

Significance
Many theories regarding the causes of insulin resistance in skeletal muscle center on the ability of muscle to oxidize fat, with evidence supporting either decreased or increased fatty acid oxidation (FAO) as causal to insulin resistance. Inhibition of fatty acid transport into mitochondria specifically in mouse muscle results in a rather remarkable phenotype. Despite an accumulation of lipids in muscle, insulin sensitivity is maintained. The muscle responds to decreased FAO by adapting muscle metabolism to use other fuel sources, and by an increased reliance upon peroxisomal fat oxidation. There is also an increase in mitochondrial biogenesis. At the whole-body level, the mice seem to enter an energy conservation mode with reduced activity, energy expenditure, and resistance to diet-induced obesity.
Keywords: carnitine palmitoyltransferase, muscle, fatty acid, lipid, carbohydrate
Abstract
The correlations between intramyocellular lipid (IMCL), decreased fatty acid oxidation (FAO), and insulin resistance have led to the hypothesis that impaired FAO causes accumulation of lipotoxic intermediates that inhibit muscle insulin signaling. Using a skeletal muscle-specific carnitine palmitoyltransferase-1 KO model, we show that prolonged and severe mitochondrial FAO inhibition results in increased carbohydrate utilization, along with reduced physical activity; increased circulating nonesterified fatty acids; and increased IMCLs, diacylglycerols, and ceramides. Perhaps more importantly, inhibition of mitochondrial FAO also initiates a local, adaptive response in muscle that invokes mitochondrial biogenesis, compensatory peroxisomal fat oxidation, and amino acid catabolism. Loss of its major fuel source (lipid) induces an energy deprivation response in muscle coordinated by signaling through AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) to maintain energy supply for locomotion and survival. At the whole-body level, these adaptations result in resistance to obesity.
Considerable evidence suggests that when oversupply of dietary fat exceeds the storage capacity of adipose tissue, ectopic lipids accumulate in skeletal muscle, leading to “metabolic stress” that induces insulin resistance. One prevailing theory is that impaired skeletal muscle fatty acid oxidation (FAO) (1–4) leads to cytosolic accumulation of lipotoxic intermediates that are directly linked to defects in insulin signaling (5–11). Recent findings counter this premise because they have shown that models of insulin resistance consistently exhibit enhanced (not reduced) FAO, as demonstrated by elevated incomplete β-oxidation and accumulation of excess lipid-derived acylcarnitines (12, 13). Supporting evidence was obtained using a whole-body genetic approach to elevate levels of the endogenous carnitine palmitoyltransferase-1 (Cpt1) inhibitor, malonyl-CoA (12). These studies have led to the contrasting theory that lipid overload and incomplete FAO within the mitochondria accelerate the progression of insulin resistance (14). Faced with a plethora of studies supporting both hypotheses, a crucial question remains: “Is inhibition of mitochondrial FAO in skeletal muscle sufficient to initiate development of insulin resistance?”
Cpt1 is essential for long-chain acyl-CoA transport into the mitochondria, and lies at the nexus of both the lipotoxicity and the mitochondrial overload hypotheses. If decreased FAO is a root cause of lipotoxicity, then muscle-specific ablation of Cpt1b activity should lead to impaired FAO, intramyocellular lipid (IMCL) accumulation, and insulin resistance. In stark contrast, the mitochondrial overload hypothesis suggests that decreased Cpt1b activity would preserve insulin sensitivity by preventing unbalanced overfueling of β-oxidation.
Characterization of the rare genetic disorders of Cpt1 and Cpt2 has focused almost exclusively on the functional, rather than metabolic, implications of sustained decrements to FAO. However, dyslipidemia and insulin resistance have been observed (15, 16). There have been a handful of studies that have used pharmacological inhibition of Cpt1 to target insulin resistance. The results are inconsistent and, in some cases, completely contradictory due to species/model differences and methods of assessing insulin sensitivity (17–23). These studies also lack tissue specificity and are generally of short duration and/or initiated after development of insulin resistance/diabetes. The lack of tissue specificity is most problematic. Decreased hepatic gluconeogenesis as a result of liver Cpt1 inhibition may have effects on whole-body insulin response independent of muscle (18, 22). Furthermore, Cpt1 inhibition is associated with both cardiac hypertrophy and hepatic steatosis (24–26). There have been attempts to inhibit the muscle isoform, Cpt1b (23), specifically; however, long-term risks of cardiac hypertrophy and mortality (27) suggest that pharmacological strategies must target specifically skeletal muscle.
Two of the studies have directly examined the impact of Cpt1 inhibition on the link between decreased FAO, increased IMCL, and lipotoxicity-induced insulin resistance. Using the muscle-selective Cpt1b inhibitor, oxfenicine, Keung et al. (23) showed decreased diacylglycerol (DAG) and ceramide levels following short-term treatment of obese mice. In contrast, Timmers et al. (21) showed increased IMCL and DAG accumulation following 8 d of treatment with the nonselective global Cpt1 inhibitor, etomoxir. These studies revealed short-term benefits in treatment of insulin resistance but contradictory conclusions with respect to the role of lipotoxicity. In combination with the studies by Conti et al. (22) using the liver-specific Cpt1 inhibitor, teglicar, it is clearly important to understand the detailed mechanisms underlying the impact of tissue-specific and isoform-selective Cpt1 inhibition to develop therapeutic inhibitors with maximum metabolic benefits and minimal side effects.
On the whole, the impact of prolonged inhibition of fatty acid transport into skeletal muscle mitochondria has not been studied. To address this issue directly, we created mice with skeletal muscle-specific deficiency of Cpt1b (Cpt1bm−/−).
Results
To determine the effects of impaired skeletal muscle mitochondrial FAO, we targeted the muscle-specific isoform, Cpt1b. We crossed mice bearing floxed alleles of Cpt1b to Mlc1f-Cre transgenic mice for specific ablation of Cpt1b in skeletal muscle, but not cardiac tissue (28). Details of the Cpt1b targeting construct and crossing scheme to generate floxed mice are shown in Fig. S1. Cpt1bm−/− mice have normal levels of Cpt1b expression in the heart, but there is near-complete knockdown of both gene expression and Cpt activity in skeletal muscle (Fig. 1 A and B). Mitochondrial FAO in Cpt1bm−/− mice is decreased significantly to levels comparable to control mitochondria treated with the inhibitor etomoxir (Fig. 1C).
Fig. 1.
