P3 in Table 1); d = 33; e = 37; f = 41; g = 44; h = 45; i = 49; j = 60; k = 61; l = 62; m = 85; n = 101
Supplemental Fig. 2. Muscle mitochondrial DNA content of paediatric patients with myopathic disorder (N = 18). The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 53; b = 67; c = 80; d = 96; e = 99
Supplemental Fig. 3. Muscle mitochondrial DNA content of paediatric patients with hepatoencephalopathic disorder (N = 9). Patients with reversible hepatoencephalopathy are marked with a cross. The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 1; b = 30
Supplemental Fig. 4. Muscle mitochondrial DNA content of paediatric patients presenting with OXPHOS defect associated with encephalomyopathy, myopathy or hepatoencephalopathy (N = 15). The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 30; b = 60; c = 67; d = 85
Supplemental Table 1. The clinical phenotypes, mitochondrial DNA content in the muscle, results of the genetic studies and results of respiratory chain activity measurements of all the patients in the study (N = 104). In the supplemental table, the patients’ IDs are given in numerical order. The patients with mtDNA depletion or deletion (P1–P5) are highlighted. mo month, y years
Acknowledgements
The authors thank Ms. Anja Heikkinen and Ms. Pirjo Keränen for their expert assistance. The work was supported by grants from the Research Council for Health of the Academy of Finland (JU, Decision number 138566; KM, Decision number 127764; RH, Decision number 266498 and 273790), the Sigrid Juselius Foundation, the Finnish Medical Foundation, the Arvo ja Lea Ylppö Foundation, the Foundation for Pediatric Research, the Alma and K.A. Snellman Foundation, the Emil Aaltonen Foundation, National Graduate School of Clinical Investigation (CLIGS), a Marie Curie International Outgoing Fellowship of the European Union’s Seventh Framework Programme under the grant agreement number 273669 (BioMit), Special State Grants for Health Research in the Department of Pediatrics and Adolescence and the Department of Neurology at Oulu University Hospital, Oulu, Finland.
Abbreviations
- CK
Creatine kinase
- ER
Endoplasmic reticulum
- mtDNA
Mitochondrial DNA
- MDDS
Mitochondrial DNA depletion syndrome
- nDNA
Nuclear DNA
- PCIAA
Phenol-chloroform-isoamyl alcohol extraction
- qRT-PCR
Real-time quantitative PCR
- XL-PCR
Long-range PCR
Synopsis
Novel phenotypes associated with mtDNA depletion and deletions without mutations in the known mtDNA maintenance genes.
Compliance with Ethics Guidelines
Conflict of Interest
Tuomas Komulainen, Milla-Riikka Hautakangas, Reetta Hinttala, Salla Pakanen, Vesa Vähäsarja, Petri Lehenkari, Päivi Olsen, Päivi Vieira, Outi Saarenpää-Heikkilä, Johanna Palmio, Hannu Tuominen, Pietari Kinnunen, Kari Majamaa, Heikki Rantala and Johanna Uusimaa declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients or their guardian for being included in the study.
Authors’ Contributions
Tuomas Komulainen is responsible for the study design, gathering, analysing and interpreting the data and drafting the manuscript. Reetta Hinttala and Johanna Uusimaa are responsible for the study design, gathering, analysing and interpreting the data and revising the manuscript for important intellectual content. Milla-Riikka Hautakangas, Reetta Hinttala, Salla Pakanen, Vesa Vähäsarja, Petri Lehenkari, Päivi Olsen, Päivi Vieira, Outi Saarenpää-Heikkilä, Johanna Palmio, Hannu Tuominen, Pietari Kinnunen, Kari Majamaa and Heikki Rantala are responsible for gathering, analysing and interpreting the data and revising the manuscript for important intellectual content.
Contributor Information
Tuomas Komulainen, Email: mikakomu@student.oulu.fi.
Collaborators: Johannes Zschocke
References
- Acham-Roschitz B, Plecko B, Lindbichler F, Bittner R, Mache CJ, Sperl W, Mayr JA. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–304. doi: 10.1016/j.ymgme.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Alberio S, Mineri R, Tiranti V, Zeviani M. Depletion of mtDNA: syndromes and genes. Mitochondrion. 2007;7:6–12. doi: 10.1016/j.mito.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Bornstein B, Area E, Flanigan KM, et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord. 2008;18:453–459. doi: 10.1016/j.nmd.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4(118):118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130:862–874. doi: 10.1093/brain/awl389. [DOI] [PubMed] [Google Scholar]
- Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787:312–319. doi: 10.1016/j.bbabio.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, DiMauro S, Shanske S, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–596. doi: 10.1016/S0140-6736(04)16851-7. [DOI] [PubMed] [Google Scholar]
- Dimmock DP, Zhang Q, Dionisi-Vici C, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008;29:330–331. doi: 10.1002/humu.9519. [DOI] [PubMed] [Google Scholar]
- Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem. 2010;56:1119–1127. doi: 10.1373/clinchem.2009.141549. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10:186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elpeleg O, Miller C, Hershkovitz E, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Chinnery PF, Durham SE, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30 doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsetos CD, Koutzaki S, Melvin JJ. Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr Neurol. 2013;20:202–215. doi: 10.1016/j.spen.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Kollberg G, Darin N, Benan K, et al. A novel homozygous RRM2B missense mutation in association with severe mtDNA depletion. Neuromuscul Disord. 2009;19:147–150. doi: 10.1016/j.nmd.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol. 2013;25:443–448. doi: 10.1016/j.ceb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Greaves LC, Reeve AK, Turnbull DM. Mitochondrial DNA mutations and aging. Ann N Y Acad Sci. 2007;1100:227–240. doi: 10.1196/annals.1395.024. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Lesko N, Naess K, Wibom R, et al. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul Disord. 2010;20:198–203. doi: 10.1016/j.nmd.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Lezza AM, Cormio A, Gerardi P, et al. Mitochondrial DNA deletions in oculopharyngeal muscular dystrophy. FEBS Lett. 1997;418:167–170. doi: 10.1016/S0014-5793(97)01374-4. [DOI] [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- Morten KJ, Ashley N, Wijburg F, et al. Liver mtDNA content increases during development: a comparison of methods and the importance of age- and tissue-specific controls for the diagnosis of mtDNA depletion. Mitochondrion. 2007;7:386–395. doi: 10.1016/j.mito.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Larner AJ, Sweeney MG, et al. Multiple mitochondrial DNA deletions in monozygotic twins with OPMD. J Neurol Neurosurg Psychiatry. 2008;79:68–71. doi: 10.1136/jnnp.2006.112250. [DOI] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- Odoardi F, Rana M, Broccolini A, et al. Pathogenic role of mtDNA duplications in mitochondrial diseases associated with mtDNA deletions. Am J Med Genet A. 2003;118A:247–254. doi: 10.1002/ajmg.a.20006. [DOI] [PubMed] [Google Scholar]
- Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, Wibrand F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29 doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J. Duplications of mitochondrial DNA: implications for pathogenesis. J Inherit Metab Dis. 1992;15:487–498. doi: 10.1007/BF01799607. [DOI] [PubMed] [Google Scholar]
- Poulton J, Harley HG, Dasmahapatra J, Brown GK, Potter CG, Sykes B. Mitochondrial DNA does not appear to influence the congenital onset type of myotonic dystrophy. J Med Genet. 1995;32:732–735. doi: 10.1136/jmg.32.9.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child. 2009;94:3–5. doi: 10.1136/adc.2008.147983. [DOI] [PubMed] [Google Scholar]
- Remes AM, Majamaa-Voltti K, Karppa M, et al. Prevalence of large-scale mitochondrial DNA deletions in an adult Finnish population. Neurology. 2005;64:976–981. doi: 10.1212/01.WNL.0000154518.31302.ED. [DOI] [PubMed] [Google Scholar]
- Renaldo F, Amati-Bonneau P, Slama A et al (2012) MFN2, a new gene responsible for mitochondrial DNA depletion. Brain 135:e223, 1–4, author reply e224, 1–3 [DOI] [PubMed]
- Sahashi K, Tanaka M, Tashiro M, Ohno K, Ibi T, Takahashi A, Ozawa T. Increased mitochondrial DNA deletions in the skeletal muscle of myotonic dystrophy. Gerontology. 1992;38:18–29. doi: 10.1159/000213303. [DOI] [PubMed] [Google Scholar]
- Sulonen AM, Ellonen P, Almusa H, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes–many genes, common mechanisms. Neuromuscul Disord. 2010;20:429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Uusimaa J, Remes AM, Rantala H, et al. Childhood encephalopathies and myopathies: a prospective study in a defined population to assess the frequency of mitochondrial disorders. Pediatrics. 2000;105:598–603. doi: 10.1542/peds.105.3.598. [DOI] [PubMed] [Google Scholar]
- Uusimaa J, Evans J, Smith C, et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur J Hum Genet. 2014;22:184–191. doi: 10.1038/ejhg.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LJ, Perng CL, Hsu CH, et al. Compensatory amplification of mtDNA in a patient with a novel deletion/duplication and high mutant load. J Med Genet. 2003;40 doi: 10.1136/jmg.40.11.e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Chinnery PF. Dysfunctional mitochondrial maintenance: what breaks the circle of life? Brain. 2012;135:9–11. doi: 10.1093/brain/awr352. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Bresolin N, Gellera C, et al. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. Am J Hum Genet. 1990;47:904–914. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Muscle mitochondrial DNA content of paediatric patients with encephalomyopathic disorder (N = 77). Patients 1–2 and 4–5 are marked in the graph. The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 3; b = 20; c = 24 (patient P3 in Table 1); d = 33; e = 37; f = 41; g = 44; h = 45; i = 49; j = 60; k = 61; l = 62; m = 85; n = 101
Supplemental Fig. 2. Muscle mitochondrial DNA content of paediatric patients with myopathic disorder (N = 18). The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 53; b = 67; c = 80; d = 96; e = 99
Supplemental Fig. 3. Muscle mitochondrial DNA content of paediatric patients with hepatoencephalopathic disorder (N = 9). Patients with reversible hepatoencephalopathy are marked with a cross. The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 1; b = 30
Supplemental Fig. 4. Muscle mitochondrial DNA content of paediatric patients presenting with OXPHOS defect associated with encephalomyopathy, myopathy or hepatoencephalopathy (N = 15). The patients with identified genetic aetiology are marked in the graph with black and marked alphabetically according to the patient ID number in the Supplemental Table 1 as follows: a = 30; b = 60; c = 67; d = 85
Supplemental Table 1. The clinical phenotypes, mitochondrial DNA content in the muscle, results of the genetic studies and results of respiratory chain activity measurements of all the patients in the study (N = 104). In the supplemental table, the patients’ IDs are given in numerical order. The patients with mtDNA depletion or deletion (P1–P5) are highlighted. mo month, y years