

Significance
This study fundamentally alters our understanding of how TLR4 drives breast cancer. Although TLR4 was previously considered a tumor promoter, we demonstrate a complex, TP53-dependent role for TLR4 in regulating tumor growth. TP53 is a tumor suppressor commonly inactivated across cancer types. In TP53 wild-type cancer cells, TLR4 activation causes secretion of IFN-γ into the microenvironment, resulting in induction of p21 and inhibition of cell growth. Conversely, TLR4 activation in TP53 mutant cells promotes cancer cell growth by regulating CXCL1 and CD154 secretion. In this paper, we identify a previously unidentified role for TLR4 in modulating tumor cell growth and microenvironment. The TLR4–TP53 association likely extends across cancer types, suggesting the need to determine the TP53 status of any tumor before implementing anti-TLR4 therapy.
Keywords: TLR4, breast cancer, TP53, microenvironment, IFN-γ
Abstract
Breast cancer is a leading cause of cancer-related death, and it is important to understand pathways that drive the disease to devise effective therapeutic strategies. Our results show that Toll-like receptor 4 (TLR4) drives breast cancer cell growth differentially based on the presence of TP53, a tumor suppressor. TP53 is mutationally inactivated in most types of cancer and is mutated in 30–50% of diagnosed breast tumors. We demonstrate that TLR4 activation inhibits growth of TP53 wild-type cells, but promotes growth of TP53 mutant breast cancer cells by regulating proliferation. This differential effect is mediated by changes in tumor cell cytokine secretion. Whereas TLR4 activation in TP53 mutant breast cancer cells increases secretion of progrowth cytokines, TLR4 activation in TP53 wild-type breast cancer cells increases type I IFN (IFN-γ) secretion, which is both necessary and sufficient for mediating TLR4-induced growth inhibition. This study identifies a novel dichotomous role for TLR4 as a growth regulator and a modulator of tumor microenvironment in breast tumors. These results have translational relevance, demonstrating that TP53 mutant breast tumor growth can be suppressed by pharmacologic TLR4 inhibition, whereas TLR4 inhibitors may in fact promote growth of TP53 wild-type tumors. Furthermore, using data generated by The Cancer Genome Atlas consortium, we demonstrate that the effect of TP53 mutational status on TLR4 activity may extend to ovarian, colon, and lung cancers, among others, suggesting that the viability of TLR4 as a therapeutic target depends on TP53 status in many different tumor types.
Breast cancer has one of the highest incidence rates of cancer in women worldwide, with more than 1.5 million women diagnosed with the disease in 2012. Owing to its high incidence, breast cancer is also one of the leading causes of cancer-related deaths, with 40,000 women predicted to die of the disease in 2014 in the US alone. The diagnosis and treatment of breast cancer has been significantly improved by the identification of three major subtypes of the disease based on receptor expression: estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-positive, and triple-negative [tumors lacking ER, progesterone receptor (PR), and HER2]. Of these subtypes, ER-positive breast cancer accounts for 70–80% of all diagnosed breast tumors.
ER-positive breast cancer is largely responsive to endocrine therapy; however, intrinsic or acquired resistance occurs in one-third of cases and contributes significantly to breast cancer-associated mortality. Therefore, identifying therapeutic targets to prevent ER-positive breast cancer mortality is a major focus of scientific investigation. ER-positive breast tumors with a high mutation load are associated with poor patient survival, and a high mutation load likely affects the response to endocrine therapy (1). Because known drivers of endocrine resistance (e.g., PR negativity and HER2 amplification) are not enriched in this subset, the identification of novel drivers is critical to the discovery of prognostic/predictive markers and generation of targeted therapies. In a screen for preferentially mutated genes, we identified Toll-like receptor 4 (TLR4) as a likely driver of this poorly surviving ER-positive subset.
TLR4 is a member of the Toll-like family of proteins, which localizes to both the cell membrane and the cytoplasm and is studied primarily in immune cells. TLR4 is activated by a variety of ligands: DNA, RNA, and viral particles; chemotherapeutic agents; and lipopolysaccharides (LPS). TLR4 induction in immune cells can activate numerous cancer-associated signaling cascades, including the MAP kinase and NFkB pathways (2, 3). These pathways transcriptionally activate the secretion of either proinflammatory cytokines, such as IL-6 and IL-8, or anti-inflammatory type I IFNs, including IFN-γ.
TLR4 activity in tumor-recruited immune cells induces antitumor immunity by modifying secreted cytokines in the tumor microenvironment, thereby regulating T-cell maturation (4). TLR4 also has been identified at the protein level in breast epithelial tumor cells (5). In contrast to its role in tumor-associated immune cells, TLR4 promotes growth (6) and chemotherapeutic resistance (7, 8) in ER-negative breast cancer cell lines, in accordance with studies of ovarian cancer (9, 10). Based on these studies, therapies targeting TLR4 appear to be novel viable strategies with significant potential for treating cancer, and have in fact been proposed as such (6–8).
In this study, we demonstrate that TLR4 promotes cell growth in TP53 mutant breast cancer, but inhibits cell growth in TP53 wild-type breast cancer. Moreover, we demonstrate TP53-dependent differential cytokine secretion by breast cancer cells on TLR4 activation, resulting in the secretion of proinflammatory cytokines in TP53 mutant cells and the tumor antagonistic cytokine, IFN-γ, in TP53 wild-type cells. Finally, we show a similar association between TLR4 and TP53 across different cancer types. Our results indicate that TLR4 may serve as a druggable target specifically in TP53 mutant tumors, whereas TLR4 inhibition in TP53 wild-type tumors can have adverse effects. Therefore, these data demonstrate the need to identify the TP53 mutational status of any tumor before administering anti-TLR4 therapy.
Results
TLR4 Is Frequently Mutated in ER-Positive High Mutation Load Breast Tumors, and TLR4 Loss Is Associated with Poor Survival.
A subset of ER-positive breast cancers have a high mutation load and are associated with poor patient outcome (1). To ascertain the pathways underlying this poor-outcome phenotype, we performed gene set enrichment analyses, and identified TLR4 as one of the most frequently mutated genes in this subset. TLR4 is mutated in ∼1% of all breast cancers, both ER-positive and ER-negative, but is mutated at >5% frequency in the poor- outcome subset of ER-positive tumors. Although TLR4 has been proposed to be a breast cancer oncogene (6–8), the TLR4 mutations in breast tumors from The Cancer Genome Atlas (TCGA) dataset appear to disrupt gene function, and manifest as either early truncating or missense mutations in protein interaction regions (Fig. 1A). Both truncating mutations observed in breast tumors occur early in the gene and most likely result in deletion-like or gene-silencing phenotypes; the missense mutations identified also are likely to inhibit gene function. The first 608 amino acids of TLR4 constitute its extracellular domain (2); consequently, missense mutations within this region potentially inhibit the binding of TLR4 to its cognate partner, myeloid differentiation protein 2 (MD2), which is essential for ligand recognition. Mutations in this region have been reported to disrupt MD2–TLR4 interactions (2). The intracellular region of TLR4 also contains clusters of missense mutations within the Toll/IL-1 receptor (TIR) domain (Fig. 1A), which are essential for the recruitment of intracellular effectors of TLR4 signaling, including myeloid differentiation primary response 88 and TIR domain-containing adaptor protein (2, 3). This mutational profile suggests that TLR4 gene function is disrupted in some breast tumors in which TLR4 may function as a tumor suppressor gene.
Fig. 1.