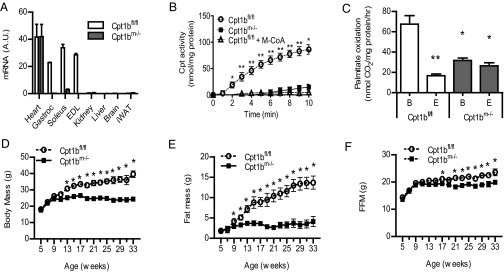
Mice with skeletal muscle-specific Cpt1b deficiency have impaired mitochondrial FAO in muscle and decreased whole-body adiposity. (A) Tissue-specific deletion in Cpt1bm−/− mice was confirmed by qRT-PCR analysis of Cpt1b mRNA (n = 5–7). Gastroc, gastrocnemius; iWAT; inguinal white adipose tissue. (B) Skeletal muscle effects were confirmed in isolated mitochondria by Cpt activity assay. Cpt1bm−/− mitochondria were identical to normal mitochondria treated with the Cpt1 inhibitor malonyl-CoA (M-CoA). (C) Impaired FAO was shown in isolated mitochondria using the [1-14C]palmitate-radiolabeled assay. B, basal conditions; E, treated with the Cpt1 inhibitor etomoxir. Ablation of Cpt1b decreases weight gain (D) and fat accumulation (E), with maintenance of lean mass (F) (n = 8–10 per group), in mice on a 25% fat chow diet. *P < 0.05 or **P < 0.01, genotype effect detected by ANOVA for multiple groups or by Student’s t test. Results shown are representative of multiple independent experiments, and are expressed as mean ± SEM.
Impairment of Mitochondrial FAO in Muscle Attenuates Development of Adiposity.
Cpt1bm−/− mice and control mice were fed a moderate fat (25% kcal) breeder chow diet. Cpt1bm−/− mice are indistinguishable from controls at weaning and continue to gain weight normally until 8–10 wk of age. Unexpectedly, Cpt1bm−/− mice reach maximum body weight by 12–13 wk of age (Fig. 1D). The difference in body mass is mainly attributed to changes in fat mass. Control mice continue to accrue fat, whereas the fat mass of Cpt1bm−/− mice is held at postpuberty levels (Fig. 1E). Importantly, lean mass is similarly maintained at young adult levels in Cpt1bm−/− mice, resulting in a slight difference from control littermates by 20 wk of age (Fig. 1F).
To determine if food intake was causing the differences in fat mass, food intake and body weight were measured over the period when the fat mass between the control and Cpt1bm−/− mice is diverging. Body weight and fat mass become significantly less in Cpt1bm−/− mice compared with control mice (Fig. 2 A and B) well before food intake becomes significantly less in Cpt1bm−/− mice compared with control mice (Fig. 2C). Fat-free mass does not become significantly less in Cpt1bm−/− mice compared with control mice until about 100 d of age (Fig. 2D). Overall, these small differences in daily food intake lead to a cumulative difference in food intake of almost −15 g in the Cpt1bm−/− mice over the 8-wk period (Fig. 2F), whereas body weight and fat mass were about 3 g less in Cpt1bm−/− mice compared with control mice over the 8-wk period (Fig. 2E).
Fig. 2.
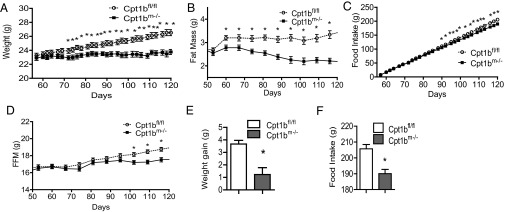
Cumulative food intake is altered by skeletal muscle-specific ablation of Cpt1b. (A) Body weight diverges in Cpt1bm−/− mice by 70 d of age. Fat mass is significantly less by 63 d of age (B), but cumulative food intake is not significantly reduced until 88 d of age (C). (D) Significant divergence of lean mass does not occur until 100 d of age. Cumulative (56–119 d of age) weight gain (E) and food intake (F) are significantly reduced in Cpt1bm−/− mice. Data are expressed as mean ± SEM (n = 9–10 per group). *P < 0.05 by Student’s t test.
To see if there were direct effects in adipose tissue, we measured markers of mitochondrial number, lipolysis, β-oxidation, adipocyte beiging/browning, and inflammation at 10 wk of age (just before divergence of fat pad mass) and at 16 wk (a few weeks after divergence of fat pad mass) in inguinal and epididymal fat depots. There were no significant effects observed in the epididymal fat depot. In inguinal adipose tissue, Pnpla2/Atgl trended higher in 10-wk-old Cpt1bm−/− mice but was significantly higher by 16 wk. The β-oxidation enzyme, 3-hydroxyacyl–CoA dehydrogenase, was significantly elevated at 10 and 16 wk in Cpt1bm−/− mice. Citrate synthase (Cs) trended higher in 10-wk-old Cpt1bm−/− mice but was significantly higher by 16 wk (Fig. S2A). Ucp-1, Cidea, and Elovl3 were all significantly elevated in Cpt1bm−/− mice at 10 and 16 wk (Fig. S2B). TNF-α mRNA levels were not different at 10 wk of age but TNF-α mRNA levels were significantly less in Cpt1bm−/− mice by 16 wk (Fig. S2C).
Given the up-regulation of markers for FAO and uncoupling in adipose tissue, one might expect increased energy expenditure to account for decreased weight gain. However, Cpt1bm−/− mice have overall reduced energy expenditure and activity (Fig. 3 A and B). Analysis of covariance (ANCOVA) suggests that the genotype effect on energy expenditure results from the reduced activity of Cpt1bm−/− mice (Fig. 3C). In addition, the respiratory exchange ratio (RER) is significantly higher in Cpt1bm−/− mice, suggesting increased carbohydrate oxidation (Fig. 3D).
Fig. 3.

Abrogation of Cpt1b decreases activity but increases carbohydrate use. Indirect calorimetry shows decreased energy expenditure (A) and activity level (B) in Cpt1bm−/− mice (n = 7–8 at 5 mo of age). (C) ANCOVA analysis suggests that the genotype effect on energy expenditure is due to the decreased activity of Cpt1bm−/− mice. Red symbols indicate Cpt1bfl/fl, and black symbols indicate Cpt1bm−/−. (D) RER values are higher, reflecting increased glucose utilization. D, dark period; FFM, fat-free mass; FM, fat mass; L, light period; VO2, oxygen consumption.
Cpt1bm−/− Mice Exhibit Moderate Myodegeneration and Impaired Exercise Performance.
