

Abstract
Rhabdomyolysis is an acute syndrome due to extensive injury of skeletal muscle. Recurrent rhabdomyolysis is often caused by inborn errors in intermediary metabolism, and recent work has suggested that mutations in the human gene encoding lipin 1 (LPIN1) may be a common cause of recurrent rhabdomyolysis in children. Lipin 1 dephosphorylates phosphatidic acid to form diacylglycerol (phosphatidic acid phosphohydrolase; PAP) and acts as a transcriptional regulatory protein to control metabolic gene expression. Herein, a 3-year-old boy with severe recurrent rhabdomyolysis was determined to be a compound heterozygote for a novel c.1904T>C (p.Leu635Pro) substitution and a previously reported genomic deletion of exons 18–19 (E766-S838_del) in LPIN1. Western blotting with patient muscle biopsy lysates demonstrated a marked reduction in lipin 1 protein, while immunohistochemical staining for lipin 1 showed abnormal subcellular localization. We cloned cDNAs to express recombinant lipin 1 proteins harboring pathogenic mutations and showed that the E766-S838_del allele was not expressed at the RNA or protein level. Lipin 1 p.Leu635Pro was expressed, but the protein was less stable, was aggregated in the cytosol, and was targeted for proteosomal degradation. Another pathogenic single amino acid substitution, lipin 1 p.Arg725His, was well expressed and retained its transcriptional regulatory function. However, both p.Leu635Pro and p.Arg725His proteins were found to be deficient in PAP activity. Kinetic analyses demonstrated a loss of catalysis rather than diminished substrate binding. These data suggest that loss of lipin 1-mediated PAP activity may be involved in the pathogenesis of rhabdomyolysis in lipin 1 deficiency.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_440) contains supplementary material, which is available to authorized users.
Introduction
Mutations in the gene encoding lipin 1 (LPIN1) have been identified as a cause of recurrent, early-onset, pediatric rhabdomyolysis and myoglobinuria (OMIM#268200) (Zeharia et al. 2008; Michot et al. 2010, 2012). Rhabdomyolysis is an acute syndrome due to extensive injury of skeletal muscle resulting in the release of intracellular metabolites and proteins, including creatine kinase and myoglobin, into the systemic circulation. Clinical features may include myalgia, weakness, pigmenturia, renal failure, and secondary injury to other organ systems. Untreated rhabdomyolysis can result in death from renal, cardiac, or hematologic dysfunction.
Although there are many common acquired causes of acute rhabdomyolysis, hereditary etiologies should be considered in the setting of recurrent rhabdomyolysis or in the setting of rhabdomyolysis with positive family history. Classes of inherited metabolic disorders associated with recurrent rhabdomyolysis include glycogen storage diseases, fatty acid oxidation defects, and mitochondrial disorders (Tonin et al. 1990; Kelly and Strauss 1994; Bennett 2010; Zutt et al. 2014). Recently, homozygous mutations in LPIN1 were identified as a common cause of recurrent rhabdomyolysis in pediatric patients, and more than 50% of infants and children with unexplained severe rhabdomyolysis in a European population may have pathologic mutations in LPIN1 (Zeharia et al. 2008; Michot et al. 2010, 2012).
Lipin 1 regulates intermediary metabolism by multiple mechanisms. Lipin 1 is a lipid phosphatase converting phosphatidic acid (PA) to diacylglycerol (DAG) (PAP activity) (Han et al. 2006) through a catalytic site and other critical accessory haloacid dehalogenase (HAD) domains in the protein’s C-terminus (Figs. 1a and S1). Lipin 1 also traffics to the nucleus to interact with DNA-bound transcription factors regulating expression of genes encoding mitochondrial fatty acid oxidation enzymes (Finck et al. 2006) through a nuclear receptor interaction domain (NRID) also in the protein’s C-terminus (Figs. 1a and S1). Of the 19 distinct mutations in LPIN1 associated with rhabdomyolysis, most are frameshift mutations leading to loss of expression or generating truncated proteins lacking both HAD catalytic sites and the NRID. It is unknown whether defects in lipin 1 PAP activity or transcriptional regulatory function, or both, underlie the pathogenesis of rhabdomyolysis in people with LPIN1 mutations.
Fig. 1.
