

Significance
This study describes new and promising electrode materials, Li3NbO4-based electrode materials, which are used for high-energy rechargeable lithium batteries. Although its crystal structure is classified as a cation-disordered rocksalt-type structure, lithium ions quickly migrate in percolative network in bulk without a sacrifice in kinetics. Moreover, the large reversible capacity originates from the participation of oxide ions for a charge compensation process, which has been confirmed by first-principles calculations combined with X-ray absorption spectroscopy. This finding can be further expanded to the design of innovative positive electrode materials beyond the restriction of the solid-state redox reaction based on the transition metals used for the past three decades.
Keywords: battery, lithium, anion redox, positive electrode
Abstract
Rechargeable lithium batteries have rapidly risen to prominence as fundamental devices for green and sustainable energy development. Lithium batteries are now used as power sources for electric vehicles. However, materials innovations are still needed to satisfy the growing demand for increasing energy density of lithium batteries. In the past decade, lithium-excess compounds, Li2MeO3 (Me = Mn4+, Ru4+, etc.), have been extensively studied as high-capacity positive electrode materials. Although the origin as the high reversible capacity has been a debatable subject for a long time, recently it has been confirmed that charge compensation is partly achieved by solid-state redox of nonmetal anions (i.e., oxide ions), coupled with solid-state redox of transition metals, which is the basic theory used for classic lithium insertion materials, such as LiMeO2 (Me = Co3+, Ni3+, etc.). Herein, as a compound with further excess lithium contents, a cation-ordered rocksalt phase with lithium and pentavalent niobium ions, Li3NbO4, is first examined as the host structure of a new series of high-capacity positive electrode materials for rechargeable lithium batteries. Approximately 300 mAh⋅g−1 of high-reversible capacity at 50 °C is experimentally observed, which partly originates from charge compensation by solid-state redox of oxide ions. It is proposed that such a charge compensation process by oxide ions is effectively stabilized by the presence of electrochemically inactive niobium ions. These results will contribute to the development of a new class of high-capacity electrode materials, potentially with further lithium enrichment (and fewer transition metals) in the close-packed framework structure with oxide ions.
To realize sustainable energy development in the future, it is widely admitted that the substitution of energy sources for fossil fuels must be considered. An efficient energy storage system using an electrochemical method, such as rechargeable lithium batteries (Li-ion batteries, LIBs), potentially provides the solution to meet these tough challenges. Now, electric vehicles equipped with an electric motor and LIB have been launched in the market, and LIBs are starting to substitute for fossil fuels as power sources in the transportation system using the technology of internal combustion engines. Since their first appearance as power sources for portable electronic devices in 1991, the technology of LIBs has now become sufficiently sophisticated. Nevertheless, the demands for a further increase in energy density are still growing to extend the driving distance for electric vehicles.
In 1980, LiCoO2 with a cation-ordered rocksalt structure (layered type) was first proposed as a positive electrode material for LIBs (1) and is still widely used for high-energy mobile applications. After this finding, lithium insertion materials with cation-ordered rocksalt-type structures, LiMeO2 (Me = Co3+, Ni3+, etc.) have been extensively studied as electrode materials. In the past decade, Li-enriched materials, Li2MeO3-type layered materials (Me = Mn4+, Ru4+, etc.), which are also classified as having cation-ordered rocksalt-type structures (2), have been extensively studied as potential high-capacity electrode materials, especially for the Mn4+ system (Li2MnO3) (3–7). Li2MnO3 had been originally thought to be electrochemically inactive because oxidation of Mn ions beyond the tetravalent state is difficult. However, the fact is that Li2MnO3 is electrochemically active, presumably because of the contribution of oxide ions for redox reaction. Although the oxidation of oxide ions in Li2MnO3 results in partial oxygen loss with irreversible structural changes (5, 6), solid-state redox reaction of oxide ions is effectively stabilized in the Li2Ru1-xSnxO3 system (8). Nearly 1.6 mol of Li+ are reversibly extracted from Li2Ru0.75Sn0.25O3 with excellent capacity retention, indicating that unfavorable phase transition is effectively suppressed in this system.
Historically, such charge compensation by nonmetal anions has already been evidenced in sulfides before 1990 (9). Because sulfide ions are relatively soft and polarizable anion compared with oxide ions, oxidation of sulfide ions to persulfide ions is easier than that of oxide ions. The voltage as positive electrode materials is also much more attractive for the oxide ions [∼2 V vs. Li for S2−/S22− (9) and more than 3 V vs. Li for O2−/O22− (8)]. The possibility of charge compensation by oxide ions on lithium extraction was also discussed for late-transition metal oxides before 2000 (10, 11). Similarly, such rearrangement of charge was known when metal 3d orbital was heavily hybridized with oxygen 2p orbital, for instance Fe4+ in SrFeO3 (12). Nevertheless, a clear and unambiguous contribution of the oxide ions for charge compensation on “electrochemical lithium extraction” was first evidenced in 2013 on the basis of an arsenal of characterization techniques with a concept of a reductive coupling mechanism (8).
The use of oxide ion redox is an important strategy to further increase the reversible capacity of positive electrode materials for LIBs because the lithium content is potentially further enriched with a lower amount of transition metals in the framework structure. Reversible capacity as electrode materials is not limited by the absence of oxidizable transition metals as a redox center. Negatively charged oxide ions can potentially donate electrons instead of transition metals. However, oxidation without transition metals inevitably results in the release of oxygen molecules (e.g., electrochemical decomposition of Li2O2) (13).