Given that the Cpt1bm−/− mice are hypoactive, we checked for muscle damage and detected increased myoglobin in serum of Cpt1bm−/− mice (Fig. 4A). H&E and phosphotungstic acid-hematoxylin (PTAH) staining shows multifocal degeneration and loss of cross-striations in some fibers by 3 mo of age (Fig. 4B). Mild functional implications appear to manifest as mice age. We initially used rotarod performance, a measure of neuromuscular coordination and muscle function, to test for impairments, and found no difference in young (12-wk-old) Cpt1bm−/− mice (Fig. 4C). We next used computerized gait analysis to probe for more subtle alterations. No differences in gait were detected in young Cpt1bm−/− mice, although mild gait disturbances were apparent by 1 y of age (Fig. 4D). Despite the decrement in mitochondrial FAO, treadmill-tested endurance is only moderately decreased in Cpt1bm−/− mice at both young (12-wk-old) and old (1-y-old) ages (Fig. 4E).
Fig. 4.
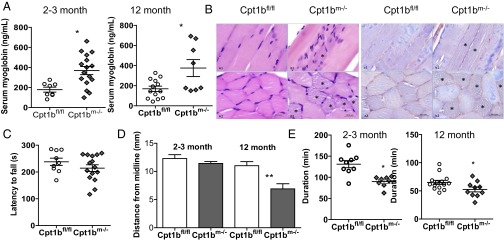
Cpt1b depletion causes mild impairment of muscle function. (A) Serum levels of myoglobin are elevated in Cpt1bm−/− mice (n = 8–12). (B) H&E and PTAH staining of gastrocnemius shows multifocal myodegeneration and loss of cross-striations in some fibers (in 3-mo-old mice). *Sites of myodegeneration. (Scale bars: 50 μm.) (C) Rotarod performance is normal in 3-mo-old mice. (D) Computerized gait analysis reveals mild impairment in rear leg extension in 1-y-old Cpt1bm−/− mice. (E) Treadmill endurance is significantly decreased in Cpt1bm−/− mice. *P < 0.05 by Student’s t test or ANOVA (n = 8–12 for behavioral assays).
Impaired Muscle Mitochondrial FAO Increases Both Systemic and IMCLs.
Plasma nonesterified fatty acid (NEFA) and triacylglycerols/triglycerides (TAGs) are significantly elevated, both prior (2–3 mo) and subsequent (12 mo) to weight divergence (Fig. 5A). Bodipy staining of gastrocnemius reveals marked IMCL accumulation in Cpt1bm−/− mice (Fig. 5B), which is visible as large lipid droplets by EM (Fig. 5C). We next examined different lipid species in soleus and extensor digitorum longus (EDL) muscle. Intramuscular TAGs are more than tripled in Cpt1bm−/− soleus but are not statistically elevated in EDL (Fig. 5D). Several of the DAG species were significantly higher in Cpt1bm−/− soleus (Fig. 5E). Likewise, most of the DAG species were significantly elevated in Cpt1bm−/− EDL (Fig. 5E). Total ceramides are approximately doubled in Cpt1bm−/− soleus, with the C16, C18, and C18:1 species all being significantly increased (Fig. 5F), whereas C16 is significantly increased in EDL (Fig. 5G). Lastly, gene expression of fatty acid transport (Cd36, Fatp1), fatty acid binding (Fabp3), and lipid droplet forming (Plin5) proteins is coordinately increased in all fiber types examined (soleus, EDL, red quadriceps, and white quadriceps; Fig. 5H and Table S1). Taken together, we can conclude that skeletal muscle-specific Cpt1b depletion successfully recapitulates a model of FAO impairment and lipid accumulation.
Fig. 5.
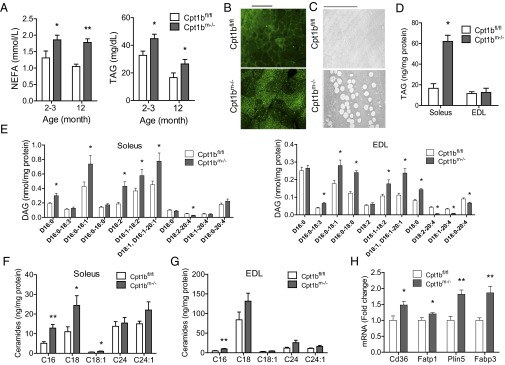
Skeletal muscle-specific Cpt1b ablation alters local and systemic lipid homeostasis. (A) Plasma NEFA and TAG levels are increased in both young and aged Cpt1bm−/− mice (n = 8–12). (B) Bodipy staining of gastrocnemius muscle shows IMCL accumulation (representative images, n = 4). (Scale bar: 100 μm.) (C) EM imaging of soleus shows enlarged lipid droplets. (Scale bar: 10 μm.) (D) IMCL accumulation is accompanied by increased levels of TAGs in soleus, but not in EDL (n = 4 per group). (E) Individual DAG species are also increased in soleus and EDL (n = 8–10 per group). Individual ceramide species are also increased in soleus (F) and EDL (G) (n = 4 per group). (H) Expression of fatty acid transport, binding, and lipid droplet proteins is increased in Cpt1bm−/− muscle. *P < 0.05 or **P < 0.01, differences detected by Student’s t test. Data are shown as mean ± SEM. Plasma was collected from mice that had been fasted for 4 h.
Impaired Muscle Mitochondrial FAO Does Not Inhibit Insulin Response.
Fasting insulin and glucose are significantly lower in Cpt1bm−/− mice than control mice at 2 mo of age, when there is no difference in body composition between Cpt1bm−/− mice and controls (Fig. 6 A and B). Insulin values in control mice increase with age, but Cpt1bm−/− mice maintain low insulin values with age (Fig. 6A), which is in agreement with less body fat. Although plasma NEFA and TAGs and intramyocellular DAGs and ceramides are considered classic hallmarks of insulin resistance, there is no difference in response to a bolus of insulin. Insulin tolerance tests (ITTs) show an equivalent insulin response in weight-matched Cpt1bm−/− and control 10- to 12-wk-old mice (Fig. 6C) and 18- to 20-wk-old Cpt1bm−/− mice (Fig. 6D). The older Cpt1bm−/− mice have lower baseline glucose values, but the slopes after insulin administration are essentially the same. Because results from the ITT studies provide a picture of whole-body glucose handling in response to insulin, we elected to perform studies using isolated muscle strips to test the direct impact of Cpt1b deficiency on insulin responsiveness. Consistent with the whole-body ITT results, basal and insulin-stimulated glucose uptake in isolated EDL and soleus muscle from Cpt1bm−/− mice was not significantly different from control mice (Fig. S3). Consistent with the whole-body and isolated muscle results, insulin-stimulated pAkt (Ser473) is similar between Cpt1bm−/− and Cpt1bfl/fl mice after injection of an insulin bolus (Fig. 6E). Likewise, insulin-stimulated translocation of Glut4 to the plasma membrane is similar in both genotypes (Fig. 6F). Despite high levels of DAG in Cpt1bm−/− muscle, membrane localization of protein kinase C theta (PKCΘ) is not significantly increased (Fig. 6G). Taken together, these data strongly suggest that insulin signaling is intact in Cpt1bm−/− mice.