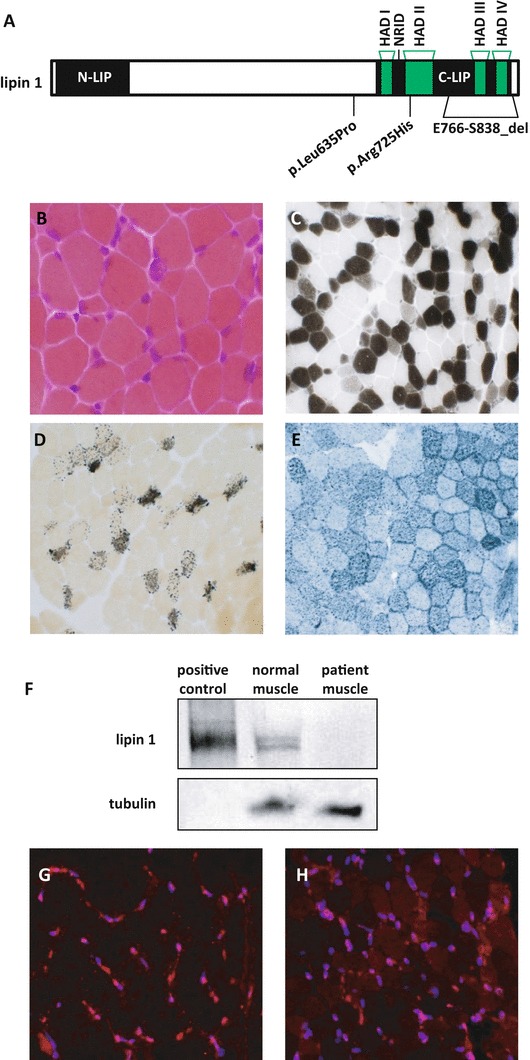
LPIN1 mutations in a boy with recurrent rhabdomyolysis. (a) Diagram of LPIN1 gene. The haloacid dehalogenase (HAD) domains that are important for regulating PAP activity are noted. The nuclear receptor interaction domain (NRID) is important for transcriptional regulatory function. Locations of LPIN1 mutations of the patient (p.Leu635Pro and E766-S838_del) are included with the location of another reported mutation (p.Arg725His). (b–h) A muscle biopsy was performed 6 weeks after the patient’s second episode of rhabdomyolysis and processed for routine histochemical staining. All photomicrographs were obtained at 20× magnification. (b) H&E demonstrated moderate variation in fiber size with occasional basophilic fibers; (c) ATPase 4.3 revealed an excessive number of Type IIC muscle fibers; (d) alkaline phosphatase with increased number of positive staining fibers; (e) Sudan black with moderately increased lipid. (f) Western blot demonstrating near absence of lipin 1 expression in the same patient. (g) Lipin 1 immunostaining in a pediatric control muscle shows strong nuclear staining of lipin 1. (h) Lipin 1 immunostaining in the p.Leu635Pro; E766-S838_del compound heterozygote muscle shows significantly reduced lipin 1 staining with an abnormal stippled or perinuclear staining pattern for the small amount of protein that is present (Blue: DAPI, Red: lipin 1)
In the present study, we detected a novel missense mutation and a known exon deletion in LPIN1 in a young boy presenting to our institution with severe recurrent rhabdomyolysis, which may be the first report of LPIN1 mutations in a North American patient. We assessed the expression and molecular function of recombinant human lipin 1 proteins harboring rhabdomyolysis-associated mutations. We found that single amino acid substitution mutations in lipin 1 known to be present in patients with rhabdomyolysis led to loss of PAP activity via loss of catalysis, but did not always affect transcriptional regulatory function. These findings link the loss of lipin 1-mediated PAP activity to the etiology of recurrent rhabdomyolysis.
Materials and Methods
Detailed methods and source material are described in Supplementary Materials.
Generation and Overexpression of Mutant Alleles
Human lipin 1 cDNA in the pENTR™ vector was a gift of Andrew Morris. Changes in nucleotide sequence corresponding to known lipin 1 mutations were introduced into the cDNA by using site-directed mutagenesis. WT and mutant cDNAs were transferred to pcDNA3.1/nV5-DEST™ fusing a V5 epitope tag to the N-terminus of the protein (Life Technologies/Invitrogen, Carlsbad, CA). HEK293 cells were transfected with vectors driving expression of WT or mutant proteins for analysis of mRNA and protein expression, subcellular localization, protein half-life, and PAP activity. For kinetic analyses, WT, p.Leu635Pro, and p.Arg725His lipin 1 cDNA were transferred to pCMVTAG2 (Agilent) and then inserted into the pAdTRACK-CMV vector for adenoviral production using the AdEasy system.