Based on these considerations, we have decided to investigate the rocksalt phase with pentavalent niobium ions (i.e., Li3NbO4). Approximately 300 mAh⋅g−1 of reversible capacity at 50 °C is experimentally observed for a manganese-substituted Li3NbO4-based sample. Large reversible capacity is seen to partly originate from charge compensation by reversible solid-state redox of oxide ions, coupled with conventional redox reactions of transition metals. From these results, we will discuss the possibility of a new class of high-capacity electrode materials, potentially with further lithium enrichment in the close-packed framework structure with oxide ions.
Results and Discussion
Fig. 1A shows an Li-Me-O ternary phase diagram in which LiMe3+O2, Li2Me4+O3, and Li3Me5+O4 are found on a tie-line between MeO and LiO. An increase in oxidation numbers of transition metals from “trivalent to tetravalent” and “tetravalent to pentavalent” states (or even higher than pentavalent) along this tie-line allows us to enrich lithium content in the close-packed framework structure of oxide ions with fewer transition metals. Similar to Li2MeO3, Li3NbO4 is also classified as one of the cation-ordered rocksalt structures (Fig. 1B, Inset), and consists of four edge-shared NbO6 octahedra (Nb4O16 tetramer) (14). Lithium ions are accommodated in a body-centered cubic lattice of the Nb4O16 tetramers. Although Li3NbO4, white powder, crystallized as the lithium-enriched rocksalt-type phase, it is electrochemically inactive because of its insulating character without electrons in a conduction band (4d 0 configuration for Nb5+) as shown in Fig. 1B. Therefore, to induce electron conductivity in Li3NbO4, transition metals are partly substituted for Nb5+ and Li+. Because Li3Nb5+O4, LiMe3+O2, and Me2+O have a structural compatibility as the rocksalt-type structures with a common cubic close-packed (ccp) oxygen lattice, synthesis of binary intermediate phases of Li3NbO4-Me2+O and Li3NbO4-LiMe3+O2 is targeted. After several trials, it is found that Co2+, Ni2+, Fe3+, and Mn3+, which have similar ionic radii (all ranges in between Li+, 0.76 Å and Nb5+, 0.64 Å), can be substituted as shown in Fig. 2A. These metal substitutions influence the clustering/ordering of niobium in the ccp oxygen array, leading to the formation of cation-disordered rocksalt phases. For the binary phases, xLi3NbO4-(1‒x)Me2+O and xLi3NbO4-(1‒x)LiMe3+O2, all compositions (0 ≤ x ≤ 1) are prepared as single phases. (One example, the Mn3+ system, is shown in SI Appendix, Fig. S1. In this study, samples with the same lithium and transition metal contents were prepared for comparisons; x = 0.43 for LiMe3+O2 and x = 0.62 for Me2+O systems.) The Mn3+-substituted sample (0.43Li3NbO4–0.57LiMnO2) is reformulated as Li1.3Nb0.3Mn0.4O2. Similarly, the Ni2+-substituted sample (0.62Li3NbO4–0.38NiO) is reformulated as Li1.3Nb0.43Ni0.27O2. Herein, amounts of Li and the sum of Nb and Me are the same for both phases, and oxygen content is normalized to two, which is the same with the conventional layered oxides, LiMeO2.
Fig. 1.
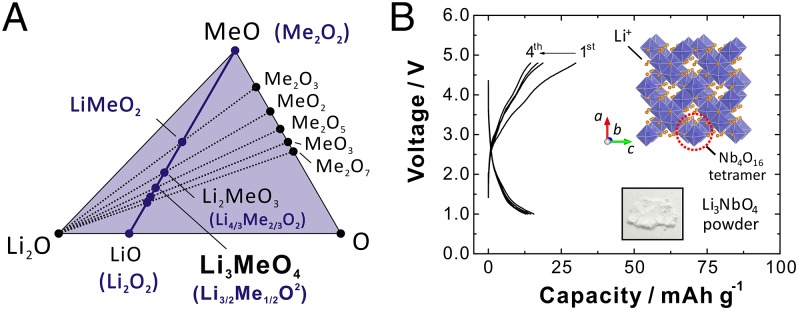
(A) An expanded view of a Li-Me-O triangular phase diagram. Many cation ordered/disordered rocksalt phases (2) are located on a tie-line (solid blue line) between MeO and LiO. Li3Nb5+O4 is found on the tie-line and lithium content is highly enriched compared with those of LiMe3+O2 and Li2Me4+O3. (B) Electrode performance of Li3NbO4 and a schematic illustration of the crystal structure of Li3NbO4. (Inset) A photograph of Li3NbO4 powder, which is an electrically insulating material.
Fig. 2.

Li3NbO4-based electrode materials. (A) SXRD patterns. (Inset) A schematic illustration of the crystal structure. (B) Particle morphology observed by SEM, as-prepared and ball-miled samples with 20 wt% of carbon, and photographs of the sample powders with different colors. (C) Electrode performance of the ball-milled samples in Li cells at room temperature at a rate of 10 mA⋅g−1 in the voltage range of 1.0 and 4.8 V (capacity retention of the samples is shown in SI Appendix, Fig. S3). (D) Electrode performance of Li1.3Nb0.3Mn0.4O2 at 60 °C at a rate of 10 mA⋅g−1 in the voltage range of 1.5 and 4.8 V.