Fig. 6.
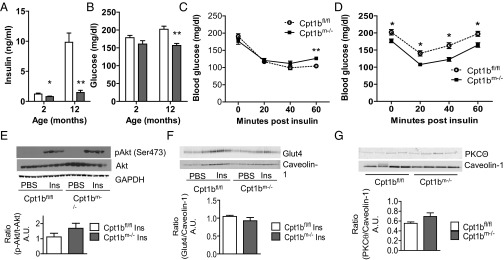
Depletion of Cpt1b in skeletal muscle does not impair whole-body insulin response or muscle insulin signaling. Blood glucose (A) and insulin levels (B) are decreased in Cpt1bm−/− mice after a 4-h fast. There is no difference in response to an ITT in Cpt1bm−/− mice (n = 8–10) at 10–12 wk (C) or 18–20 wk (D). (E) Insulin (Ins)-stimulated phosphorylation of Akt is unchanged in Cpt1bm−/− and Cpt1bfl/fl muscle (n = 4). (F) Similarly, insulin-stimulated translocation of Glut4 to the membrane is unaffected by lack of Cpt1b (n = 3). (G) Membrane-associated levels of PKCΘ are not increased in Cpt1bm−/− mice (n = 4). Data are shown as mean ± SEM. *P < 0.05 or **P < 0.01, Student’s t test. A.U., arbitrary units.
Cpt1bm−/− Mice Have Increased Carbohydrate Utilization.
A consistent hallmark of studies that have found beneficial effects of acute Cpt1 inhibition is a natural shift in substrate selection that favors glucose utilization. Although we did not observe any improvements or detriments to insulin-stimulated glucose uptake in Cpt1bm−/− mice, glucose utilization appears to be enhanced. Blood glucose levels are significantly lower in the Cpt1bm−/− mice compared with controls during an i.p. glucose tolerance test in both 10- to 12-wk-old mice and 18- to 20-wk-old mice (Fig. 7 A and B). Likewise, we detected increased pyruvate dehydrogenase (PDH) subunit-Pdha1 mRNA expression (Fig. 7C), increased PDH activity (Fig. 7D), and increased complete oxidation of pyruvate via the tricarboxylic acid (TCA) cycle in muscle homogenates (Fig. 7E). The increased carbohydrate oxidation is further supported at the whole-body level, because the RER is higher in Cpt1bm−/− mice (Fig. 3D). Furthermore, glycogen levels are significantly reduced in red quadriceps but only moderately reduced in white quadriceps (Fig. 7F). Taken together, these data indicate that there is normal insulin action and increased glycolytic flux in Cpt1bm−/− mouse muscle.
Fig. 7.

Glucose utilization is increased by abrogation of Cpt1b. Glucose disposal capacity is significantly improved in Cpt1bm−/− mice, as measured by a glucose tolerance test, at both 10–12 wk (A) and 18–20 wk (B) (n = 8–10). (C) Expression of Pdha1 is increased. Both PDH (assayed with [1-14C]pyruvate) (D) and complete pyruvate oxidation (Ox.) (measured with 3-[14C]pyruvate) (E) are significantly increased (n = 3–6, representative of multiple repeated experiments). (F) Glycogen content is decreased, suggesting reliance on glucose utilization, in red quadriceps (R.Q.), but not in white quadriceps (W.Q.) (n = 4). *P < 0.05 or **P < 0.01, differences detected by Student’s t test.
Impaired Mitochondrial FAO Leads to Mitochondrial Biogenesis.
To gain further insight into how impaired mitochondrial FAO affects skeletal muscle metabolism in a chronic setting, we examined the activation of the energy sensor AMP-activated protein kinase (AMPK) and mRNA expression of genes involved in the regulation of energy and lipid metabolism, mitochondrial biogenesis, and function. Phosphorylation of the α-subunit of AMPK at Thr-172 is significantly increased in Cpt1bm−/− muscle (Fig. 8A). We found robust increases in expression of the AMPK target peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α) in both red and white muscles without changes in Pgc1β or PPARα mRNA (Fig. 8B and Table S1). PGC1α is known as a master regulator of mitochondrial biogenesis. Consistent with AMPK/PGC1α activation, EM in soleus muscle reveals large bands of mitochondria surrounding enlarged lipid droplets in Cpt1bm−/− mice (Fig. S4A). Mitochondria appear larger, with denser cristae. Measurement of mitochondrial copy number (as the ratio of DNA copy number of the mitochondrial genes, Cytb and Cox2, to the nuclear genes, glucagon and β-globin) shows a 50% increase in mitochondrial content in Cpt1bm−/− muscle compared with control mice (Fig. 8C). Additionally, we found a coordinate increase in expression of genes associated with citric acid cycle and oxidative phosphorylation of mitochondria, including Cs, Ndufs8 (complex I), Cox5a (complex IV), and SdhB (complex II) in Cpt1bm−/− muscle (Fig. 8D and Table S1). Expression of genes related to mitochondrial FAO, such as Hadha, Ech1, Cpt2, and Cact, is significantly up-regulated in muscle of Cpt1bm−/− mice compared with control mice (Fig. 8E). Additionally, a proteomic analysis of muscle of Cpt1bm−/− mice compared with control mice confirmed much of the mRNA analysis in Table S1, including a number of mitochondrial proteins up-regulated in Cpt1bm−/− mice (Table 1). Furthermore, upstream pathway analysis predicts activation of PGC1α (Fig. 8F). Importantly, despite lifelong deficiency of the mitochondrial enzyme Cpt1b, isolated mitochondria respire normally (Fig. S4B). Collectively, the EM, quantitative RT-PCR (qRT-PCR), mitochondrial gene copy number, proteomics, and mitochondrial function studies indicate adaptive biogenesis of functional mitochondria in skeletal muscle of Cpt1bm−/− mice.
Fig. 8.