Immunohistochemistry/Fluorescent Staining
For human tissue, 10 μm sections of frozen muscle biopsies were placed on microscope slides, fixed, blocked, stained with lipin 1 antibody against the C-terminal region (Harris et al. 2007), stained with appropriate secondary antibodies, and coverslip mounted with DAPI. For human muscle histochemistry, cryostat sections of rapidly frozen muscle were processed as previously described (Mozaffar and Pestronk 2000). For cultured cell staining and cellular distribution analysis, Cos7 cells were plated and lipin 1 WT and mutant expression vectors were transfected. Twenty-four hours later, cells were fixed and incubated with DAPI, anti-V5, and anti-calnexin (cytoplasmic marker). Cells were stained with appropriate secondary antibodies and visualized by fluorescence for subcellular protein distribution analysis as described previously (Ren et al. 2010).
Protein and RNA Analysis
Protein from cultured cells was isolated and subjected to SDS-PAGE. After blotting, WT and lipin 1 mutant protein was determined from an antibody against lipin 1, and alpha-tubulin antibody was used to demonstrate equal loading. Total RNA was isolated from cultured cells and subjected to reverse transcription, and lipin 1 gene expression was determined and normalized to 36B4 expression.
Protein Half-Life
Pulse-chase labeling of the HEK293cells containing WT and lipin 1 mutant expression vectors was achieved by incubating the cells with [35S]Cys/Met followed by chase media consisting of DMEM containing 10 mM Met and 3 mM Cys for 0, 1, 2, or 4 h. Lysates were collected in homogenization buffer as described in Supplementary Material and incubated with lipin 1 antibody overnight at 4°C. The protein-antibody complex was pulled down, subjected to SDS-PAGE, and analyzed by autoradiography.
Transcriptional Regulation
The ability of lipin 1 to coactivate overexpressed Gal4-PGC-1α and MEF2A was assayed by cotransfection of Cos7 cells with WT and mutant lipin proteins with the Gal4-reponsive UAS thymidine kinase luciferase construct.
PAP Activity
PAP-1 activity was assessed as described (Martin et al. 1987) with modifications. In brief, transfected cultured cells were incubated with 14C-phosphatidic acid (PA), PA salt, phosphatidylcholine, and MgCl2 without or with N-ethylmaleimide to determine PAP-1 and PAP-1-independent activity, respectively.
Kinetic Analysis
FLAG-tagged WT, p.Leu635Pro, and p.Arg725His lipin 1 proteins were expressed in HeLa cells with 72 h adenoviral infection, harvested, lysed, incubated with anti-FLAG beads for 2–4 h, and column purified. Lipin 1 proteins were quantified, and PAP assays were performed at pH 7.5 with Triton X-100/PA mixed micelles as previously described (Eaton et al. 2013).
Statistical analysis
Data were expressed as means ± SEM. Unpaired t-tests were used to determine significant differences. P values of <0.05 was considered statistically significant. The Michaelis-Menten equation kcat was calculated by kcat = Vmax/Et, where Et = enzyme catalytic site concentration.
Results
Patient with Novel c.1904T>C (p.Leu635Pro) Mutation
A 3-year-old boy with normal motor and cognitive development was evaluated at our institution for severe, recurrent rhabdomyolysis with myoglobinuria. His first episode of rhabdomyolysis occurred at age 16 months. After several hours of play, he developed an unsteady gait with eventual refusal to walk and tea-colored urine. Examination showed that he was afebrile and in obvious discomfort, with no evidence of hepatosplenomegaly or scleral icterus. Neurologic examination showed generalized weakness with retention of antigravity strength in all muscle groups. He would not sit or stand. During the hospital course, he remained afebrile, his discomfort slowly resolved, and his strength steadily improved with a complete return to his normal baseline within 1 month of presentation.