Electrode performance of as-prepared samples was examined in Li cells. Although well-crystallized and several micrometer-sized samples are electrochemically active, reversible capacities are limited to 50–150 mAh⋅g−1 (SI Appendix, Fig. S2). Reversible capacities observed correspond to expected values based on redox reaction of Me3+/Me4+ (118 mAh⋅g−1) and Me2+/Me4+ (150 mAh⋅g−1) for Li1.3Nb0.3Me3+0.4O2 and Li1.3Nb0.43Me2+0.27O2, respectively, suggesting that the reversible capacity originates mainly from the solid-state redox reaction of transition metals without contribution of oxide ions. To reduce the particle size (and surface area) and to improve the electrical conductivity as the composite electrode, as-prepared samples and carbon materials were thoroughly mixed by mechanical ball milling. The electrode performance of Li2MnO3-based electrode materials is also known to depend on the primary particle size, which originates from the slow kinetics for the oxidation of oxide ion (15). Particle size of the samples is effectively reduced to submicrometer size (Fig. 2B). Electrode performance is significantly improved by ball milling with carbon and large reversible capacities of 250–300 mAh⋅g−1 are obtained beyond the limit of redox for transition metals. The Mn-substituted sample, Li1.3Nb0.3Mn0.4O2, delivers large reversible capacity with relatively high operating voltage among four samples in Fig. 2C. Electrode performance of Li1.3Nb0.3Mn0.4O2 is further improved at elevated temperatures. Charge/discharge curves of Li1.3Nb0.3Mn0.4O2 for first five cycles are shown in Fig. 2D. Reversible capacity observed at 60 °C reaches 300 mAh⋅g−1 with high reversibility for the initial five cycles. Electrode performance of Li1.3Nb0.3Mn0.4O2 samples are further examined in a later section.
In general, well-crystallized cation-disordered rocksalt phases are known to be electrochemically inactive (16). Nevertheless, Li1.3Nb0.3Mn0.4O2 delivers large reversible capacity with small initial irreversible capacity at 60 °C (Fig. 2D). Recently, it has also revealed the formation of a percolating network for lithium diffusion in Li1.211Mo0.467Cr0.3O2 with a lithium-excess cation-disordered phase, and a lithium diffusion barrier calculated by density functional theory (DFT) also suggests that facile macroscopic diffusion in the lithium-percolating network is energetically feasible in the lithium-excess cation-disordered rocksalt-type structure (17). Additionally, Li2VO3, which was obtained by electrochemical reduction of LiVO3 in lithium cells, has also been reported to be electrochemically active as lithium-enriched cation-disordered rocksalt phase (18). Similar to both samples, Li+ are highly mobile in the percolation network formed in Li1.3Nb0.3Mn0.4O2 and large reversible capacity is, therefore, observed with the cation-disordered rocksalt-type structure. Moreover, Li amount is enriched in Li1.3Nb0.3Mn0.4O2, compared with Li1.211Mo0.467Cr0.3O2, indicating that the accessible probability of Li by the site percolation is also enhanced. According to the calculation in the literature (17), ∼90% of Li in Li1.3Nb0.3Mn0.4O2 can migrate throughout the particle by site percolation. Such site percolation probability is further increased by the increase in Li contents using the transition metals with higher oxidation states, potentially leading to better electrode performance with cation-disordered rocksalt phase.
To further clarify the role of oxide ions for the charge compensation, first we have theoretically analyzed by ab initio calculations in the generalized gradient approximation to density functional theory (GGA DFT). The methodology and models used for the DFT calculations is described in SI Appendix. Formation energies and the corresponding voltage profile of the delithiated phase of L(4-y)/3Mn1/3Nb1/3O2 (0 ≤ y ≤ 4) are shown in SI Appendix, Fig. S4 by considering various Li/vacancy arrangements. The formation energies of intermediate compositions are negative, indicating solid solution reaction or formation of intermediate superstructure phase during electrochemical delithiation. The calculated voltage profile agrees well with the experimental one. To examine the oxidation mechanisms, partial density of state (PDOS) for the Li(4-y)/3Nb1/3Mn1/3O2 at various Li composition y is shown in Fig. 3. The Mn 3d orbital formed just below the Fermi level at the composition y = 0 without significant contribution of Nb 4d and O 2p orbitals. Hence, charge compensation mainly is due to oxidation of Mn ions from +3 to +4 at the onset of Li removal from y = 0–1, which corresponds to the theoretical capacity based on Mn3+/Mn4+ (Fig. 2C). This is also supported by net-moment analysis as listed in SI Appendix, Table S1. It is clearly indicated that Mn eg orbital at octahedral MnO6 octahedra mainly contributes to charge compensation according to the calculated partial electron density for the top of valence band at y = 0 and the bottom of conduction band at y = 1 (Fig. 3C). However, in the composition range from y = 1–3, the uppermost level of valence bands is composed of Mn 3d and O 2p hybridized orbitals due to their similar PDOS profiles. In addition, the valence state of transition metals is identical because there are minor changes in net-moment around Nb and Mn ions (SI Appendix, Table S1). Accordingly, oxygen mainly acts as the redox center for the electrochemical delithiation from y = 1–3. This fact is also supported by the partial electron density analysis, where the top of valence band at y = 1 (before delithiation) and the bottom of conduction band at y = 2 (after delithiation) are mainly composed of dumbbell-shaped O-2p orbitals, and both partial electron distributions are almost identical, as shown in Fig. 3C. The PDOS profile changes largely at y = 4, showing symmetrical configuration in terms of up and down spin states as indicated in the net-moment analyses (SI Appendix, Table S1). This charge may stem from empty electrons at the Mn 3d orbital, that is, the nominal charge of Mn is +7 (3d0 configuration). Therefore, an abrupt increase of the calculated voltage at y = 3–4 would be related to the oxidation of Mn ion accompanying electronic structural change over y > 3 (SI Appendix, Fig. S4).