TLR4 correlates with patient survival in a TP53-dependent manner. (A) Mutational profile of TLR4 in breast cancer modified from cBio and COSMIC indicating the frequency of nonsense mutations resulting in truncation (red) and missense (blue) mutations in the context of protein interaction domains. (B and C) Boxplots depicting decreased TLR4 gene expression in invasive breast cancer relative to normal breast tissue in two publically available datasets (Curtis, B; TCGA, C). The Student t test identified P values. (D, F, and G) Kaplan–Meier survival curves of TLR4-low (red) and TLR4-high (black) subsets (delineated based on gene expression) of the Curtis dataset for all breast cancer subtypes (D), TP53 mutant breast tumors (F), and TP53 wild-type breast tumors (G). The log-rank test determined P values. Accompanying information is presented in Fig. S1 and Tables S1 and S2. (E) Paired comparison of TLR4 gene expression between normal tissue and associated TP53 mutant (Left) and TP53 wild-type (Right) invasive breast cancer tissue from the same patient in the TCGA dataset. The paired t test determined P values. Error bars in B and C represent SD; black lines in E indicate samples from the same patient. LRR, leucine-rich repeat; TIR, Toll/IL-1R resistance domain.
To determine whether TLR4 gene function is dysregulated in breast tumors, we analyzed TLR4 gene expression in two public datasets (Curtis and TCGA), and found that significantly lower TLR4 gene expression in many breast tumors relative to normal breast tissue (Fig. 1 B and C). Moreover, analysis of the cBio database (Computational Biology Center, Memorial Sloan-Kettering Cancer Center) indicates that TLR4 loss through homozygous/heterozygous deletion, mutation, and/or RNA-based disruption occurred in ∼20% of breast tumors in the TCGA dataset. These data led us to assess whether TLR4 loss is associated with poor clinical outcome (both overall and disease-free survival) in breast cancer patients. Using the same two publically available breast cancer datasets (TCGA and Curtis), we found that low TLR4 gene expression did indeed associate with worse overall survival (Curtis, Fig. 1D; TCGA, Fig. S1A) and disease-free survival (TCGA, Fig. S1B; Curtis, Fig. S1C) relative to patients with high TLR4 gene expression. The association between TLR4 and overall survival remained significant even after multivariate analysis (Curtis; Table S1). We then examined survival based on ER status, and found that the significant association between TLR4 loss and poor patient survival was restricted to ER-positive, largely TP53 wild-type, breast cancers (Fig. S1 D and E).
Fig. S1.

TLR4 gene expression associates with patient survival. (A–E) Kaplan–Meier survival curves of TLR4-low (red) and TLR4-high (black) subsets (delineated based on gene expression) describing overall survival (A) and disease-free survival (B and C) associated with all breast tumors from the TCGA (A and B) and Curtis (C) datasets, and with ER-positive and ER-negative breast tumors individually from the Curtis dataset (D and E). Log-rank test determined P values. (F) Lolliplots of TP53 mutations in TLR4-low and TLR4-high ER-positive breast tumors from the TCGA dataset. DBD, DNA binding domain; TA, transactivation domain; Tetra, tetramerisation domain. Accompanying data presented in Fig. 1 D, F, and G and Table S1.
Table S1.
Proportional hazards table identifying TLR4 as an independent prognostic factor for breast cancer (Curtis dataset)
| Factor | Hazard ratio | 95% CI | P value |
| TLR4 gene expression | |||
| High | Reference | ||
| Low | 1.24 | 1.02–1.51 | 0.03* |
| ER status | |||
| Positive | Reference | ||
| Negative | 0.93 | 0.77–1.12 | 0.44 |
| PR status | |||
| Negative | Reference | ||
| Positive | 0.80 | 0.44–1.82 | 0.008** |
| HER2 status | |||
| Negative | Reference | ||
| Positive | 1.39 | 1.13–1.70 | 0.002** |
| Tumor grade | |||
| Grade I | Reference | ||
| Grade II | 1.18 | 0.84–1.68 | 0.34 |
| Grade III | 1.33 | 0.93–1.89 | 0.11 |
| Tumor stage | |||
| Stage NA | Reference | ||
| Stage 0 | 1.06 | 0.88–1.29 | 0.54 |
| Stage I | 0.72 | 0.54–0.95 | 0.02* |
| Stage II | 0.97 | 0.79–1.19 | 0.79 |
| Stage III | 1.76 | 1.26–2.46 | <0.001*** |
| Stage IV | 4.84 | 2.25–10.39 | <0.001*** |
| Tumor size, mm | 1.00 | 1.00–1.01 | <0.001*** |
P values generated by Cox regression analysis for proportional hazards. *P < 0.05; **P < 0.01; ***P < 0.001.
Clinical Relevance of TLR4 Expression Is TP53 Dependent.
The frequency of TP53 mutagenesis varies between ER-positive and ER-negative breast cancers; ER-negative cancers are predominantly TP53 mutant, whereas ER-positive cancers are largely TP53 wild-type. For this reason, we next investigated TLR4 gene expression based on TP53 mutational status. In paired samples of normal breast and associated invasive breast cancer tissues, TLR4 expression levels were significantly lower in invasive breast tumors from women with TP53 wild-type tumors compared with women with TP53 mutant breast tumors (TCGA; Fig. 1E). Although almost one-half of the TP53 mutant tumors analyzed demonstrated increased TLR4 gene expression relative to normal breast tissue, <16% of TP53 wild-type tumors showed increased TLR4 expression. Furthermore, >80% of the TP53 wild-type tumors had lower TLR4 gene expression than the associated normal breast tissue. These data indicate that TP53 wild-type breast tumor cells are more likely than TP53 mutant breast tumor cells to lose TLR4 expression, and thus suggest the hypothesis that TLR4 functions as a TP53-dependent tumor suppressor.
To test whether specific TP53 mutations associate with TLR4 gene expression, we next classified ER-positive breast tumors into TLR4-low (lower than the mean value) and TLR4-high (higher than the mean value) based on gene expression, and assessed whether the type and site of TP53 mutations differs between these two groups (Fig. S1F). We found no significant site-specific differences in TP53 mutations (R175H: P = 0.60; R273C/H: P = 0.20) between the TLR4-low and TLR4-high tumors, but this may be a reflection of the small sample size and relatively low incidence of repeated mutations. We also classified ER-positive tumors based on their TP53 mutations into dominant-negative, gain-of-function, and loss-of-function types (International Agency for Research on Cancer database) and assessed whether TLR4 gene expression differed between these tumors, but found no significant differences.
We next examined whether the effect of TLR4 expression on breast cancer survival depended on TP53 mutational status. We found that, in contrast to our previous results, women with TLR4-low TP53 mutant tumors had better overall survival than women with TLR4-high TP53 mutant tumors (Curtis; Fig. 1F). Conversely, consistent with our previous results across all breast tumors and all ER-positive tumors, women with TLR4-low TP53 wild-type tumors, had significantly worse overall survival than women with TLR4-high TP53 wild-type tumors (Curtis; Fig. 1G). In addition, when TP53 status was included in the multivariate survival analysis, the prognostic value of TLR4 no longer was independently significant (Curtis; Table S2), supporting the hypothesis that these two factors jointly affect patient outcome.
Table S2.