His laboratory studies included a markedly elevated plasma creatine kinase (CK, peak 498,800 U/L), urine myoglobin (peak 8,277,000 ng/mL), aspartate aminotransferase (peak > 8000 IU/L), and alanine aminotransferase (peak 3762 IU/L). The remainder of the laboratory testing was normal and included cerebrospinal fluid (CSF) analysis, serum and CSF lactate/pyruvate, serum ammonia, total and free plasma carnitine, acylcarnitine profile, serum and CSF amino acids, urine organic acids, leukocytic coenzyme Q10, and peroxisomal profile. Pathogenic mutations were not present in RYR1 or PYGM. Cardiac and abdominal ultrasounds were normal. Muscle ultrasound demonstrated diffusely increased echogenicity in the bilateral hip adductor compartments with a relative increase in signal heterogeneity on the right side. The findings were not specific to any one etiology, but were deemed consistent with the clinical history of rhabdomyolysis. A muscle biopsy was obtained 6 weeks after a second episode of rhabdomyolysis and revealed changes consistent with recent rhabdomyolysis. Increased lipid was detected with normal mitochondrial stains (Fig. 1b–e) and enzymatic activities (data not shown). He had two additional episodes of myoglobinuria (one associated with 9 h of continuous play outside his home and the other associated with an upper respiratory tract infection) and once again made complete recoveries after each episode. Serum CK values with the subsequent episodes peaked at 477,791 U/L. Between episodes, when asymptomatic, his CK nadir was 164 U/L (normal < 300 U/L).
LPIN1 genetic testing revealed compound heterozygosity for a novel c.1904T>C (p.Leu635Pro; Figs. 1a and S1) variant and a previously reported pathogenic genomic deletion of exons 18–19 (E766-S838_del). The p.Leu635Pro variant was classified as “likely pathogenic” based on Polymorphism Phenotyping (PolyPhen) and Sorting Intolerant From Tolerant (SIFT) software (Ng and Henikoff 2001; Ramensky et al. 2002). Indeed, whereas lipin 1 protein was readily detectable by immunohistochemical staining and by western blotting using muscle homogenate from a control pediatric patient (Fig. 1f, g), very little lipin 1 protein was detected in the patient with LPIN1 mutations by western blot (Fig. 1f). Immunohistochemical staining with a lipin 1 antibody showed reduced staining with abnormal subcellular localization in the patient section (Fig. 1h). Whereas lipin 1 staining in the normal patient was distributed throughout the cell with relatively greater signal in the nucleus, the patient section exhibited perinuclear aggregation of lipin 1 with reduced nuclear staining.
LPIN1 Exon Deletion Impairs Expression of the Lipin 1 Protein
The most common lipin 1 mutation in Caucasians (Michot et al. 2012) is an exon deletion that is predicted to delete amino acids E766-S838 in the C-terminus of the lipin 1 protein (Figs. 1a and S1). When the analogous deletion was made in a human V5-tagged lipin 1 cDNA in an expression vector (V5 tag on the N-terminus), very little V5-tagged lipin 1 protein was expressed when the expression vector was transfected into HEK293 cells (Fig. 2a). The mRNA encoded by this mutant allele was also poorly expressed (Fig. 2b), and very little 35S-methionine-containing protein was synthesized in pulse-chase experiments (Fig. 2c). This indicates that the mutation resulting in the E766-S838_del is likely a complete loss of function allele.
Fig. 2.
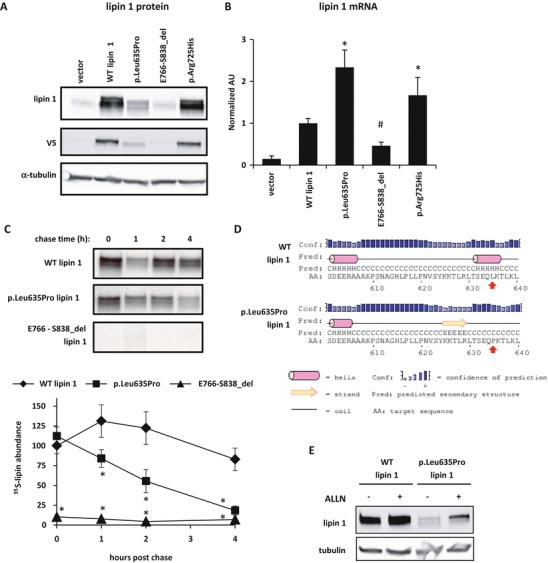