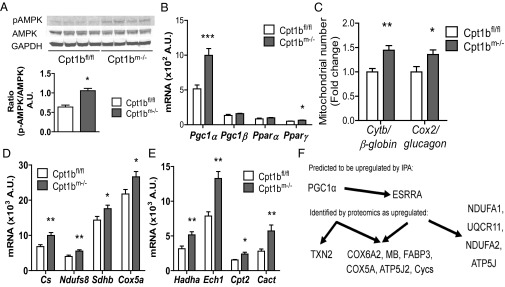
PGC1α pathway leads to mitochondrial biogenesis in Cpt1bm−/− muscle. (A) Phosphorylation of AMPKα at Thr172 is increased by FAO deficiency. (B) Gene expression of PGC1α and PPARγ, but not PGC1β or PPARα mRNA, is increased in Cpt1bm−/− muscle. Mitochondrial proliferation is induced, as shown by increased mtDNA copy number (quadriceps, n = 11) (C), coordinate up-regulation of mitochondrial gene expression (D), and increased expression of mitochondrial FAO genes (E). (F) Proteomics analysis (n = 7–8) detected significant overrepresentation of mitochondrial genes, and mechanistic network analysis predicts activation of a PGC1α signaling pathway. *P < 0.05 or **P < 0.01, differences detected by Student’s t test. Data are shown as mean ± SEM.
Table 1.
Quantitative proteomics results from Cpt1bm−/− muscle
| GO biological process | No. of identified proteins | Proteins significantly up-regulated in Cpt1bm−/− | Expected value in Cpt1bm−/− | Process overrepresented (+) or underrepresented (−) in Cpt1bm−/− | P value of statistical overrepresentation test |
| Respiratory electron transport chain | 44 | 22 | 6.37 | + | 2.65E-05 |
| Generation of precursor metabolites and energy | 46 | 22 | 6.66 | + | 5.71E-05 |
| Oxidative phosphorylation | 33 | 18 | 4.78 | + | 1.39E-04 |
| Cellular process | 161 | 10 | 23.32 | − | 5.08E-02 |
| Primary metabolic process | 216 | 17 | 31.29 | − | 6.65E-02 |
Quantitative proteomics results from Cpt1bm−/− muscle were evaluated using the Panther statistical overrepresentation test for Gene Ontology (GO) Biological Processes (www.pantherdb.org, version 8.0). The top five identified pathways are shown.
Metabolic Remodeling Leads to Alternative Substrate Utilization by Mitochondria.
To probe further the implications of decreased mitochondrial FAO on metabolism within the muscle, we used muscle homogenates to examine oxidation of other substrates. Oxidation of the amino acid Leu is significantly increased in Cpt1bm−/− muscle homogenate (Fig. 9A). Gene expression of the Bcat2 and Bckdha enzymes is elevated (Fig. 9B), and metabolomics analysis reveals significant elevations in muscle levels of nearly all amino acids tested (Fig. 9 C and D). This effect appears to be confined to muscle, because only Gly and citrulline are elevated in serum (Fig. S5). Furthermore, propionylcarnitine (C3) and succinylcarnitine (C4-DC) acylcarnitines are significantly increased in Cpt1bm−/− muscle (Fig. S6). Production of C3 is commonly used as an indicator of amino acid catabolism, because the breakdown of Val, Leu, Ile, and Met provides a significant source of propionyl-CoA. Additionally, propionyl-CoA enters the TCA cycle after it is carboxylated to form succinyl-CoA. As such, the increase in C4-DC and C3 in the present study is quite likely a result of increased amino acid catabolism in Cpt1bm−/− muscle. Globally, these analyses provide compelling evidence that Cpt1b deficiency in muscle induces a localized signal promoting mobilization and subsequent utilization of amino acids as metabolic fuel.
Fig. 9.
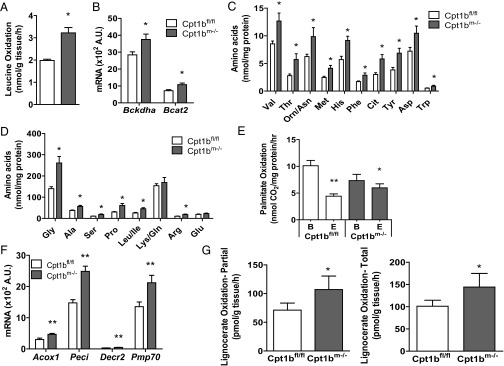
Cpt1b deficiency in skeletal muscle remodels energy metabolism, leading to compensatory amino acid catabolism and peroxisomal FAO. Cpt1bm−/− muscles use amino acids as an alternative energy source, as shown by enhanced Leu oxidation in muscle homogenates (A) and increased expression of genes involved in amino acid metabolism (B). Leu oxidation was measured from [U-14C]Leu in gastrocnemius homogenate (n = 4). (C and D) Consistent with amino acid catabolism in Cpt1bm−/− muscle, levels of nearly all amino acids are elevated. (E) Although mitochondrial FAO is decreased, there is no reduction in FAO in quadriceps muscle homogenates (measured with [1-14C]palmitate; n = 3, representative of multiple independent experiments). B, basal; E, treated with the Cpt1 inhibitor etomoxir. (F) Up-regulation of peroxisomal activity is suggested by increased expression of peroxisomal FAO genes (n = 5–7). (G) Increased peroxisomal capacity for FAO is shown by enhanced oxidation of lignoceric acid (C24:0), which can only be oxidized by peroxisomes. Lignoceric acid (partial and total) oxidation was measured from [1-14C]lignoceric acid in gastrocnemius homogenate (n = 4). *P < 0.05 or **P < 0.01, differences detected by Student’s t test. Data are shown as mean ± SEM.
Impaired Mitochondrial FAO Leads to Compensatory Peroxisomal FAO.
In addition to measuring FAO in mitochondria, we measured FAO in the whole-muscle homogenate. FAO in whole-muscle homogenate is only slightly reduced in Cpt1bm−/− muscle compared with control muscle (Fig. 9E). Peroxisomes are capable of processing a variety of long-chain and very-long-chain fatty acids, and tend to be up-regulated in response to elevated fatty acids, such as the environment in Cpt1b-deficient mice. Expression of peroxisomal FAO genes, including Acox1, Peci, Decr2, Pmp70, Pex19, and the medium-chain carnitine O-octanoyltransferase, Crot, is up-regulated in muscles from Cpt1bm−/− mice (Fig. 9F and Table S1). We used extracellular flux analysis to confirm that Cpt1bm−/− mitochondria are unable to oxidize long-chain acyl-CoA esters but can respirate on long-chain acylcarnitines, which could theoretically be supplied by peroxisomes (Fig. S4C). To confirm changes to peroxisomal function, we measured oxidation of lignoceric acid (C24:0), which requires peroxisomes to be catabolized (29). Results show that both total and partial lignoceric acid oxidation is increased in Cpt1bm−/− homogenates (Fig. 9G). Taken together, the data strongly suggest that Cpt1bm−/− mice partially compensate for reduced mitochondrial FAO by induction of peroxisomal FAO.