Proportional hazards table identifying TLR4 as a TP53 linked prognostic factor for breast cancer (Curtis dataset)
| Factor | Hazard ratio | 95% CI | P value |
| TLR4 gene expression | |||
| High | Reference | ||
| Low | 1.21 | 0.99–1.47 | 0.05’ |
| ER status | |||
| Positive | Reference | ||
| Negative | 0.92 | 0.76–1.11 | 0.39 |
| PR status | |||
| Negative | Reference | ||
| Positive | 0.80 | 0.45–1.85 | 0.008** |
| HER2 status | |||
| Negative | Reference | ||
| Positive | 1.39 | 1.13–1.70 | 0.002** |
| Tumor grade | |||
| Grade I | Reference | ||
| Grade II | 1.20 | 0.84–1.70 | 0.31 |
| Grade III | 1.31 | 0.92–1.87 | 0.13 |
| Tumor stage | |||
| Stage NA | Reference | ||
| Stage 0 | 1.02 | 0.82–1.27 | 0.84 |
| Stage I | 0.70 | 0.52–0.93 | 0.02* |
| Stage II | 0.96 | 0.77–1.19 | 0.70 |
| Stage III | 1.72 | 1.22–2.41 | 0.002** |
| Stage IV | 4.40 | 2.02–9.59 | <0.001*** |
| Tumor size, mm | 1.00 | 1.00–1.01 | <0.001*** |
| TP53 status | |||
| Wild type | Reference | ||
| Mutant | 1.53 | 1.13–2.07 | 0.005** |
P values generated by Cox regression analysis for proportional hazards. Asterisks denote significance; apostrophe denotes trend; ‘P < 0.10; *P < 0.05; **P < 0.01; ***P < 0.001.
TLR4 Inhibits Growth in TP53 Wild-Type Breast Cancer Cells but Promotes Growth in TP53 Mutant Breast Cancer Cells.
To determine whether TLR4 is a TP53 dependent tumor suppressor, we studied the effect of loss of TLR4 (by both knockdown and pharmacologic inhibition) on breast cancer cell growth using a panel of TP53 mutant (BT20, MDA-MB-231, MDA-MB-361, and T47D) and TP53 wild-type (MCF7 and ZR75) breast cancer cell lines. We used pooled siRNA to knock down TLR4 expression (reduced RNA and protein expression levels confirmed in Fig. S2 A–C), and then tested the effect of TLR4 inhibition on in vitro growth (Fig. 2A). We found that TLR4 knockdown inhibited the growth of TP53 mutant breast cancer cell lines irrespective of ER status (Fig. 2A). In contrast, TLR4 suppression in TP53 wild-type ER-positive breast cancer cell lines dramatically promoted growth (Fig. 2A). These results suggest that cooperation between TLR4 and TP53 to inhibit cell growth can be lost in cells with either loss-of-function (MDA-MB-231, BT20) or dominant-negative (T47D) TP53 mutations. We independently confirmed these results using TAK242, a pharmacologic inhibitor of TLR4, in both TP53 mutant (Fig. 2B) and TP53 wild-type (Fig. 2C) breast cancer cell lines. TAK242 treatment inhibited TP53 mutant cell growth but stimulated TP53 wild-type cell growth, as seen with knockdown of TLR4.
Fig. S2.

TLR4 is inhibited by siRNA against TLR4 and is activated by LPS* treatment in both TP53 mutant and TP53 wild-type breast cancer cells. TLR4 mRNA (C, D, and F) and protein (A, B, and E–I) levels were detected using quantitative RT-PCR and immunofluorescence (A, B, E, and G), and NFkB protein (H and I) levels were detected using both Western blot (H) and immunofluorescence (I) analyses in specified cell lines treated with siRNA against either luciferase (siLuc) or TLR4 (siTLR4) (A–C), or with ultrapure LPS* to activate TLR4 signaling (D–I). Three different siRNA oligonucleotides against TLR4 were used in combination. Accompanying growth curves are presented in Fig. 2 A, D, and E. The Student t test generated all P values. Columns represent the mean, and error bars denote SD. (Scale bars: 20 μm.)
Fig. 2.

TLR4 inhibits growth of TP53 wild-type, and promotes growth of TP53 mutant, breast cancer cells. (A) Growth curves of TP53 mutant ER-negative (Left), TP53 mutant ER-positive (Middle), and TP53 wild-type ER-positive (Right) breast cancer cells treated with either siRNA against TLR4 (red) or control siRNA against luciferase (black). Validation of siRNA treatment is presented in Fig. S2 A–C. (B and C) Dose-dependence curves of TP53 mutant (B) and TP53 wild-type (C) breast cancer cells treated with TAK242, a TLR4 inhibitor, at specified concentrations. Growth was determined on day 3 and day 5 posttreatment and represented as growth relative to control (DMSO-treated) cells. (D and E) Growth curves of TP53 mutant (D) and TP53 wild-type (E) breast cancer cells treated with a TLR4 agonist, LPS* (10 ng/mL; blue) or vehicle (black). Validation of TLR4 activation by LPS* treatment is presented in Fig. S2 D–I. The Student t test generated the P values. Error bars represent SD.
We next attempted to activate TLR4 signaling using a TLR4 agonist, a mutant form of LPS (LPS*). We found that LPS* treatment up-regulated TLR4 RNA and protein expression (Fig. S2 D–G), and induced NFkB (Fig. S2 H and I), indicating activation of TLR4 signaling. Using LPS* treatment to activate TLR4 signaling, we performed growth assays, and found that LPS* treatment significantly promoted the growth of TP53 mutant breast cancer cells (Fig. 2D) while inhibiting the growth of TP53 wild-type cells (Fig. 2E).
Using three independent approaches, we determined that the effect of LPS* on breast cancer cell growth is mediated by TLR4. First, Western blot analysis showed that LPS* treatment of MCF7 cells induced NFkB only in the presence of TLR4 (Fig. S2H). In a second analysis, preincubation with a monoclonal neutralizing antibody against TLR4 disrupted LPS*-mediated growth promotion of TP53 mutant cells and LPS*-induced growth inhibition of TP53 wild-type cells (Fig. 3A). Finally, siRNA-mediated TLR4 inhibition before treatment with LPS* abrogated the growth effect of LPS* on both TP53 mutant and wild-type cells (Fig. 3B). These data suggest that activation of TLR4 inhibits TP53 wild-type breast cancer cell growth while promoting the growth of TP53 mutant cells.
Fig. 3.

TLR4 inhibits proliferation of TP53 wild-type, and promotes proliferation of TP53 mutant, breast tumor cells. (A) Fold change in growth (day 3) of TP53 mutant (Left) and TP53 wild-type (Right) breast cancer cells treated with control antibody either against IgG (αIgG) or against TLR4 (αTLR4) and subsequently treated with either vehicle or LPS* to activate TLR4 signaling. (B) Fold change in growth of TP53 mutant (Left) and TP53 wild-type (Right) breast cancer cells treated with either siRNA against luciferase as control (siLuc) or against TLR4 (siTLR4) and subsequently treated with vehicle or LPS* to activate TLR4 signaling. (C) Percentage of cells in mitosis as assayed by immunofluorescence for pH3 in TP53 mutant (Left) and TP53 wild-type (Right) cells treated with the TLR4 inhibitor (TAK242; red), the TLR4 agonist (LPS*, blue), or vehicle controls. (D) Cell count at 48 h after vehicle/LPS* treatment of cells pretreated with siRNA targeted to luciferase (siLuc, control) or to TP53 (siTP53). Supporting data are presented in Figs. S3 and S4. The Student t test generated all P values. Columns represent the mean, and error bars denote SD.
TLR4 Inhibits Breast Cancer Cell Proliferation in a TP53-Dependent Manner.