Lipin 1 proteins with disease-associated mutations have reduced protein abundance despite increased RNA levels and reduced protein stability. (a) Representative western blots using lysates from HEK293 cells transfected with V5-tagged lipin 1 (wild-type or mutant) or vector-only control are shown using antibodies against lipin 1, V5, and α-tubulin. (b) Lipin 1 mRNA expression of HEK293 cells transfected with V5-tagged lipin 1 (wild type or mutant) or vector only. Compared to WT lipin 1, p.Leu635Pro and p.Arg725His have significantly greater (*P < 0.05, t-test) and E766-S838 has significantly lower mRNA (#P < 0.05, t-test) expression. (c) A representative autoradiograph and average percent of radiolabeled lipin 1 abundance relative to WT lipin 1 from pulse-chase studies calculated from three separate gels are shown. Protein half-life of p.Leu635Pro was significantly lower than WT lipin 1 at 1, 2, and 4 h (*P < 0.05, t-test). (d) The panel depicts the results of PSIPRED prediction of the secondary structures of the affected region of lipin 1 proteins. The red arrows indicate the affected amino acids. p.Leu635Pro mutation results in disruption of a predicted α-helix and possible formation of a β-strand. (e) Representative western blots of HEK293 cells with overexpressed V5-tagged lipin 1 and p.Leu635Pro are shown. Cells were treated for 4 h with 26 μM ALLN prior to harvest
Single Amino Acid Substitutions in Lipin 1
We also examined the expression and activity of two single amino acid substitution mutations in lipin 1 associated with recurrent rhabdomyolysis in children, including the novel p.Leu635Pro variant identified in the child described above. The protein abundance of p.Leu635Pro was lower than WT lipin 1 protein (Fig. 2a), though the p.Leu635Pro mRNA was more abundant (Fig. 2b). The lower abundance of the protein was due to reduced protein stability as pulse-chase analyses demonstrated diminished protein half-life (Fig. 2c). Predictions of protein secondary structure generated by using the online PSIPRED tool (Jones 1999; Buchan et al. 2013) suggested that Leu635 is located within an α-helix (Fig. 2d). The p.Leu635Pro substitution was predicted to disrupt this α-helix and may cause formation of a β-strand just upstream of the mutation. Incubation with the proteasomal inhibitor, ALLN, promoted the accumulation of the mutant protein (Fig. 2e). This suggests that lipin 1 p.Leu635Pro protein is expressed, but is less stable, may be misfolded, and is targeted for proteasomal degradation.
Another previously reported lipin 1 single amino acid substitution associated with rhabdomyolysis (p.Arg725His) (Michot et al. 2012) was well expressed at the RNA and protein level (Fig. 2a, b). Since the p.Arg725His and p.Leu635Pro proteins were expressed, functional analysis of these mutant lipin 1 alleles might yield clues to the molecular basis for disease in lipin 1-deficient patients.
Analysis of the Subcellular Localization and Transcriptional Regulatory Function of Lipin 1 Mutant Proteins
Lipin 1 protein has been visualized across a variety of cellular compartments. Within a given cell, lipin 1 may be predominantly cytoplasmic (very little in the nucleus), equally distributed in the nucleus and cytoplasm, or predominantly nuclear (Fig. 3a). Comparison of the subcellular distribution WT and p.Arg725His protein detected no differences in the proportions of cells with lipin 1 staining in cytoplasmic, nuclear, or both compartments (Fig. 3a). Compared to WT lipin 1, the ability of p.Arg725His to coactivate MEF2A and PGC-1α, which are transcriptional regulators relevant to skeletal muscle, was also not affected (Fig. 3b). However, p.Leu635Pro protein was predominantly localized to the cytoplasm (Fig. 3a) and did not significantly coactivate MEF2A or PGC-1α activity (Fig. 3b). The perinuclear staining pattern of p.Leu635Pro is also consistent with the immunohistochemical staining results of the patient biopsy (Fig. 1h).
Fig. 3.