Fig. 3.

Partial densities of states with electrochemical charging in L(4-y)/3Mn1/3Nb1/3O2 as calculated with the DFT method. The calculated partial electron density is also shown. Li removal at y ≥ 1.0 indicates oxidation of oxide ions due to the formation of unoccupied oxygen p-states above the Fermi level (EFermi) as shown in the light blue circle in the magnification.
Charge compensation mechanisms of Li1.3Nb0.3Mn0.4O2 in Li cells were further experimentally examined by synchrotron X-ray diffraction (SXRD) combined with X-ray absorption spectroscopy (XAS) with both hard and soft X-ray. Crystal structural analysis by SXRD in Fig. 4A reveals that reversible solid-state redox reaction occurs with continuous shift of diffraction lines on charge/discharge cycles. The lattice parameter of the sample, a = 4.187 Å (after ball milling with carbon), decreases into a = 4.105 Å after charge to 4.8 V, corresponding to ∼6% shrinkage of volume. For the layered materials, interlayer distance is significantly reduced when a large fraction of lithium ions is extracted from the crystal lattice. Uniform distribution of transition metals in the cation-disordered rocksalt phase is beneficial to suppress anisotropic volume shrinkage observed in layered oxides. Although some extent of peak broadening is observed after fully charged (320 mAh⋅g−1, extraction of 1.1 mol of Li from Li1.3Nb0.3Mn0.4O2), peak position and profile recover to the original ones after discharge to 2.0 V. Upon further reduction (Li insertion) down to 1.0 V, an additional shoulder appears at the lower diffraction angles for all diffraction lines, suggesting that strain is induced into the crystal lattice for the fully discharged state. Note that destruction of core structures is not observed after continuous cycles as shown in a later section, suggesting that electrochemical lithiation/delithiation is highly reversible.
Fig. 4.

Reaction mechanisms of Li1.3Nb0.3Mn0.4O2 as electrode materials in Li cells. (A) Changes in the SXRD patterns on lithium extraction. (B) Soft and hard X-ray absorption spectra (XAS), Mn LII,III-edge and Mn K-edge, collected for the charged/discharged samples. Mn oxidation from trivalent to tetravalent is found in the early stage of charge (80 mAh⋅g−1, x = 0.27 in Li1.3-xNb0.3Mn0.4O2) whereas no change in the spectra is found by further charge to 4.8 V at 60 °C (350 mAh⋅g−1, x = 1.19 in Li1.3-xNb0.3Mn0.4O2). (C) Changes in O K-edge XAS spectra on lithium extraction. Highlighted pre-edge peaks for the selected samples are also shown (Middle). The evolution of area for pre-edge peaks is calculated and plotted as a function of charge capacity (Right). A clear increase in the pre-edge peaks is noted as decrease in the lithium content, and the area is returned to its original area after lithium insertion (plotted as the filled circle).
XAS spectra of Mn and Nb at K-edge and L-edge have been collected for the samples with different oxidation/reduction conditions to examine the redox species on oxidation/reduction process. Because soft X-ray absorption spectra were collected using the fluorescence yield mode, electronic structures of each element can be obtained from the inside of particles [in general, >100 nm from the surface (19)]. From Mn K-edge (mainly dipole transition from 1s core level to empty 4p level) and LII, III-edge (dipole transition from filled 2p to empty 3d level) spectra as shown in Fig. 4B, energy position and profile of spectra for the as-prepared sample are found to be consistent to those of trivalent manganese ions (20). In the early stage of Li extraction (80 mAh⋅g−1, x = ∼0.3 in Li1.3-xNb0.3Mn0.4O2), a clear energy shift (∼4.5 eV) in Mn K-edge spectra is found. Similarly, a profile of Mn LII, III-edge spectra changes until x reaches 0.4 (∼120 mAh⋅g−1), and no change in the spectra is found by further Li extraction, indicating that charge compensation in this region (x > 0.4) is not achieved by the oxidation of Mn 3d level. Note that manganese ions are reversibly reduced into the trivalent state after discharge. No change in oxidation state of Nb is found, as shown in SI Appendix, Fig. S5. From these results, including theoretical/experimental data, it is concluded that Mn are reversibly oxidized from trivalent to tetravalent state in the early stage of Li extraction (x = ∼0.4), and transition metals (Mn/Nb) are not responsible for the large capacity as suggested by the DFT calculations (Fig. 3).