To understand the mechanism by which TLR4 regulates breast cancer cell growth, we assayed the effect of TLR4 on apoptosis and proliferation. We found no effect of TLR4 perturbation, either by siRNA-mediated inhibition (Fig. S3A) or LPS*-induced activation (Fig. S3B) on apoptosis in TP53 wild-type cells; however, activation or suppression of TLR4 signaling differentially affected proliferation, depending on TP53 mutation status. In TP53 mutant breast cancer cells, pharmacologic TLR4 inhibition using the inhibitor TAK242 decreased phosphohistone H3 (pH3) positivity, which is consistent with reduced mitotic activity and reduced cell growth, whereas TLR4 activation using LPS* promoted pH3 positivity (Fig. 3C), which is similarly consistent with increased growth. Conversely, pharmacologic TLR4 inhibition with TAK242 in TP53 wild-type cells induced a twofold increase in pH3 positivity (concomitant with increased cell growth), whereas TLR4 activation with LPS* resulted in a 50% decrease in pH3-positive cells (Fig. 3C), in accordance with the observed inhibition in growth. As an independent verification, cell cycle analyses also detected significant accumulation of TP53 wild-type breast cancer cells, but not TP53 mutant cells, in G0/G1 within 48 h of TLR4 activation, along with a concomitant decrease in the G2/M population (Fig. S3C). Finally, after siRNA knockdown of TLR4 in TP53 wild-type cells, LPS* treatment no longer could induce this proliferative block (Fig. S3D).
Fig. S3.
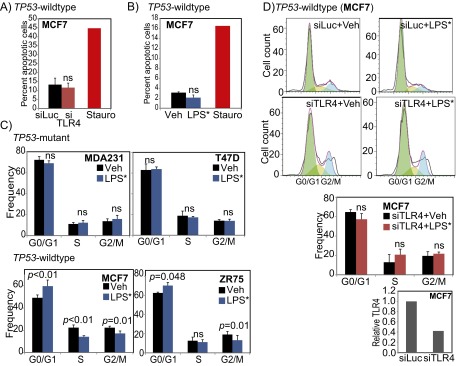
TLR4 alters TP53 wild-type breast cancer cell cycle. (A and B) Annexin V analysis for apoptosis after siRNA mediated inhibition of TLR4 (A), and LPS* treatment (B) of TP53 wild-type breast tumor cells with staurosporine-treated cells (Stauro) serving as a positive control. (C and D) Cell cycle analysis of TP53 mutant and TP53 wild-type (C) breast cancer cells along with representative figures and accompanying quantification from cell cycle analyses of MCF7 cells treated with siRNA against luciferase (siLuc; control) or against TLR4 (siTLR4; validated by qRT-PCR), and subsequently treated with vehicle (Veh) or EB-LPS (LPS*) (D). Supports the data presented in Fig. 3C. The Student t test generated all P values. Columns represent the mean, and error bars denote SD.
To directly test whether TP53 is required for the growth-inhibitory effect of TLR4, we knocked down TP53 in TP53 wild-type breast cancer cells and then treated them with LPS* (TP53 inhibition confirmed in Fig. S4). Treatment of control TP53 wild-type cells with LPS* significantly inhibited growth by day 2; however, after TP53 knockdown, LPS* treatment no longer inhibited growth (Fig. 3D). Indeed, LPS* treatment subsequent to TP53 inhibition promoted growth, similar to its effect on TP53 mutant breast tumor cells. These data indicate that TLR4 and TP53 together mediate growth, with TLR4 promoting proliferation in the absence of TP53 activity and inhibiting growth in the presence of wild-type TP53.
Fig. S4.
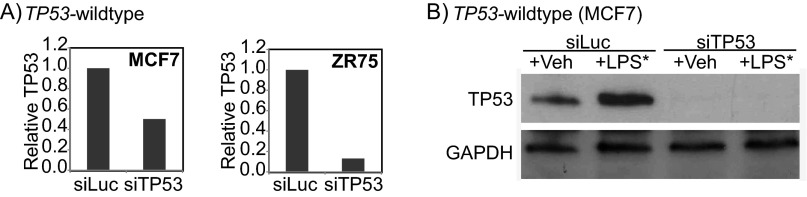
Validation of siRNA knockdown of TP53. Validation of siRNA treatment against TP53 using quantitative RT-PCR at the RNA level (A) and Western blot analysis at the protein level (B). Accompanies the data presented in Fig. 3D.
TLR4 Regulates IFN-γ Secretion by Breast Cancer Cells.
Based on our results and previous studies on the role of TP53 in immune cells (11), we hypothesized that the dichotomous effect of TLR4 on breast cancer cell growth is driven by TP53-dependent differential secretion of cytokines by tumor cells. To test this hypothesis, we first investigated whether the growth-inhibitory effect of TLR4 is extracellular (e.g., via factors secreted by cancer cells) or intracellular (via direct activation of the cell cycle or proliferation pathways within the cell). We first treated TP53 wild-type (MCF7) and TP53 mutant (MDA-MB-231) breast cancer cells with conditioned media from LPS*-treated MDA-MB-231 cells. If TLR4 induces proliferation in TP53 mutant breast cancer cells through extracellular factors, then media from LPS*-treated TP53 mutant breast cancer cells would be expected to promote the growth of both TP53 mutant and TP53 wild-type cells. However, if TLR4-mediated cell proliferation occurs through intracellular signaling, then conditioned media from TP53 mutant cells treated with LPS* should promote growth of only TP53 mutant cells, and not of TP53 wild-type cells. Our results show that conditioned media from LPS*-treated TP53 mutant MDA-MB-231 cells promoted the growth of both TP53 mutant (MDA-MB-231) and TP53 wild-type (MCF7) cells (Fig. S5 A and B), indicating that TLR4-induced cell proliferation in TP53 mutant breast cancer cells is mediated through extracellular factors. We then repeated the experiment using conditioned media from LPS*-treated MCF7 cells. Again, we observed that conditioned media from these LPS*-treated TP53 wild-type breast cancer cells significantly inhibited the growth of both TP53 mutant and TP53 wild-type breast cancer cells (Fig. S5 C and D). These data indicate that TLR4 activation regulates the profile of factors secreted by a breast cancer cell, and that those factors secreted by TP53 wild-type cells are able to inhibit the proliferation of any adjacent cell irrespective of its TP53 mutational status.
Fig. S5.

Effect of TLR4 activation on breast cancer cell growth is extracellular. Bar graphs representing cell counts of TP53 mutant (MD-MB-231) (A and C) and TP53 wild-type (MCF7) (B and D) breast cancer cells treated with conditioned media from TP53 mutant (A and B) and TP53 wild-type (C and D) breast cancer cells with or without exposure to EB-LPS (LPS*) for 48 h. Supports the data presented in Fig. 4 A and B. The Student t test generated all P values. Columns represent the mean, and error bars denote SD.
To identify the factors underlying this phenotype, we used an array to detect secreted cytokines in conditioned media from both TP53 wild-type (MCF7) and TP53 mutant (MDA-MB-231) breast cancer cells treated with (i) either vehicle or LPS* and (ii) either siRNA against luciferase (control) or TLR4. We observed a striking difference in baseline chemokine secretion between the two cell lines. TP53 mutant ER-negative cells produced significantly higher levels of cytokines overall (Fig. S6), in accordance with our previous findings (12). Most of the cytokines secreted by MDA-MB-231 were proinflammatory and known to promote tumor cell growth. TLR4 inhibition in MDA-MB-231 cells decreased the secretion of several progrowth cytokines, including IL-6, IL-8 (7), and chemokine (C-X-C motif) ligand 1 (CXCL1), suggesting one mechanism by which cell growth is inhibited after siRNA-mediated knockdown of TLR4. Simultaneously, the low baseline level secretion of the growth-inhibitory cytokine CD154 (13) by MDA-MB-231 cells was increased on TLR4 knockdown but further decreased after LPS* treatment (Fig. S6), suggesting additional means by which TLR4 signaling can mediate TP53 mutant breast cancer cell growth. Collectively, these data suggest that both the growth inhibition associated with TLR4 suppression and the growth promotion after LPS* treatment of TP53 mutant breast cancer cells are regulated by a delicate balance of progrowth and antigrowth cytokines secreted into the extracellular microenvironment.