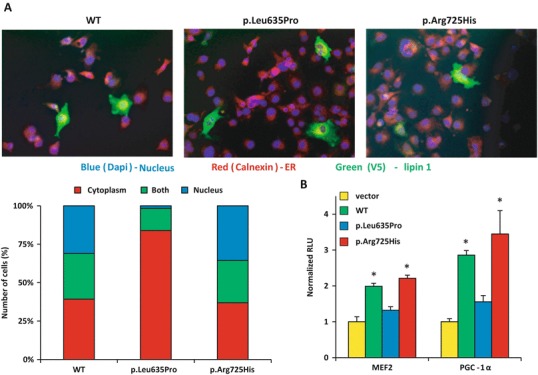
Nuclear localization and transcriptional regulatory function of lipin 1 mutant proteins. (a) COS7 cells transfected with V5-tagged lipin 1 (wild type or mutant) to examine its subcellular localization with the nuclear marker (DAPI) and the endoplasmic reticulum marker (calnexin). The percentage of the number of cells analyzed that localized to the cytoplasm, nucleus, or both was measured in at least 60 cells in at least 15 distinct fields. L365P had very little nuclear localization compared to WT and p.Arg725His. (b) Coactivation of Gal4-MEF2A and PGC-1α in COS7 cells cotransfected with a Gal4-responsive UAS-TK-luciferase reporter construct and cDNAs to express lipin 1 (WT or p.Arg725His), Gal4-MEF2A, Gal4-PGC-1α, or vector control. *p < 0.05 vs. vector control
PAP Activity of Lipin 1 Mutant Proteins
We compared the intrinsic PAP activity of lipin 1 mutant proteins to the activity of WT lipin 1 protein. Overexpressing the WT lipin 1 allele in HEK-293 cells markedly increased cellular PAP activity (Fig. 4a). Transfection of the p.Leu635Pro expression construct did not increase PAP activity compared to vector control-transfected cells, with the caveat that the p.Leu635Pro protein expressed at lower levels compared to than WT lipin 1. Treating p.Leu635Pro-transfected cells with ALLN caused p.Leu635Pro lipin 1 protein accumulation, but did not increase PAP activity in these cells (Fig. 4b). Transfection of p.Arg725His lipin 1, which was well expressed, also did not increase PAP activity. These results indicate that the two lipin 1 mutant proteins are deficient in PAP activity. To further investigate the defect in lipin 1 activity in the amino acid substitution mutants, the kinetic activities of purified WT, p.Leu635Pro, and p.Arg725His were examined. We found that the maximum velocity of the p.Arg725His and p.Leu635Pro proteins was markedly reduced, whereas the affinity for substrate was unaffected, compared to WT lipin 1 protein (Fig. 4c). This suggests that the defect in PAP activity of the p.Arg725His mutant is due to loss of catalysis and not substrate binding. Collectively, these data suggest that disease-associated mutations in lipin 1 lead to deficiency in intrinsic PAP activity.
Fig. 4.
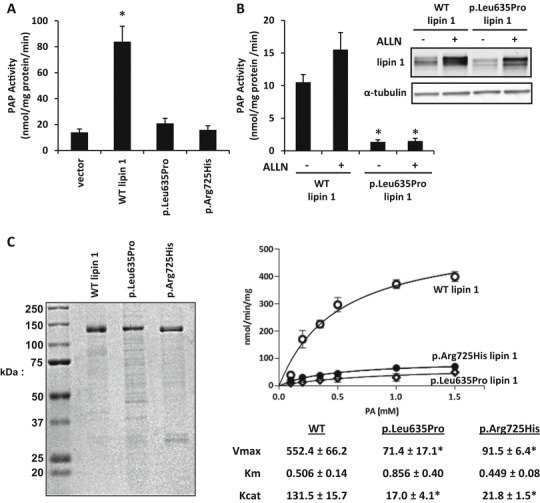
Lipin 1 proteins with disease-associated mutations lack PAP activity. (a) PAP activity of HEK293 cells overexpressing lipin 1 and lipin 1 mutants. PAP activity of WT lipin 1 was significantly greater than vector, p.Leu635Pro, and p.Arg725His (*P < 0.05, t-test). (b) PAP activity of HEK293 cells overexpressing lipin 1 and p.Leu635Pro incubated with ALLN. PAP activity of untreated WT lipin 1 was significantly greater than ALLN-treated and ALLN-untreated p.Leu635Pro (*P < 0.05, t-test). (c) Coomassie stain and PAP activity of purified recombinant WT lipin 1, p.Leu635Pro, and p.Arg725His. PAP activity was measured using Triton X-100/PA mixed micelles at pH 7.5. WT lipin 1 had approx. sixfold higher V max than p.Leu635Pro and p.Arg725His with no change in K m
Discussion
The molecular basis for the pathogenesis of recurrent rhabdomyolysis associated with LPIN1 mutations in humans is poorly understood. Previous work showed that PAP activity was markedly reduced in isolated myocytes from three patients with lipin 1 mutations, including the E766-S838_del mutation studied herein (Michot et al. 2013). However, those cells exhibited a complete loss of lipin 1 mRNA expression, and a distinction regarding impaired transcriptional regulatory function or PAP activity could not be made. In this study, by using recombinant lipin 1 proteins, we show that pathogenic, single amino acid substitutions in human lipin 1 disrupt PAP activity, but may not always affect nuclear transcriptional regulator function. A caveat to this conclusion is that only two single amino acid substitutions in LPIN1 linked to rhabdomyolysis, including the novel mutation identified herein, have been characterized, and thus, broad generalizations regarding this conclusion should be tempered accordingly.