Discussion
There have been decades of conflicting literature regarding pharmacological inhibition of Cpt for treatment of insulin resistance. Given this ongoing confusing literature and the contrasting theories of the role of impaired vs. increased muscle FAO on development of insulin resistance, we developed a skeletal muscle-specific model of Cpt1b deficiency. As many would expect, inhibition of mitochondrial FAO causes the muscle to rely more heavily on carbohydrate for fuel. Enhanced glucose disposal and elevated pyruvate oxidation as a result of restricted FAO are consistent with substrate competition models between glucose and fatty acids and the allosteric regulation of hexokinase and PDH regulating both glucose uptake and oxidation (14, 30). However, as with any homeostatic system, perturbations in one component cause compensatory adaptations. Our results suggest that impaired skeletal muscle mitochondrial FAO has complex effects that go beyond the regulation of glucose uptake and oxidation.
These adaptations are vital to sustain energy supply to muscle and are the result of several interrelated pathways. We suggest that an energy deficit signal is induced that elicits a suite of effects, including mitochondrial biogenesis, compensatory oxidation of fat by peroxisomes, and the use of amino acids for fuel. To our knowledge, this study is the first report of enhanced peroxisomal FAO in muscle following decreased mitochondrial FAO, and the implications of prolonged reliance on peroxisomal fat catabolism are unknown.
Another adaptation that appears to be directly affected by reduced mitochondrial FAO is alterations in the cellular response to accumulation of lipotoxic species. The loss of mitochondrial FAO induces up-regulation of virtually every gene examined related to fatty acid uptake and transport, lipolysis, carnitine/acylcarnitine shuttle, and β-oxidation in Cpt1bm−/− muscle (Table S1). However, the long-chain acyl-CoAs cannot enter the mitochondria, leading to a massive accumulation of IMCL. It has been previously suggested that IMCL, specifically TAGs, are not detrimental, per se, but that it is rather the accumulation of lipotoxic DAGs and ceramides that impairs insulin signaling. However, our results clearly show substantial increases in both DAGs and ceramides, and yet no detriments to insulin signaling. This result is at first counterintuitive; however, other reports have found similar increases in lipotoxic species without induction of insulin resistance, including the well-known athlete’s paradox (31–34). Elevated DAGs have been found in endurance athletes with high insulin sensitivity (35). In these studies, insulin sensitivity was associated both with specific DAG species and with higher oxidative capacity (35), suggesting that alterations to nutrient and energy balance may play a role in determining the response to a lipotoxic environment. As a result, one possible explanation for our findings is that activation of energy/nutrient deprivation pathways is capable of overriding the traditional lipid-induced decrements in insulin-stimulated glucose uptake as a means to rescue energy production; however, this possibility remains untested. It is also possible that as yet unknown differences between specific species of DAGs and ceramides and their subcellular locations are responsible for the discrepancy (33, 36). Recent studies in humans have correlated both individual DAG species, particularly saturated DAG, and their subcellular localization to insulin resistance in humans (37, 38). In whole-muscle homogenates, we observe increases and a few decreases in saturated, unsaturated, and mixed DAG species in the Cpt1bm−/− muscle. Nevertheless, whether or not altered distribution of lipotoxic species plays a role, our results argue strongly that impaired mitochondrial FAO, even coupled with chronic cellular lipid oversupply, is not sufficient to induce insulin resistance.
In summary, to our knowledge, the first and almost foregone results from decreased transport of fatty acids into the mitochondria are impaired mitochondrial FAO and the concomitant increase in carbohydrate oxidation. More important are the observed adaptations in muscle. To our knowledge, these results provide the first observation in which decreased mitochondrial FAO elicits a signal indicating a shortage of lipid in the muscle causing the up-regulation of genes governing fatty acid uptake, transport, storage, and oxidation, yet the lipids cannot enter the mitochondria. However, the increased lipids are uncoupled to their usual association with the interference in the insulin signaling cascade. Understanding these uncoupling mechanisms may be important in the treatment of type 2 diabetes. One could conclude that the increased number of mitochondria and oxidative capacity offset the lipid accumulation similar to the athlete’s paradox, but because the mitochondria have reduced FAO capacity, there may be other unexplored explanations. Perhaps the most striking phenotype is that Cpt1bm−/− mice do not gain or lose fat mass beyond 10–12 wk of age. Careful measurements of food intake and body composition during the divergence in fat mass between control and Cpt1bm−/− mice implicate decreased food intake as a partial contributor. The finding that inhibition of mitochondrial FAO in muscle increases oxidative and uncoupling capacity in inguinal fat suggests that energy may be dissipated as heat. Elucidating the exact mechanisms that prevent gain of fat mass in Cpt1bm−/− mice could have wide-ranging implications in the obesity field.
Materials and Methods
Animal Studies.
Animal studies were conducted at Pennington Biomedical Research Center’s American Association for the Accreditation of Laboratory Animal Care-approved facility, with mice receiving a standard chow diet, composed of 20% (wt/wt) protein, 25% (wt/wt) fat, and 55% (wt/wt) carbohydrate (Purina Rodent Chow no. 5015; Purina Mills), and were approved by the Institutional Animal Care and Use Committee.
Animal Procedures.
Body composition was measured using a Bruker NMR Minispec (Bruker Corporation). Food intake was measured as described by Kruger et al. (39), with slight modifications. Plasma collections were performed by submandibular bleed. Glucose tolerance tests were performed following a 4-h fast by i.p. injection of 20% d-glucose (40 mg of glucose per mouse). ITTs were done in the fed state, using an i.p. dose of 0.04 units (U) per mouse. For insulin signaling studies, mice were fasted overnight (∼16 h) and given an i.p. injection of insulin (1.0 U/kg of body weight) or saline, and tissues were collected 10 min later. Tissue processing and isolated muscle experiments are described in SI Materials and Methods. Indirect calorimetry was done in a 16-chamber Oxymax system (Columbus Instruments) as described by Albarado et al. (40). Behavioral studies were performed with slight modifications from the method of Wicks et al. (41). Endurance was measured following three habituation sessions; speed was increased from 6 m⋅min−1 by 0.5 m⋅min−1 at 5-min intervals until exhaustion.
Metabolic Profiling.