Fig. S6.

TLR4 regulates breast tumor cell cytokine secretion. Protein expression levels of all cytokines included in the cytokine array, normalized by subtracting background measured from negative control spots, detected in conditioned media from specified breast cancer cells treated with siRNA against luciferase (siLuc, control) or TLR4 (siTLR4), followed by treatment with vehicle or LPS*. Selected subsets of cytokines are presented in Fig. 4 A and B, and fold changes are specified in Table S3.
To determine the specific cytokines responsible for TLR4/TP53-mediated breast cancer cell growth inhibition, we focused on the cytokines differentially regulated by TLR4 signaling in TP53 wild-type breast cancer cells, using three approaches as outlined in Table S3. Because TLR4 inhibition in TP53 wild-type breast cancer cells stimulates growth, we first identified the subset of cytokines whose secretion decreased by ≤0.5-fold or increased by ≥2-fold in TP53 wild-type (MCF7) cells treated with siRNA against TLR4. Similarly, we identified a second subset of cytokines whose secretion decreased by ≤0.5-fold or increased by ≥2-fold in TP53 wild-type (MCF7) cells treated with LPS* to activate TLR4. From these two subsets, we then selected those cytokines that changed reciprocally in response to TLR4 inhibition and activation, i.e., exhibited ≤0.5-fold decrease after TLR4 knockdown and α ≥2-fold increase with LPS* treatment or vice versa. To ensure that we limited further analysis to only those cytokines specifically regulated by TLR4, we excluded all cytokines whose secretion was similarly altered by LPS* treatment in both the presence and absence of TLR4 in MCF7 cells. Those cytokines that remained after these three screens constituted our final working set of cytokines for further investigation.
Table S3.
Fold change in cytokines secreted by MCF7 cells
| Cytokine | siTLR4/siLuc | Vehicle/LPS* | siTLR4+vehicle/siTLR4+LPS* |
| CD154 | ≤0.00 | NA | >316 |
| GM-CSF | ≤0.00 | NA | >370 |
| CCL4 | ≤0.00 | ≤0.00 | NA |
| IL-1RA | ≤0.00 | 0.15 | >355 |
| CCL1 | ≤0.00 | 6.72 | >43 |
| IL-17E | 0.09 | 0.15 | 7.89 |
| G-CSF | 0.13 | NA | 3.79 |
| sTREM-1 | 0.17 | ≤0.00 | ≤0.00 |
| IL-23 | 0.20 | NA | 0.25 |
| IFN-γ | 0.24 | 2.14 | NA |
| CXCL1 | 0.27 | NA | 25.77 |
| IL-12p70 | 2.13 | ≤0.00 | ≤0.00 |
| MCP-1 | 2.32 | 3.16 | NA |
| PAI-1 | 3.53 | 5.40 | NA |
| TNF-α | 5.24 | NA | 0.42 |
| CXCL12 | 6.24 | 41.04 | NA |
| IL-4 | 30.79 | ≤0.00 | ≤0.00 |
| CD54 | >8 | >355 | 30.25 |
| IL-13 | >24 | >922 | 48.10 |
| IL-1B | >29 | NA | ≤0.00 |
| IL-6 | >44 | NA | ≤0.00 |
| IL-10 | >44 | NA | ≤0.00 |
| CXCL11 | >90 | >335 | NA |
| IL-5 | >111 | NA | ≤0.00 |
| IL-2 | >119 | NA | ≤0.00 |
| CCL5 | >173 | >419 | 6.49 |
| IL-32A | >185 | NA | ≤0.00 |
| IL-27 | NA | NA | ≤0.00 |
| IL-17 | NA | ≤0.00 | ≤0.00 |
| CXCL10 | NA | 0.17 | NA |
| CCL3 | NA | 2.10 | NA |
| IL-8 | NA | >419 | >313 |
| IL-1A | NA | >730 | NA |
Cytokines in italic type change reciprocally between TLR4 knockdown and activation; cytokines in bold italic type change reciprocally and respond specifically to TLR4 activation. NA, not applicable.
Of the cytokines that we identified through this screening process, IFN-γ was the sole cytokine exhibiting significant reciprocal changes in expression with TLR4 inhibition and activation, i.e., a ≤0.5-fold decrease after TLR4 inhibition and a ≥2-fold increase in secretion after LPS* treatment of TP53 wild-type MCF7 cells (Fig. 4A). This suggests that IFN-γ is the best candidate for mediating the observed growth phenotype; however, the other cytokines identified in our screen may well contribute to the growth phenotype individually or in combination.
Fig. 4.

TLR4 activation increases IFN-γ secretion from TP53 wild-type breast tumor cells, but not from TP53 mutant cells. (A) Venn diagram representing subsets of cytokines from TP53 wild-type breast cancer cells (Fig. S6 presents results of complete array in TP53 wild-type and TP53 mutant cells) whose secretion changes by ≥2-fold (pink) or ≤0.5-fold (blue) in culture supernatant of cells treated with siRNA against TLR4 vs. luciferase or treated with LPS* vs. vehicle. The black arrow indicates cytokine that is reciprocally altered by siTLR4 and LPS* treatment. Fold change for each cytokine is presented in Table S3. (B and C) Changes in secreted IFN-γ from culture supernatant of TP53 wild-type and TP53 mutant breast cancer cells treated with LPS* (B), as well as from TP53 wild-type breast cancer cells treated with siRNA against either TLR4 or TP53 and subsequently with LPS* (C) as assayed by ELISA. (D and E) Plots of means depicting gene expression level of IFN-γ in TLR4-high vs. TLR4-low TP53 mutant and TP53 wild-type breast tumors in the TCGA (D) and Curtis (E) datasets. Error bars represent SEM. Associated data are presented in Fig. S7A. The Student t test generated all P values. Columns represent the mean, and error bars denote SD, except where noted otherwise.
IFN-γ is known to inhibit growth of breast cancer cells by inducing a p21-mediated G0/G1 arrest (14), similar to the phenotype reported here in TP53 wild-type breast cancer cells (Fig. S3 C and D). Using ELISA, we confirmed that LPS* treatment induced a <2-fold increase in IFN-γ secretion in the extracellular microenvironment of TP53 wild-type breast cancer cells (MCF7, ZR75) but not in that of TP53 mutant breast cancer cells (MDA-MB-231, T47D) (Fig. 4B). Furthermore, in the absence of either TLR4 or TP53, LPS* treatment could not increase IFN-γ secretion by TP53 wild-type breast cancer cells (Fig. 4C). To test whether IFN-γ in human tumors is involved in mediating TLR4-TP53–orchestrated growth, we assessed correlations between TLR4 and IFN-γ gene expression in both the Curtis and TCGA datasets. In TLR4-high TP53 wild-type tumors, we found a significant increase in IFN-γ gene expression relative to TLR4-low tumors, but not in TP53 mutant tumors (Fig. 4 D and E).
We next tested whether specific types of TP53 mutation are associated with IFN-γ expression in ER-positive breast tumors. Using TCGA data, we examined the level of IFN-γ RNA expression in ER-positive TLR4-low and TLR4-high breast tumors with TP53 loss-of-function, gain-of-function, or dominant-negative mutations (Fig. S7A). This analysis showed that in TLR4-low tumors, IFN-γ expression is low for all TP53 mutant tumors, whereas in TLR4-high tumors, IFN-γ expression is significantly greater in tumors with TP53 gain-of-function mutations, but not in tumors with TP53 loss-of-function or dominant-negative mutations. A larger sample size is needed to understand the true nature of the IFN-γ–TLR4 association in the context of the various classes of TP53 mutations.
Fig. S7.