The data presented herein suggest that abnormalities in PAP activity, and potentially resulting effects on glycerolipid content or partitioning, play a role in myocyte injury and necrosis leading to the rhabdomyolysis of lipin 1 deficiency. Clear links between the loss of lipin 1-mediated PAP activity and myocyte damage can be drawn. Conversion of PA to DAG is required for synthesis of triglyceride and phospholipids that are major constituents of cell membranes. Mutations in lipin 1 PAP activity would be predicted to increase the cellular PA concentration, which may be toxic and/or activate inflammatory MAPK signaling cascades (Nadra et al. 2008). PA also activates mTORC1 kinase (Sun and Chen 2008; Mitra et al. 2013), which has been linked to development of muscle injury and myopathy (Castets et al. 2013). A number of mitochondrial defects lead to recurrent rhabdomyolysis (Bennett 2010; Zutt et al. 2014), and lipin 1 PAP activity impacts mitochondrial function by regulating fission and fusion (Huang et al. 2011). Very recently, it was reported that lipin 1 deficiency in muscle of mice caused myopathy via impaired autophagy, and a link between lipin 1-mediated PAP activity and autolysosome maturation was defined (Zhang et al. 2014). Further exploration of the molecular mechanisms involved will undoubtedly be the focus of future work.
Our molecular characterization of the E766-S838_del and p.Leu635Pro alleles in vitro agrees with the characterization of lipin 1 in the patient biopsy. Neither allele was well expressed at the protein level in cultured cells. Consistent with this, we had difficulty detecting lipin 1 protein in the patient biopsy by western blotting analysis. The recombinant p.Leu635Pro lipin 1 protein that was expressed tended to aggregate in the cytosol, particularly in the perinuclear region, of Cos7 fibroblasts. Immunohistochemical staining of a section of the patient biopsy showed a remarkably similar pattern of staining. This mislocalization of the p.Leu635Pro lipin 1 protein may also prevent it from trafficking to its substrate, PA, which is embedded in the ER membrane, though this remains to be determined. We believe that this is the first characterization of recombinant lipin 1 proteins harboring pathogenic mutations, and our work provides a clear mechanistic basis for the loss of lipin 1 activity in this patient with recurrent rhabdomyolysis.
Our current understanding of lipin 1 structure-function relationships provides clues regarding why these single amino acid substitutions lead to loss of lipin 1 function. The loss of PAP activity in the p.Arg725His mutant, which was well expressed, is consistent with the location of this highly conserved arginine in the HAD II domain (Figs. 1a and S1), which is a canonical component of classic HAD domains (Kok et al. 2012). Kinetic analyses suggest that this mutant lacks catalytic activity rather than the ability to bind substrate, which is consistent with its location in a HAD domain rather than the polybasic region required for binding (Eaton et al. 2013). The p.Leu635Pro substitution falls outside of the highly conserved C-lipin domain (Peterfy et al. 2001). The proline insertion in a predicted α-helix domain disrupts the α-helix and may lead to protein misfolding and degradation. Pulse-chase studies demonstrated a reduced protein half-life for p.Leu635Pro protein, and proteosomal inhibition caused accumulation of the WT and mutant allele. This is consistent with recent work indicating that the yeast and mouse homologues of lipin 1 are degraded via a proteosomal pathway (Pascual et al. 2014; Zhang et al. 2014).
In conclusion, we believe that this is the first report of a pathogenic p.Leu635Pro mutation in LPIN1 as well as the first report of a LPIN1 mutation in a North American patient with recurrent myoglobulinuria. We present evidence that single amino acid substitutions in lipin 1, including the novel variant, that are associated with recurrent rhabdomyolysis result in loss of PAP catalytic activity. This may facilitate the identification of the pathogenic mechanisms leading to myocyte cell death and suggest targeted therapies to treat afflicted patients.
Electronic Supplementary Material
Fig. S1. Alignment of mutant lipin 1 with lipin 2 and 3 protein sequences. The catalytic site (DIDGT) for phosphatidic acid phosphohydrolase (PAP) activity and other haloacid dehalogenase (HAD) domains that are important for regulating PAP activity are noted. The nuclear receptor interaction domain (NRID) is important for transcriptional regulatory function. Amino acid substitutions or deletions in lipin 1 mutant alleles are in red font
One Sentence
Herein, we show that recurrent rhabdomyolysis in a pediatric patient was associated with mutations in the bifunctional lipin 1 protein and that pathogenic mutations in lipin 1 led to loss of intrinsic phosphatidic acid phosphohydrolase activity.