To examine the contribution of the oxide ions for the charge compensation, oxygen K-edge spectra have been, therefore, collected. Fig. 4C compares normalized O K-edge (dipole transition from 1s core-level to empty 2p level) spectra with different charge/discharge conditions. As shown in Fig. 4C, a clear change in the profile of O K-edge spectra is found with the increase in the lithium extraction amount, especially for the low-energy-region ranges from 527 to 533 eV (a pre-edge region). Highlighted O K-edge spectra in the pre-edge region are also plotted in Fig. 4C. The change in the profile is found after charge to 80 mAh⋅g−1 (x = ∼0.3); a single absorption peak centered at 530.3 eV for the as-prepared sample splits into two peaks centered at 528.7 and 530.6 eV after charge (black dotted line in Fig. 4C). This change would be expected because of the influence of the oxidation of Mn. Indeed, a single absorption peak at 530 eV is again observed after full discharge with Mn3+. The change in the area of normalized absorption spectra in the pre-edge region is also plotted in Fig. 4C. Normalized area slightly increases (∼12%) after charge to 80 mAh⋅g−1 with the charge in the profile of spectra. After charge from 80 to 350 mAh⋅g−1 of charge capacity, a profile of the spectra is not changed. However, a clear increase in the area of absorption spectra is found with the increase in the amount of lithium extraction. The area of spectra in O K-edge (pre-edge region) is enlarged by >40% on charge from 150 to 350 mAh⋅g−1, whereas no change is found in this region in Mn LII, III-edge spectra. This fact suggests that a new component formed by oxidation appears at around 530.2 eV (red dotted line in Fig. 4C), and its fraction increases as a function of charge capacity. Additionally, the peak at around 530 eV is in good agreement with that of Li2O2 (21). Such systematic change in O K-edge spectra was not found in conventional lithium insertion materials [e.g., LiNi1/2Mn1/2O2 (22) and LiCo1/3Ni1/3Mn1/3O2 (23)], and more pronounced changes are observed for Li1.3-xNb0.3Mn0.4O2 in comparison with Li1.16Ni0.15Co0.19Mn0.50O2 in the fluorescence yield mode (24). In the literature for these layered materials, the changes in the spectra were mainly found in the early stage of charge, and these changes can be partly contributed by the oxidation of transition metals, as also observed in this study. Similar to this observation by XAS, clear and reversible change in oxygen 1s spectra by XPS has been also reported for Li2Ru1−yMnyO3(7) and Li2Ru1−ySnyO3(8). From these results, it is concluded that large reversible capacity of Li1.3-xNb0.3Mn0.4O2 originates from the reversible compensation of charge by oxide ions. When it is assumed that all charges (except the redox of Mn3+/Mn4+) are consumed by oxide ions, formal oxidation state of Li1.3Nb5+0.3Mn3+0.4O2−2 changes into Li0.25Nb5+0.3Mn4+0.4O1.675−2 (4.8 V charge at 60 °C).
To further evaluate the feasibility of the concept, solid-state redox reaction of oxide ions as electrode materials, electrochemical properties of Li1.3Nb0.3Mn0.4O2 were examined for a modified sample (Fig. 5). Synthesis and characterization of the modified samples are described in SI Appendix, Figs. S6 and S7. Electrode performance of modified Li1.3Nb0.3Mn0.4O2 is shown in Fig. 5A in the voltage range of 1.5–4.8 V at 50 °C. Although a lesser amount of carbon (see SI Appendix, Fig. S6) and a lower operating temperature is used compared with the sample in Fig. 2D, polarization for charge/discharge is clearly reduced with the modified sample, leading to an increase in the energy density. The energy density of the sample, estimated from initial discharge capacity and operating voltage, reaches 950 mWh⋅g−1. Cyclability as electrode materials with galvanostatic charge/discharge is, however, insufficient as electrode materials, as shown in Fig. 5A.
Fig. 5.

Electrode performance of modified Li1.3Nb0.3Mn0.4O2 in Li cells at 50 °C. (A) Galvanostatic charge/discharge curves in the range of 1.5–4.8 V, initial cycle at a rate of 10 mA⋅g-1 and cycles 2–20 at a rate of 25 mA⋅g-1. (B) Charge/discharge curves with constant charge capacity (100, 150, 200, and 250 mAh⋅g−1). (C) Charge/discharge curves with constant charge capacity of 250 mAh⋅g−1 at a rate of 25 mA⋅g−1. (Inset) Discharge capacity retention with constant current and constant charge capacity (250 mAh⋅g−1) is compared. (D) Rate capability of the sample; the cell was charged to 4.6 V at 50 mA⋅g−1 and held at 4.6 V for 90 min and then discharged to 1.5 V at different rates, 50 mA⋅g−1 to 1.6 A⋅g−1. (Inset) A Ragone plot of the sample. The sample loading used was 2.8 mg⋅cm−2.