TLR4 and TP53 coregulate p21 induction. (A C, and D) Plot of means depicting gene expression changes in IFN-γ between tumors with gain-of-function, loss-of-function, and dominant negative TP53 mutations (A), and between p27 gene expression changes in TLR4-low and TLR4-high tumors from Curtis (C) and TCGA (D) datasets. Error bars represent SEM. Student’s t test determined P values. (B) Induction of p21 RNA detected by quantitative RT-PCR of lysate from TP53 wild-type breast cancer cells (ZR75) treated with siRNA against luciferase (siLuc, control) or TP53 (siTP53), with the subsequent addition of either vehicle (Veh) or EB-LPS (LPS*) to the media for 24 h. Supports data presented in Figs. 4 C and D and 5B. Student’s t test generated P values. Columns represent the mean and error bars denote SD.
Next, we confirmed that TLR4 activation in TP53 wild-type breast cancer cells preferentially induces nuclear p21, a known downstream effector of IFN-γ signaling and mediator of growth inhibition (Fig. 5A). LPS* treatment also increased mRNA levels of p21 in TP53 wild-type breast cancer cells (MCF7, Fig. 5B; ZR75, Fig. S7B). To determine the coordinated roles of TP53 and TLR4 in regulating p21 levels, we used siRNA knockdown against TP53 and TLR4 individually in TP53 wild-type breast cancer cells. Our results show that LPS*-induced activation of p21 is dependent on both TP53 (MCF7: Fig. 5C; ZR75: Fig. S7B) and TLR4 (Fig. 5D). We also confirmed this effect at the protein level (Fig. 5E). Corroborating these data, we also found that TLR4-high TP53 wild-type tumors have more p21, as measured by gene expression (Curtis: P = 0.01; TCGA: P = 0.01), compared with TLR4-low tumors in patient tumor datasets (Fig. 5 F and G). The specificity of this correlation is indicated by the lack of correlation between TLR4 and p27 gene expression in both datasets assayed (Fig. S7 C and D). These data indicate a role for both TLR4 and TP53 signaling in regulating IFN-γ secretion and p21 regulation by TP53 wild-type cells.
Fig. 5.

LPS*-induced p21 activation requires both TLR4 and TP53. (A and B) Representative photomicrographs and accompanying quantification of nuclear localized p21 in TP53 wild-type cells (A), as well as quantification of RNA levels (B) 24 h after LPS* treatment. (C and D) Relative levels of p21 mRNA in TP53 wild-type breast tumor cells treated with siRNA against either luciferase as control, against TP53 (C) or against TLR4 (D) and subsequently treated with either vehicle or LPS*. Supporting data are presented in Fig. S7B. (E) Western blot confirming p21 induction after LPS* treatment in control TP53 wild-type cells but not in cells lacking either TP53 or TLR4. (F and G) Plots of means depicting alterations in p21 gene expression changes in TLR4-low and TLR4-high tumors from the Curtis (F) and TCGA (G) datasets. Error bars depict SEM. Accompanying data are provided in Fig. S7 C and D. The Student t test generated all P values. Columns represent the mean, and error bars represent the SD. (Scale bar: 20 µm.)
IFN-γ Secretion by TP53 Wild-Type Breast Cancer Cells Is Both Necessary and Sufficient to Mediate Growth Inhibition by TLR4.
We next investigated whether IFN-γ is both necessary and sufficient to mediate the effect of TLR4 on TP53 wild-type breast cancer cell growth. To ascertain necessity, we neutralized IFN-γ in conditioned media from LPS*-treated TP53 wild-type cells using an anti–IFN-γ antibody, and then assessed the effect of this neutralized media on cell growth. We found that conditioned media from LPS*-treated TP53 wild-type breast cancer cells significantly inhibited growth by day 2, as expected, but preincubation of the conditioned media with the IFN-γ neutralization antibody negated any growth-inhibitory effect (Fig. 6 A and C). These data indicate that TLR4 loses its growth-inhibitory effect on TP53 wild-type breast cancer cells in the absence of secreted IFN-γ, even when all other secreted cytokines remain empirically unperturbed in the microenvironment.
Fig. 6.

IFN-γ is both necessary and sufficient for mediating growth inhibition of TP53 wild-type breast cancer cells by TLR4. (A and C) Bar graphs representing the fold change in growth of TP53 wild-type breast cancer cells when exposed to conditioned media from isogenic cells pretreated with LPS* for 48 h and incubated with antibody against either IgG or a neutralizing antibody against IFN-γ. (B and D) Bar graphs representing the fold change in growth of TP53 wild-type breast cancer cells treated sequentially with TAK242 (10 nM) and recombinant IFN-γ. The Student t test generated all P values. Columns represent the mean, and error bars denote SD.
We next pharmacologically inhibited TLR4 by treating TP53 wild-type breast cancer cells with TAK242, and then added recombinant IFN-γ to the culture media. Our results show that inhibition of TLR4 in TP53 wild-type cells promotes growth by day 2; however, subsequent treatment with recombinant IFN-γ blocks TAK242-induced growth promotion (Fig. 6 B and D). This result suggests that in the absence of all other secreted cytokines, the addition of IFN-γ alone is sufficient to replicate the growth inhibition induced by TLR4 activation. These data indicate that secreted IFN-γ is both necessary and sufficient to mediate the growth-inhibitory effect of TLR4 on TP53 wild-type breast cancer cells.
To test whether this TP53-dependent role for TLR4 in regulating growth is restricted to breast cancer, we assessed for a correlation between TP53 mutational status and TLR4 alterations (including mutations, genomic alterations, and gene expression changes) across multiple cancer types using publically available TCGA datasets. This analysis identified an inverse correlation between the incidence of TLR4 and TP53 alterations across all cancer types included in the analysis (Fig. 7A). Cancer sites more prone to TP53 loss, such as ovarian serous, head and neck, and bladder cancers, are more likely to retain TLR4, in keeping with our hypothesis that TLR4 retention provides a growth advantage to tumor cells in the absence of TP53 function. On the other hand, tumors that tend to maintain wild-type TP53 have a higher incidence of TLR4 alterations, indicating that these tumors cells tolerate, and likely even select for, the loss of TLR4. Among lung cancers alone, lung adenocarcinomas have a higher incidence of wild-type TP53 than lung squamous carcinomas, and also exhibit a greater propensity for TLR4 loss (Fig. 7A). These observations indicate that there likely exists a complex interaction between TLR4 and TP53 across many different histological tumor types.
Fig. 7.

IFN-γ, TLR4, and TP53 coordinately regulate growth of breast tumor cells. (A) Regression analysis indicates inverse correlation between alterations (mutations, genomic loss, and gene expression change) in TLR4 and TP53 (mutations, genomic loss) across multiple cancer types. R2 values were generated using a generalized linear model. (B) Working model. (Left) In TP53 wild-type cells, TLR4-mediated inhibition of cell growth occurs through (1) TLR4 activation by ligands; (2) TLR4-mediated induction of IFN-γ secretion; (3) autocrine/juxtacrine activation of IFN-γgamma signaling; (4) IFN-γ/TP53 coordinated induction of p21; (5) formation of p21-cyclin/CDK complexes; and (6) growth inhibition. (Right) In TP53 mutant cells, TLR4-mediated up-regulation of cell growth occurs through (1) TLR4 activation by ligands; (2) TLR4-mediated induction of proinflammatory cytokines, including CXCL1; (3) autocrine growth promotion by cytokine; and (4) TLR4-mediated inhibition of anti-growth cytokines, such as CD154.