ELISA kits were used for measurement of insulin (Ultra Sensitive Mouse Insulin ELISA kit; Crystal Chem, Inc.), NEFA, and TAG (NEFA and triglyceride-H; Wako Diagnostics). Isolation of lipid and quantification of TAGs and ceramides were performed as described by Obanda et al. (42). DAGs were measured as described by Wang et al. (43). Glycogen was measured using a commercial kit (65620; Abcam) according to the manufacturer’s instructions. Measurement of acylcarnitines, amino acids, and proteomics is described in SI Materials and Methods.
qRT-PCR.
Total RNA from mouse tissue was isolated using an RNeasy Mini Kit (Qiagen) and DNase digested. cDNA synthesized with the iScript cDNA synthesis kit was used for qRT-PCR with the SYBR Green system (Bio-Rad). DNA was isolated using a DNA/RNA Mini Kit (Qiagen). Primer details are provided in Table S2.
Substrate Oxidation Assays.
Red quadriceps or mixed gastrocnemius muscle homogenates were prepared as previously described (44). Mitochondrial isolation was performed as described by Frezza et al. (45); the IBm2 buffer was modified to 70 mM sucrose, 200 mM mannitol, 5 mM EGTA/Tris (pH 7.4), and 10 mM Tris⋅HCl (pH 7.4). Fatty acid and pyruvate oxidation was assessed using whole-muscle homogenates or isolated skeletal muscle mitochondria as in the study by Hulver et al. (46); etomoxir was used at a final concentration of 100 μM. Peroxisomal FAO was measured from [1-14C]lignoceric acid (20 μM) as previously shown (44). Capture of 14CO2 from [U-14C]Leu (100 μM) was measured over the course of 2 h in reaction media (pH 7.8) consisting of the following: 100 mM sucrose, 10 mM Tris⋅HCl, 10 mM K2HPO4, 80 mM KCl, 1 mM MgCl2⋅6H2O, 2 mM l-carnitine, 0.2 mM malate, 0.08 mM pyridoxal phosphate, 0.4 mM α-ketoglutarate, 1 mM DTT, 0.1 mM nicotinamide-adenine dinucleotide, 2 mM ATP, and 0.05 mM CoA.
CPT Activity Assay.
CPT activity was assayed using isolated mitochondria (120 μg per well) according to principles outlined by Bieber et al. (47). Mitochondrial assays are described in SI Materials and Methods.
Immunofluorescence and Bodipy Staining.
Gastrocnemius muscles were frozen in a mixture of Optimal Cutting Temperature Compound (Sakura) and Tragacanth (Spectrum). Sectioned samples were fixed in 4% paraformaldehyde solution (Invitrogen), incubated in a 1:100 dilution of Bodipy in PBS (Invitrogen), and mounted in Citifluor (Ted Pella, Inc.). PTAH and H&E staining was performed by standard protocols in formalin-fixed tissues at the Louisiana State University-AgCenter Histology Core.
EM.
EM was performed at the Socolofsky Microscopy Center at Louisiana State University. Materials were fixed in 2% (wt/vol) glutaraldehyde/1% (wt/vol) formaldehyde and then postfixed in 2% (wt/vol) osmium tetroxide, en bloc stained in 0.5% uranyl acetate, and embedded in Epon-NMA. Ultrathin sections (70 nm) were mounted on collodion-coated copper grids, stained with Reynolds lead citrate, and imaged with a JEOL 100CX transmission electron microscope.
Statistical Analysis.
Data were analyzed by t test, ANOVA/repeated measures ANOVA, with Bonferroni posttests using GraphPad Prism 5 software. ANCOVA analysis was performed using JMP software from SAS. The P value was set at <0.05 a priori.
Supplementary Material
Acknowledgments
We thank Tamra Mendoza, Estrellita Bermudez, Steven Bond, Dieyun Ding, Jeffrey Covington, Sudip Bajpeyi, Krisztian Stadler, David Burke, Ryan Grant, and Eric Ravussin for critical advice, reagents, and/or technical assistance. We also thank the Socolofsky Microscopy Center, the Department of Agricultural Chemistry, and the W. A. Callegari Environmental Center at Louisiana State University (LSU) for measurement of lipid species and EM. We thank the Duke University School of Medicine for the use of the Duke Proteomics Core Facility, and Drs. Matthew Foster and Arthur Moseley for performing the proteomic analysis. Dr. Nobuko Wakamoto (LSU-Agricultural Center Histology Department) is also acknowledged for assistance with muscle pathology staining and interpretation. This work used Pennington Biomedical Research Center core facilities (Proteomics and Metabolomics, Genomics, Cell Biology and Bioimaging, Transgenic and Animal Phenotyping, and Animal Metabolism and Behavior) that are supported, in part, by Center of Biomedical Research Excellence (COBRE) (NIH Grant 8P20-GM103528) and Nutrition Obesity Research Center (NORC) (NIH Grant 2P30-DK072476) center grants from the National Institutes of Health. This research was supported by American Diabetes Association Grant 1-10-BS-129 and NIH Grant R01DK089641 (to R.L.M.). S.E.W. is supported by Fellowship T32 AT004094, and received a pilot and feasibility grant from NORC (NIH Grant 2P30-DK072476). R.C.N. is supported as a project primary investigator on the COBRE grant (NIH Grant 9P20-GM103528) and as primary investigator on NIH Grant R01DK103860.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.R. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1418560112/-/DCSupplemental.