Improvement of cyclability as electrode materials is realized by charge with a constant capacity mode. Charge capacities were stepwisely increased from 100 to 250 mAh⋅g−1 by 50 mAh⋅g−1 after each five cycles. Typical discharge curves at 25 mA⋅g−1 with different charge capacities are shown in Fig. 5B. Good capacity retention is observed even at 250 mAh⋅g−1, and the reversible limit of Li1.3Nb0.3Mn0.4O2 as electrode materials is estimated to be ∼250 mAh⋅g−1 in this experimental condition. Therefore, a continuous cycle test was conducted by charge with constant capacity of 250 mAh⋅g−1. As shown in Fig. 5C, cyclability is significantly improved by constant capacity charge. The sample delivers ∼245 mAh⋅g−1 of reversible capacity (98% of coulombic efficiency) for 20 continuous cycles (Fig. 5C, Inset) even though initial discharge capacity is slightly low (210 mAh⋅g−1) and a gradual increase in the polarization is observed. To examine the possibility of damage to bulk crystal structure, an XRD pattern was collected for the electrode after 20 cycles with the constant capacity charge condition (250 mAh⋅g−1). As shown in SI Appendix, Fig. S8, an XRD pattern is observed similar to that in Fig. 3A, indicating that good structural reversibility on continuous cycles.
Finally, rate capability of the sample, which influences power density as electrode materials, was examined. As shown in Fig. 5D, Li1.3Nb0.3Mn0.4O2 with the cation-disordered rocksalt phase surprisingly shows good rate capability in Li cells at 50 °C even though slow kinetics for lithium migration in percolation network was expected. Discharge capacity reaches ∼200 mAh⋅g−1 even at 800 mA⋅g−1, which is clearly larger than that of the reversible capacity based on Mn3+/Mn4+ redox. A Ragone plot of the sample is also shown in the inset, and the plot clearly shows that the rate capability of solid-state redox reaction of oxide ions is acceptable as electrode materials for rechargeable lithium batteries. We have used the ball-milling process to reduce primary particle size and prepare carbon composite samples. Note that ball milling with carbon is not a necessary condition to use the redox reaction of oxide ions. A well-crystallized and micrometer-sized sample without ball milling shows high reversible capacity (>250 mAh⋅g−1), as shown in SI Appendix, Fig. S9. Further increase in the reversible capacity and energy density with acceptable capacity retention for Li3NbO4-based electrode materials is anticipated through systematic studies in relation to the optimization of particle morphology (surface area), chemical compositions, surface coating of the particle, and so on.
Conclusions
In this study, Nb5+ ions have been chosen to stabilize the redox reaction of oxide ions, and high-reversible capacities of 250–300 mAh⋅g−1 for Li1.3Nb0.3Mn0.4O2 have been achieved at 50 oC. Large reversible capacity originates from the solid-state redox of oxide ions coupled with transition metal redox. Although the host structure is classified as the cation-disorderd rock-salt phase, fast kinetics for Li migration in the percolation network has been experimentally evidenced. Moreover, this concept would be extended to other elements with high oxidation states. Many oxides containing Li+, which had been originally thought to be electrochemically inactive, such as Li3NbO4, can potentially be used as the new host structures for high-capacity electrode materials. Recently, Co-doped Li2O prepared by ball milling has been proposed as electrode materials with the concept of oxide ion redox even though the practical reversible capacity is limited to 200 mAh⋅g−1 (25). We believe that our finding will lead to material innovations on positive electrode materials for rechargeable batteries, beyond the restriction of the solid-state redox reaction based on the transition metals used for the past three decades.
Materials and Methods
Synthesis of Materials.
Li3NbO4 was prepared by a solid-state reaction from stoichiometric amounts of Li2CO3 (>98.5%; Kanto Kagaku) and Nb2O5 (99.9%; Wako Pure Chemical Industries) at 950 °C for 24 h in air. Li1.3Nb0.3Me0.4O2 (Me = Mn3+ and Fe3+) and Li1.3Nb0.43Me0.27O2 (Me = Co2+ and Ni2+) were also prepared from Li2CO3, Nb2O5, and precursors containing each transition metal: Mn2O3, Fe2O3 (99.9%; Wako Pure Chemical Industries), CoO (Wako Pure Chemical Industries), and Ni4CO3(OH)6(H2O)4 (Kishida Chemical). Mn2O3 was obtained by heating of MnCO3 (Kishida Chemical) at 850 °C for 5 h. The precursors were thoroughly mixed by wet mechanical ball milling and then dried in air. Thus obtained mixtures of the samples were pressed into pellets. The pellets were heated at 950 °C for 24 h in air (Fe3+ and Ni2+) or inert atmosphere (Mn3+ and Co2+). The samples were stored in an Ar-filled glove box until use.
Electrochemistry.
Electrode performance of the samples was examined for as-prepared samples and the carbon composite samples prepared by ball milling. As-prepared samples (80 wt%) were mixed using a mortar and pestle with 10 wt% acetylene black and 10 wt% poly(vinylidene fluoride) and pasted on aluminum foil as a current collector. Additionally, as-prepared Li3NbO4, Li1.3Nb0.3Me0.4O2 (Me = Mn3+ and Fe3+), and Li1.3Nb0.43Me0.27O2 (Me = Co2+ and Ni2+) samples were mixed with acetylene black (samples: AB = 80: 20 wt%) by using a planetary ball mill (PULVERISETTE 7; FRITSCH) at 300 rpm with a zirconia container and balls. Composite positive electrodes consisted of 72 wt% active materials, 18 wt% acetylene black, and 10 wt% poly(vinylidene fluoride), pasted on aluminum foil as a current collector. The electrodes were dried at 80 °C in vacuum. Metallic lithium (Honjo Metal) was used as a negative electrode. The electrolyte solution used was 1.0 mol⋅dm−3 LiPF6 dissolved in ethylene carbonate: dimethyl carbonate (1:1 by volume) (Kishida Chemical). A polyolefin microporous membrane was used as a separator. R2032-type coin cells were assembled in the Ar-filled glove box. The coin cells were cycled in the voltage ranges of 1.0‒4.8 V or 1.5‒4.8 V at a rate of 10 mA⋅g−1 at room temperature or 60 °C.