Discussion
The results presented herein suggest that TLR4 regulates breast cancer cell growth in a TP53-dependent manner. To our knowledge, this constitutes the first report of TLR4 activation of TP53 wild-type breast cancer cells causing secretion of IFN-γ into the extracellular microenvironment, resulting in activation of p21 and subsequent suppression of tumor cell proliferation (Fig. 7B). Conversely, in TP53 mutant breast cancer cells, TLR4 activation alters the balance of progrowth and antigrowth cytokines in the extracellular microenvironment, ultimately resulting in increased proliferation and growth (Fig. 7B). These data indicate a complex role for TLR4 in driving breast cancer in the context of TP53 mutational status, and provide important insight into the effect of TLR4 activation in tumor cells on both growth and modulation of the cytokine milieu in the microenvironment. This has profound implications for the proposed use of TLR4 as a druggable target in breast cancer and in other cancers, and suggests that only patients with TP53 mutant tumors can benefit from anti-TLR4 therapy. Furthermore, our results indicate that patients with TP53 wild-type tumors should not be treated with anti-TLR4 therapy, because such treatment could promote, rather than suppress, tumor growth. Previous reports have suggested that TLR4 has a systemic role as a tumor suppressor in mice (15), potentially through the immune system. Therefore, if anti-TLR4 therapy is to be used in the clinic, it will be necessary to consider the effects on the immune system in addition to direct effects on the tumor. Future studies of TLR4 antagonists will need to assess both direct and systemic effects to develop these drugs for the treatment of TP53 mutant tumors.
A previous report identified TLR4 as promoting ER-negative, TP53 mutant MDA-MB-231 breast cancer cell growth (6). That study also found that TLR4 promotes cancer cell proliferation by increasing secretion of the proinflammatory cytokines IL-6 and IL-8 (6), supporting our results reported herein. However, that study focused on a single cell line and used a candidate approach to identify IL-6 and IL-8, which might not be the principal cytokines mediating proliferation regulation by TLR4. Our present study confirms a decrease in the secretion of both these cytokines after TLR4 knockdown in MDA-MB-231 cells, but also identifies large decreases in the secretion of other progrowth cytokines, including CXCL1 (16), as well as a dramatic increase in antigrowth cytokine CD154 (13). Most importantly, the previous study did not uncover the critical role of TP53 mutational status in regulating the observed growth phenotype.
In another previous study, Yang et al. (17) reported that treatment of MDA-MB-231 and MCF7 cells with LPS promoted invasion of both cell lines. Those results appear to contradict our findings, which demonstrate a growth-suppressive role for TLR4 in TP53 wild-type MCF7 cells. It is possible that TLR4 signaling in TP53 wild-type cells suppresses tumor growth but also may stimulate invasion and metastasis of the tumor cells; however, it is important to note that Yang et al. used reagents that can activate several TLRs in their experiments, and that in both of the large-scale human datasets analyzed in this work, TLR4 is associated with improved survival of patients with TP53 wild-type breast cancer in a stage-independent manner. These results suggest that in TP53 wild-type tumors, high TLR4 is associated with a good outcome. Such results do not support the hypothesis that high TLR4 promotes metastasis.
Other studies have identified TLR4 as a promoter of chemotherapeutic resistance in MDA-MB-231 cells (7, 8). Those studies used HCC1806, another TP53 mutant, ER-negative breast cancer cell line with very low baseline levels of TLR4, as a negative control. Rajput et al. (8) reported that TLR4 knockdown in MDA-MB-231 cells sensitizes them to chemotherapy, whereas overexpression of TLR4 in HCC1806 cells renders them resistant to chemotherapy. That group has since published a follow-up study demonstrating a role for TLR4 in promoting migration on exposure to cytotoxic therapy (8). Although both studies demonstrate an important role for TLR4 as an oncogene in breast cancer, our identification of a dual role for TLR4 based on TP53 function provides important context for interpreting their results. Previous studies have shown that in human breast cancer samples, wild-type TP53 is associated with chemotherapeutic sensitivity (18). Other studies have reported that specific TP53 mutations or variants are associated with chemotherapeutic resistance (19–21). In addition, a role for TP53 in resistance to cisplatin treatment has been identified in ovarian cancer (22). Therefore, our identification of the TLR4-TP53 nexus in breast cancer suggests an important avenue for further exploration of the mechanisms involved in resistance to cytotoxic therapies.
The results presented herein validate a role for TLR4 in tumor growth regulation independent of its immune activity, which has been a primary focus of previous TLR4-based studies. Our data also indicate that the TP53 mutational context of a breast tumor epithelial cell can significantly affect the tumor microenvironment by regulating the extracellular cytokine secretions of tumor cells, and have a profound effect on response to targeted therapy. The tumor growth and immunomodulatory roles of TLR4 may be integrated, because cytokines in the tumor microenvironment play an important role in attracting or evading antitumor immunity (23). Therefore, the roles of TLR4 in regulating tumor growth and cytokine secretion may act together in promoting the growth of epithelial TP53 mutant tumor cells and in suppressing any antitumor immune response.
In this context, it is striking that the TP53-dependent differential role of TLR4 appears to occur in multiple cancer types in addition to breast cancer. The global nature of this interaction is supported by the observation that an oncogenic role for TLR4 in tumor cells has been identified primarily in cancer types known to inactivate TP53 at high frequencies, such as ovarian (24), bladder (25), and head and neck (26) cancers. Further investigation is warranted in other cancer sites to define the association between TLR4 and TP53 and its effect on tumor biology and behavior.
Materials and Methods
Cell Lines, siRNA Transfection, and Growth Assays.
Cell lines were obtained from American Type Culture Collection and maintained and validated as reported previously (12). TP53 mutant cell lines have the following TP53 mutations: K132Q (BT-20), L194F (T47D), E166* (MDA-MB-361), and R280K (MDA-MB-231). Three TLR4 and TP53 siRNA oligonucleotides each were purchased from Sigma-Aldrich. Transfection and validation were conducted as described previously (12). Growth assays were conducted in triplicate and repeated independently as described previously using Trypan blue to identify live, viable cells and manual quantification with hemocytometers (27).
Inhibitors, Neutralizing Antibodies, Recombinant Proteins, and Agonists.
TAK242 (InvivoGen) was dissolved in DMSO and used at specified concentrations, with concomitant amounts of DMSO as vehicle control. Recombinant IFN-γ (BD Biosciences) was dissolved in serum-free media and used at a concentration of 15 pg/mL. Neutralizing antibody to IFN-γ (eBiosciences) was used at a concentration of 2.5 µg/mL. Neutralization was conducted through a 2-h incubation of conditioned media with either IFN-γ or rabbit polyclonal IgG antibody (Santa Cruz Biotechnology) at 37 °C. Neutralizing antibody to TLR4 (Imgenex) was preincubated with cells for 2 h at 10 ng/mL before subsequent treatment as specified. LPS (InvivoGen) was dissolved in 1× PBS and used as described previously (7).
Immunostaining and Microscopy.
Immunofluorescence was performed in accordance with the manufacturer’s instructions. Cells were washed in PBS, fixed in 4% PFA for 20 min at room temperature, blocked in 5% goat serum and 1% Triton X-100 in 1× PBS for 1 h at room temperature, incubated with primary antibody in 1% goat serum and 1% Triton X-100 in 1× PBS antibody diluent overnight at 4 °C, incubated with secondary antibody in diluent for 1 h at room temperature, and then mounted with DAPI-containing mounting media. Primary antibodies used included pH3 (Cell Signaling Technology; 1:500), p21 (Cell Signaling Technology; 1:200), NFkB (Cell Signaling Technology; 1:200), and TLR4 (Thermo Scientific; 1:200). Fluorescent images were captured with a Nikon microscope, quantified with ImageJ, and processed with Adobe Photoshop software.
Apoptosis and Cell Cycle Analyses.