References
- 1.Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94(6):2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaster M, Rustan AC, Aas V, Beck-Nielsen H. Reduced lipid oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin: Evidence from cultured myotubes. Diabetes. 2004;53(3):542–548. doi: 10.2337/diabetes.53.3.542. [DOI] [PubMed] [Google Scholar]
- 3.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279(5):E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- 4.Mensink M, Blaak EE, van Baak MA, Wagenmakers AJ, Saris WH. Plasma free Fatty Acid uptake and oxidation are already diminished in subjects at high risk for developing type 2 diabetes. Diabetes. 2001;50(11):2548–2554. doi: 10.2337/diabetes.50.11.2548. [DOI] [PubMed] [Google Scholar]
- 5.Chavez JA, et al. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278(12):10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 6.De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry. 1997;36(42):12939–12947. doi: 10.1021/bi971157f. [DOI] [PubMed] [Google Scholar]
- 7.Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49(5):677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 9.Long SD, Pekala PH. Lipid mediators of insulin resistance: Ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem J. 1996;319(Pt 1):179–184. doi: 10.1042/bj3190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravichandran LV, Esposito DL, Chen J, Quon MJ. Protein kinase C-zeta phosphorylates insulin receptor substrate-1 and impairs its ability to activate phosphatidylinositol 3-kinase in response to insulin. J Biol Chem. 2001;276(5):3543–3549. doi: 10.1074/jbc.M007231200. [DOI] [PubMed] [Google Scholar]
- 11.Griffin ME, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48(6):1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 12.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Noland RC, et al. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem. 2009;284(34):22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muoio DM, Neufer PD. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 2012;15(5):595–605. doi: 10.1016/j.cmet.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haap M, et al. Metabolic characterization of a woman homozygous for the Ser113Leu missense mutation in carnitine palmitoyl transferase II. J Clin Endocrinol Metab. 2002;87(5):2139–2143. doi: 10.1210/jcem.87.5.8380. [DOI] [PubMed] [Google Scholar]
- 16.Hamilton-Craig I, Yudi M, Johnson L, Jayasinghe R. Fenofibrate therapy in carnitine palmitoyl transferase type 2 deficiency. Case Rep Med. 2012;2012:163173. doi: 10.1155/2012/163173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnett M, Collier GR, O’Dea K. The longitudinal effect of inhibiting fatty acid oxidation in diabetic rats fed a high fat diet. Horm Metab Res. 1992;24(8):360–362. doi: 10.1055/s-2007-1003335. [DOI] [PubMed] [Google Scholar]
- 18.Deems RO, Anderson RC, Foley JE. Hypoglycemic effects of a novel fatty acid oxidation inhibitor in rats and monkeys. Am J Physiol. 1998;274(2 Pt 2):R524–R528. doi: 10.1152/ajpregu.1998.274.2.R524. [DOI] [PubMed] [Google Scholar]
- 19.Dobbins RL, et al. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50(1):123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 20.Hubinger A, Knode O, Susanto F, Reinauer H, Gries FA. Effects of the carnitine-acyltransferase inhibitor etomoxir on insulin sensitivity, energy expenditure and substrate oxidation in NIDDM. Horm Metab Res. 1997;29(9):436–439. doi: 10.1055/s-2007-979072. [DOI] [PubMed] [Google Scholar]
- 21.Timmers S, et al. Augmenting muscle diacylglycerol and triacylglycerol content by blocking fatty acid oxidation does not impede insulin sensitivity. Proc Natl Acad Sci USA. 2012;109(29):11711–11716. doi: 10.1073/pnas.1206868109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conti R, et al. Selective reversible inhibition of liver carnitine palmitoyl-transferase 1 by teglicar reduces gluconeogenesis and improves glucose homeostasis. Diabetes. 2011;60(2):644–651. doi: 10.2337/db10-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keung W, et al. Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes. 2013;62(3):711–720. doi: 10.2337/db12-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greaves P, Martin J, Michel MC, Mompon P. Cardiac hypertrophy in the dog and rat induced by oxfenicine, an agent which modifies muscle metabolism. Arch Toxicol Suppl. 1984;7:488–493. doi: 10.1007/978-3-642-69132-4_103. [DOI] [PubMed] [Google Scholar]
- 25.Bachmann E, Weber E. Biochemical mechanisms of oxfenicine cardiotoxicity. Pharmacology. 1988;36(4):238–248. doi: 10.1159/000138390. [DOI] [PubMed] [Google Scholar]
- 26.Vickers AE, Bentley P, Fisher RL. Consequences of mitochondrial injury induced by pharmaceutical fatty acid oxidation inhibitors is characterized in human and rat liver slices. Toxicol In Vitro. 2006;20(7):1173–1182. doi: 10.1016/j.tiv.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 27.Haynie KR, Vandanmagsar B, Wicks SE, Zhang J, Mynatt RL. Inhibition of carnitine palymitoyltransferase1b induces cardiac hypertrophy and mortality in mice. Diabetes Obes Metab. 2014;16(8):757–760. doi: 10.1111/dom.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26(2):165–166. [PubMed] [Google Scholar]
- 29.Singh I, Moser AE, Goldfischer S, Moser HW. Lignoceric acid is oxidized in the peroxisome: Implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc Natl Acad Sci USA. 1984;81(13):4203–4207. doi: 10.1073/pnas.81.13.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 31.Kienesberger PC, et al. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J Biol Chem. 2009;284(44):30218–30229. doi: 10.1074/jbc.M109.047787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmers S, et al. Paradoxical increase in TAG and DAG content parallel the insulin sensitizing effect of unilateral DGAT1 overexpression in rat skeletal muscle. PLoS ONE. 2011;6(1):e14503. doi: 10.1371/journal.pone.0014503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cantley JL, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci USA. 2013;110(5):1869–1874. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86(12):5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 35.Amati F, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: Another paradox in endurance-trained athletes? Diabetes. 2011;60(10):2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15(5):585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia. 2012;55(4):1140–1150. doi: 10.1007/s00125-011-2419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szendroedi J, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci USA. 2014;111(26):9597–9602. doi: 10.1073/pnas.1409229111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruger C, Kumar KG, Mynatt RL, Volaufova J, Richards BK. Brain transcriptional responses to high-fat diet in Acads-deficient mice reveal energy sensing pathways. PLoS ONE. 2012;7(8):e41709. doi: 10.1371/journal.pone.0041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albarado DC, et al. Impaired coordination of nutrient intake and substrate oxidation in melanocortin-4 receptor knockout mice. Endocrinology. 2004;145(1):243–252. doi: 10.1210/en.2003-0452. [DOI] [PubMed] [Google Scholar]
- 41.Wicks SE, et al. Effect of intrastriatal mesenchymal stromal cell injection on progression of a murine model of Krabbe disease. Behav Brain Res. 2011;225(2):415–425. doi: 10.1016/j.bbr.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obanda DN, et al. Bioactives of Artemisia dracunculus L. mitigate the role of ceramides in attenuating insulin signaling in rat skeletal muscle cells. Diabetes. 2012;61(3):597–605. doi: 10.2337/db11-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang M, Hayakawa J, Yang K, Han X. Characterization and quantification of diacylglycerol species in biological extracts after one-step derivatization: A shotgun lipidomics approach. Anal Chem. 2014;86(4):2146–2155. doi: 10.1021/ac403798q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noland RC, et al. Peroxisomal-mitochondrial oxidation in a rodent model of obesity-associated insulin resistance. Am J Physiol Endocrinol Metab. 2007;293(4):E986–E1001. doi: 10.1152/ajpendo.00399.2006. [DOI] [PubMed] [Google Scholar]
- 45.Frezza C, Cipolat S, Scorrano L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 46.Hulver MW, et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2(4):251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bieber LL, Abraham T, Helmrath T. A rapid spectrophotometric assay for carnitine palmitoyltransferase. Anal Biochem. 1972;50(2):509–518. doi: 10.1016/0003-2697(72)90061-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.