Materials Characterization.
SXRD patterns were collected at beam line BL02B2, SPring-8 in Japan, equipped with a large Debye–Scherrer camera (26). Li1.3-yNb0.3Mn0.4O2 samples were electrochemically prepared in the coin cells at a rate of 5 mA⋅g−1. The composite electrodes were rinsed with dimethyl carbonate to remove excess electrolyte and then dried at room temperature in the Ar-filled glove box. The composites were separated from the aluminum current collectors, crushed into powders, and then poured into glass capillaries (diameter 0.5 mm). The glass capillaries were sealed using a resin in the glove box to eliminate sample exposure to air. To minimize the effect of X-ray absorption by the samples, the wavelength of incident X-ray beam was set to 0.5 Å using a silicon monochromator, which was calibrated to 0.5001(1) Å with a CeO2 standard. Structural analysis was carried out using RIETAN-FP (27). Schematic illustrations of the crystal structures of Li3NbO4-based samples were drawn using the program VESTA (28).
Hard X-ray absorption spectroscopy at Mn K-edge and Nb K-edge was performed at beam line BL-7C of the Photon Factory Synchrotron Source in Japan. Hard X-ray absorption spectra were collected with a silicon monochromator in a transmission mode. The intensity of incident and transmitted X-ray was measured using an ionization chamber at room temperature. Li3NbO4-based composite samples were prepared using the coin cells at a rate of 12 mA⋅g−1. The composite electrodes were rinsed with dimethyl carbonate and sealed in a water-resistant polymer film in the Ar-filled glove box. Normalization of the XAS spectra was carried out using the program code IFEFFIT (29). The postedge background was determined using a cubic spline procedure.
Soft X-ray absorption spectra were collected at BL-2 (O K-edge and Mn LII, III-edge) and BL-10 (Nb LIII-edge) in the synchrotron facility of Ritsumeikan University (Synchrotron Radiation Center) (30). A grazing incidence grating monochromator is equipped in BL-2 with Monk–Gillieson-type optics and the Golovchenko-type double crystal monochromator is equipped in BL-10. The absorption spectra were collected with the fluorescence yield mode. Similar to the measurements for hard X-ray absorption spectroscopy, the samples were prepared in the Ar-filled glove box, and thus prepared samples were set on the spectrometer using a laboratory-made transfer vessel without air exposure.
Supplementary Material
Acknowledgments
This research has been partly supported by Advanced Low Carbon Technology Research and Development Program of the Japan Science and Technology Agency (JST) Special Priority Research Area “Next-Generation Rechargeable Battery.” The synchrotron X-ray diffraction experiments were made possible through the support of the Japanese Ministry of Education, Science, Sports and Culture, Nanotechnology Support Project (Proposals 2012B1777 and 2013A1681) with the approval of the Japan Synchrotron Radiation (SR) Research Institute. The hard X-ray absorption work was done under the approval of the Photon Factory Program Advisory Committee (Proposals 2012G149 and 2012G594) and the soft X-ray absorption work was done at the SR Center of Ritsumeikan University under the “Project for Creation of Research Platforms and Sharing of Advanced Research Infrastructure” (Proposal R1344). M.N. is grateful for financial support from the JST Precursory Research for Embryonic Science and Technology program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504901112/-/DCSupplemental.
References
- 1.Mizushima K, Jones PC, Wiseman PJ, Goodenough JB. LiXCOO2(0 < x ≤ 1): A new cathode material for batteries of high energy density. Mater Res Bull. 1980;15(6):783–789. [Google Scholar]
- 2.Mather GC, Dussarrat C, Etourneau J, West AR. A review of cation-ordered rock salt superstructure oxides. J Mater Chem. 2000;10(10):2219–2230. [Google Scholar]
- 3.Robertson AD, Bruce PG. Mechanism of electrochemical activity in Li2MnO3. Chem Mater. 2003;15(10):1984–1992. [Google Scholar]
- 4.Johnson CS, et al. The significance of the Li2MnO3 component in ‘composite’ xLi(2)MnO(3) center dot (1-x)LiMn0.5Ni0.5O2 electrodes. Electrochem Commun. 2004;6(10):1085–1091. [Google Scholar]
- 5.Armstrong AR, et al. Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li0.2Mn0.6]O2. J Am Chem Soc. 2006;128(26):8694–8698. doi: 10.1021/ja062027+. [DOI] [PubMed] [Google Scholar]
- 6.Yabuuchi N, Yoshii K, Myung S-T, Nakai I, Komaba S. Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3-LiCo(1/3)Ni(1/3)Mn(1/3)O2. J Am Chem Soc. 2011;133(12):4404–4419. doi: 10.1021/ja108588y. [DOI] [PubMed] [Google Scholar]