Annexin V was used to assay apoptosis as described previously (12). Cell cycle analysis was conducted in triplicate using propidium iodide to stain cells, followed by cell sorting with a Gallios flow cytometer (Beckman Coulter) at 24 or 48 h posttreatment with specified reagents. Analysis of cell cycle was conducted using FlowJo software (Tree Star).
Cytokine Array, ELISA, and Western Blot Analysis.
ELISA and Western blot analysis were conducted as described previously (12, 28). Primary antibodies were incubated with the membrane overnight at 4 °C and included p21 (Cell Signaling Technology; 1:1,000), STAT1 (Cell Signaling Technology; 1:1,000), p53 (Santa Cruz Biotechnology; 1:500), p65 (Cell Signaling Technology; 1:1,000), and GAPDH (Sigma-Aldrich; 1:6,000). The cytokine array (R&D Systems) was used according to the manufacturer’s instructions. Supernatant for the ELISA and cytokine array experiments was collected 24 h posttreatment, and spun down for 5 min at 1,000 rpm. The supernatant was then stored at −20 °C, as required.
Statistical Analysis.
ANOVA or the Student t test was used for independent samples with normal distribution. Where distribution was not normal (assessed using Q-Q plots with the Wilk–Shapiro test of normality), either the Kruskal–Wallis or Wilcoxon rank-sum test was used. All experiments were conducted in triplicate, and each experiment was duplicated independently more than two times. Criteria for including datasets in the regression analysis were that the dataset comprised >100 samples, each TLR4 alteration in the dataset occurred at >1% frequency, and alterations in both TP53 and TLR4 occurred at >3% frequency. These criteria were formulated to ensure that results from each dataset were calculable within the range of sensitivity of the statistical test used. Regression analysis was performed using R and a generalized linear model. Databases used for human data mining were publically available resources: Oncomine, cBio, and COSMIC. Median cutoffs were used for gene expression stratification. All survival data were analyzed using Kaplan–Meier curves and log-rank tests. Proportional hazards were determined using Cox regression. All graphs and statistical analyses were generated in either Microsoft Excel or R.
Supplementary Material
Acknowledgments
We thank Michelle Savage for her technical editing of the manuscript and Dr. Matthew Bainbridge for his help with statistical analyses and creative input. This work was supported by Cancer Center Support Grant 5 P30 CA016672-38, a Norman Brinker Award for Research Excellence (to P.B.), a John Charles Cain Distinguished Chair Award (to P.B.), the Mariam Rogers Fund for Breast/Women’s Cancer Research (P.B.), The University of Texas M. D. Anderson Cancer Center Halliburton Fellowship in Cancer Prevention Fund (to S.H.), and Susan G. Komen Promise Grant KG081694 (to P.B.). P.B. currently serves as a Scientific Advisory Board member for Susan G. Komen for the Cure.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420811112/-/DCSupplemental.
References
- 1.Haricharan S, Bainbridge MN, Scheet P, Brown PH. Somatic mutation load of estrogen receptor-positive breast tumors predicts overall survival: An analysis of genome sequence data. Breast Cancer Res Treat. 2014;146(1):211–220. doi: 10.1007/s10549-014-2991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HM, et al. Crystal structure of the TLR4–MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130(5):906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Piao W, et al. Recruitment of TLR adapter TRIF to TLR4 signaling complex is mediated by the second helical region of TRIF TIR domain. Proc Natl Acad Sci USA. 2013;110(47):19036–19041. doi: 10.1073/pnas.1313575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin B, Sun T, Yu XH, Yang YX, Yeo AE. The effects of TLR activation on T-cell development and differentiation. Clin Dev Immunol. 2012;2012:836485. doi: 10.1155/2012/836485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.González-Reyes S, et al. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer. 2010;10:665. doi: 10.1186/1471-2407-10-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H, et al. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J Exp Clin Cancer Res. 2010;29:92. doi: 10.1186/1756-9966-29-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajput S, Volk-Draper LD, Ran S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther. 2013;12(8):1676–1687. doi: 10.1158/1535-7163.MCT-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volk-Draper L, et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014;74(19):5421–5434. doi: 10.1158/0008-5472.CAN-14-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang JM, et al. Atractylenolide-I sensitizes human ovarian cancer cells to paclitaxel by blocking activation of TLR4/MyD88-dependent pathway. Sci Rep. 2014;4:3840. doi: 10.1038/srep03840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang AC, Su QB, Wu FX, Zhang XL, Liu PS. Role of TLR4 for paclitaxel chemotherapy in human epithelial ovarian cancer cells. Eur J Clin Invest. 2009;39(2):157–164. doi: 10.1111/j.1365-2362.2008.02070.x. [DOI] [PubMed] [Google Scholar]
- 11.Dijsselbloem N, et al. A critical role for p53 in the control of NF-kappaB-dependent gene expression in TLR4-stimulated dendritic cells exposed to Genistein. J Immunol. 2007;178(8):5048–5057. doi: 10.4049/jimmunol.178.8.5048. [DOI] [PubMed] [Google Scholar]
- 12.Hartman ZC, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73(11):3470–3480. doi: 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong AW, et al. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin Cancer Res. 2001;7(3):691–703. [PubMed] [Google Scholar]
- 14.Gooch JL, Herrera RE, Yee D. The role of p21 in interferon gamma-mediated growth inhibition of human breast cancer cells. Cell Growth Differ. 2000;11(6):335–342. [PubMed] [Google Scholar]
- 15.Ahmed A, Wang JH, Redmond HP. Silencing of TLR4 increases tumor progression and lung metastasis in a murine model of breast cancer. Ann Surg Oncol. 2013;20(Suppl 3):S389–S396. doi: 10.1245/s10434-012-2595-9. [DOI] [PubMed] [Google Scholar]
- 16.Acharyya S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, et al. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS ONE. 2014;9(10):e109980. doi: 10.1371/journal.pone.0109980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertheau P, et al. Effect of mutated TP53 on response of advanced breast cancers to high-dose chemotherapy. Lancet. 2002;360(9336):852–854. doi: 10.1016/S0140-6736(02)09969-5. [DOI] [PubMed] [Google Scholar]
- 19.Bergamaschi D, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73 dependent apoptosis. Cancer Cell. 2003;3(4):387–402. doi: 10.1016/s1535-6108(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 20.Yang P, Du CW, Kwan M, Liang SX, Zhang GJ. The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci Rep. 2013;3:2246. doi: 10.1038/srep02246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aas T, et al. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996;2(7):811–814. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- 22.Reles A, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001;7(10):2984–2997. [PubMed] [Google Scholar]
- 23.Faget J, et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011;71(19):6143–6152. doi: 10.1158/0008-5472.CAN-11-0573. [DOI] [PubMed] [Google Scholar]
- 24.Wang AC, et al. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma in vitro. Oncol Lett. 2014;7(3):871–877. doi: 10.3892/ol.2013.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang YH, et al. Effect of TLR4 and B7-H1 on immune escape of urothelial bladder cancer and its clinical significance. Asian Pac J Cancer Prev. 2014;15(3):1321–1326. doi: 10.7314/apjcp.2014.15.3.1321. [DOI] [PubMed] [Google Scholar]
- 26.Ren G, et al. Rapamycin inhibits Toll-like receptor 4-induced pro-oncogenic function in head and neck squamous cell carcinoma. Oncol Rep. 2014;31(6):2804–2810. doi: 10.3892/or.2014.3134. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, et al. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009;69(23):8853–8861. doi: 10.1158/0008-5472.CAN-09-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, et al. The rexinoid bexarotene represses cyclin D1 transcription by inducing the DEC2 transcriptional repressor. Breast Cancer Res Treat. 2011;128(3):667–677. doi: 10.1007/s10549-010-1083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]