Compliance with Ethics Guidelines
Conflict of Interest
George G. Schweitzer, Sara L. Collier, Zhoujj Chen, James M. Eaton, Anne M. Connolly, Robert C. Bucelli, Alan Pestronk, Thurl E. Harris, and Brian N. Finck declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Author Contributions
G.G.S. performed experiments, analyzed data, generated figures, and wrote the manuscript. S.L.C, Z.C., J.M.E. performed experiments, analyzed data, generated figures, and edited portions of the manuscript. A.M.C. worked with the patient, analyzed data, and edited the manuscript. R.C.B performed experiments, analyzed data, generated figures, and wrote portions of the manuscript. A.P., T.E.H., and B.N.F analyzed data and wrote portions of the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Brian N. Finck, Email: bfinck@dom.wustl.edu
Collaborators: Johannes Zschocke
References
- Bennett MJ. Pathophysiology of fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:533–537. doi: 10.1007/s10545-010-9170-y. [DOI] [PubMed] [Google Scholar]
- Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, et al. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013;17:731–744. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Eaton JM, Mullins GR, Brindley DN, Harris TE. Phosphorylation of lipin 1 and charge on the phosphatidic acid head group control its phosphatidic acid phosphatase activity and membrane association. J Biol Chem. 2013;288:9933–9945. doi: 10.1074/jbc.M112.441493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin homolog is a Mg2+−dependent phosphatidate phosphatase enzyme. J Biol Chem. 2006;281:9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TE, Huffman TA, Chi A, et al. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem. 2007;282:277–286. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- Kok BP, Venkatraman G, Capatos D, Brindley DN. Unlike two peas in a pod: lipid phosphate phosphatases and phosphatidate phosphatases. Chem Rev. 2012;112:5121–5146. doi: 10.1021/cr200433m. [DOI] [PubMed] [Google Scholar]
- Martin A, Hales P, Brindley DN. A rapid assay for measuring the activity and the Mg2+ and Ca2+ requirements of phosphatidate phosphohydrolase in cytosolic and microsomal fractions of rat liver. Biochem J. 1987;245:347–355. doi: 10.1042/bj2450347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot C, Hubert L, Brivet M, et al. LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat. 2010;31:E1564–E1573. doi: 10.1002/humu.21282. [DOI] [PubMed] [Google Scholar]
- Michot C, Hubert L, Romero NB, et al. Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. J Inherit Metab Dis. 2012;35:1119–1128. doi: 10.1007/s10545-012-9461-6. [DOI] [PubMed] [Google Scholar]
- Michot C, Mamoune A, Vamecq J, et al. Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim Biophys Acta. 2013;1832:2103–2114. doi: 10.1016/j.bbadis.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra MS, Chen Z, Ren H, et al. Mice with an adipocyte-specific lipin 1 separation-of-function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc Natl Acad Sci U S A. 2013;110:642–647. doi: 10.1073/pnas.1213493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68:472–478. doi: 10.1136/jnnp.68.4.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadra K, de Preux Charles AS, Medard JJ, et al. Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 2008;22:1647–1661. doi: 10.1101/gad.1638008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual F, Hsieh LS, Soto-Cardalda AI, Carman GM (2014) Yeast Pah1p phosphatidate phosphatase is regulated by proteasome-mediated degradation. J Biol Chem [DOI] [PMC free article] [PubMed]
- Peterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Federico L, Huang H, et al. A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol Biol Cell. 2010;21:3171–3181. doi: 10.1091/mbc.E10-01-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- Tonin P, Lewis P, Servidei S, DiMauro S. Metabolic causes of myoglobinuria. Ann Neurol. 1990;27:181–185. doi: 10.1002/ana.410270214. [DOI] [PubMed] [Google Scholar]
- Zeharia A, Shaag A, Houtkooper RH, et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. 2008;83:489–494. doi: 10.1016/j.ajhg.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Verity MA, Reue K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014;20:267–279. doi: 10.1016/j.cmet.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutt R, van der Kooi AJ, Linthorst GE, Wanders RJ, de Visser M. Rhabdomyolysis: review of the literature. Neuromuscul Disord. 2014;24:651–659. doi: 10.1016/j.nmd.2014.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Alignment of mutant lipin 1 with lipin 2 and 3 protein sequences. The catalytic site (DIDGT) for phosphatidic acid phosphohydrolase (PAP) activity and other haloacid dehalogenase (HAD) domains that are important for regulating PAP activity are noted. The nuclear receptor interaction domain (NRID) is important for transcriptional regulatory function. Amino acid substitutions or deletions in lipin 1 mutant alleles are in red font