- 7.Sathiya M, et al. High performance Li2Ru1–yMnyO3 (0.2 ≤ y ≤ 0.8) cathode materials for rechargeable lithium-ion batteries: Their understanding. Chem Mater. 2013;25(7):1121–1131. [Google Scholar]
- 8.Sathiya M, et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat Mater. 2013;12(9):827–835. doi: 10.1038/nmat3699. [DOI] [PubMed] [Google Scholar]
- 9.Blandeau L, Ouvrard G, Calage Y, Brec R, Rouxel J. Transition-metal dichalcogenides from disintercalation processes. Crystal structure determination and Mossbauer study of Li 2 FeS 2 and its disintercalates Li x FeS 2 (0.2≤x≤2) J Phys C Solid State Phys. 1987;20(27):4271. [Google Scholar]
- 10.Aydinol MK, Kohan AF, Ceder G, Cho K, Joannopoulos J. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys Rev B. 1997;56(3):1354–1365. [Google Scholar]
- 11.Tarascon JM, et al. In situ structural and electrochemical study of Ni1−xCoxO2 metastable oxides prepared by soft chemistry. J Solid State Chem. 1999;147(1):410–420. [Google Scholar]
- 12.Bocquet AE, et al. Electronic structure of SrFe4+O3 and related Fe perovskite oxides. Phys Rev B Condens Matter. 1992;45(4):1561–1570. doi: 10.1103/physrevb.45.1561. [DOI] [PubMed] [Google Scholar]
- 13.Ogasawara T, Débart A, Holzapfel M, Novák P, Bruce PG. Rechargeable LI2O2 electrode for lithium batteries. J Am Chem Soc. 2006;128(4):1390–1393. doi: 10.1021/ja056811q. [DOI] [PubMed] [Google Scholar]
- 14.Ukei K, Suzuki H, Shishido T, Fukuda T. Li3NbO4. Acta Crystallogr C. 1994;50(5):655–656. [Google Scholar]
- 15.van Bommel A, Dahn JR. Kinetics study of the high potential range of lithium-rich transition-metal oxides for lithium-ion batteries by electrochemical methods. Electrochem Solid-State Lett. 2010;13(5):A62–A64. [Google Scholar]
- 16.Rougier A, Gravereau P, Delmas C. Optimization of the composition of the Li1−z Ni1+z O2 electrode materials: Structural, magnetic, and electrochemical studies. J Electrochem Soc. 1996;143(4):1168–1175. [Google Scholar]
- 17.Lee J, et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science. 2014;343(6170):519–522. doi: 10.1126/science.1246432. [DOI] [PubMed] [Google Scholar]
- 18.Pralong V, Gopal V, Caignaert V, Duffort V, Raveau B. Lithium-rich rock-salt-type vanadate as energy storage cathode: Li2–xVO3. Chem Mater. 2011;24(1):12–14. [Google Scholar]
- 19.de Groot FMF. X-ray absorption and dichroism of transition metals and their compounds. J Electron Spectrosc Relat Phenom. 1994;67(4):529–622. [Google Scholar]
- 20.Yoon W-S, Chung K-Y, Oh K-H, Kim K-B. Changes in electronic structure of the electrochemically Li-ion deintercalated LiMn2O4 system investigated by soft X-ray absorption spectroscopy. J Power Sources. 2003;119–121(0):706–709. [Google Scholar]
- 21.Qiao RM, Chuang YD, Yan SS, Yang WL. Soft X-Ray irradiation effects of Li2O2, Li2CO3 and Li2O revealed by absorption spectroscopy. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.004918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon W-S, et al. Soft X-ray absorption spectroscopic study of a LiNi0.5Mn0.5O2 cathode during charge. J Electrochem Soc. 2004;151(2):A246–A251. [Google Scholar]
- 23.Yoon W-S, et al. Investigation of the charge compensation mechanism on the electrochemically Li-ion deintercalated Li1-xCo1/3Ni1/3Mn1/3O2 electrode system by combination of soft and hard X-ray absorption spectroscopy. J Am Chem Soc. 2005;127(49):17479–17487. doi: 10.1021/ja0530568. [DOI] [PubMed] [Google Scholar]
- 24.Oishi M, et al. Charge compensation mechanisms in Li1.16Ni0.15Co0.19Mn0.50O2 positive electrode material for Li-ion batteries analyzed by a combination of hard and soft X-ray absorption near edge structure. J Power Sources. 2013;222(0):45–51. [Google Scholar]
- 25.Okuoka S-i, et al. 2014. A new sealed lithium-peroxide battery with a Co-doped Li2O cathode in a superconcentrated lithium bis(fluorosulfonyl)amide electrolyte. Sci Rep 4, 10.1038/srep05684.
- 26.Nishibori E, et al. 2001. The large Debye–Scherrer camera installed at SPring-8 BL02B2 for charge density studies. J Phys Chem Solids 62(12):467:2095–2098.
- 27.Izumi F, Momma K. Three-dimensional visualization in powder diffraction. Solid State Phenom. 2007;130:15–20. [Google Scholar]
- 28.Momma K, Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Cryst. 2011;44(6):1272–1276. [Google Scholar]
- 29.Newville M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J Synchrotron Radiat. 2001;8(Pt 2):322–324. doi: 10.1107/s0909049500016964. [DOI] [PubMed] [Google Scholar]
- 30.Ishii H, Nakanishi K, Watanabe I, Ohta T, Kojima K. 2011. Improvement of ultra soft X-ray absorption spectroscopy and photoelectron spectroscopy beamline for studies on related materials and cathodes of lithium ion batteries. e-Journal Surface Sci Nanotechnol 9:416–421.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.