

Significance
Arbuscular mycorrhizal fungi (AMF) are soil fungi associated with the majority of land plants worldwide. They supply plants with mineral nutrients in exchange for photosynthates. Most AMF harbor endobacteria from the Mollicutes class. Exploring metagenomes of endobacterial populations in three AMF species, we discovered that these endobacteria have minimal genomes and are metabolically dependent on their fungal host. Despite vertical transmission, endobacterial genomes are uniquely plastic. In addition, the endobacteria contain multiple genes horizontally transferred from fungi. Many of these genes encode products thought to interact with fungal host proteins. Overall, the endobacterial genomes reveal a tightly knit network of interactions with the fungal host and highlight the importance of associations between bacteria and fungi.
Keywords: genome contraction, genome plasticity, horizontal gene transfer, vertical transmission
Abstract
Arbuscular mycorrhizal fungi (AMF, Glomeromycota) colonize roots of the majority of terrestrial plants. They provide essential minerals to their plant hosts and receive photosynthates in return. All major lineages of AMF harbor endobacteria classified as Mollicutes, and known as mycoplasma-related endobacteria (MRE). Except for their substantial intrahost genetic diversity and ability to transmit vertically, virtually nothing is known about the life history of these endobacteria. To understand MRE biology, we sequenced metagenomes of three MRE populations, each associated with divergent AMF hosts. We found that each AMF species harbored a genetically distinct group of MRE. Despite vertical transmission, all MRE populations showed extensive chromosomal rearrangements, which we attributed to genetic recombination, activity of mobile elements, and a history of plectroviral invasion. The MRE genomes are characterized by a highly reduced gene content, indicating metabolic dependence on the fungal host, with the mechanism of energy production remaining unclear. Several MRE genes encode proteins with domains involved in protein–protein interactions with eukaryotic hosts. In addition, the MRE genomes harbor genes horizontally acquired from AMF. Some of these genes encode small ubiquitin-like modifier (SUMO) proteases specific to the SUMOylation systems of eukaryotes, which MRE likely use to manipulate their fungal host. The extent of MRE genome plasticity and reduction, along with the large number of horizontally acquired host genes, suggests a high degree of adaptation to the fungal host. These features, together with the ubiquity of the MRE–Glomeromycota associations, emphasize the significance of MRE in the biology of Glomeromycota.
Arbuscular mycorrhizal fungi (AMF) of the phylum Glomeromycota are obligate biotrophs that form mutualistic associations with the roots of the majority of terrestrial plant species (1). AMF provide essential mineral nutrients from the soil to the plant host and receive up to 20% of the plant’s photosynthetic energy in return. AMF are emerging as an integral component of sustainable agriculture, with the goal of optimizing mycorrhizal associations in crop plants as an alternative to the use of nonrenewable mineral fertilizers (2).
For almost half a century, AMF have been known to harbor morphologically diverse bacteria in their hyphae and spores (3). Recently, representatives of one of these morphotypes have been identified as novel members of the class Mollicutes (4) and are referred to as mycoplasma-related endobacteria (MRE). All Mollicutes are intimate associates of eukaryote hosts (5), acting as parasites or, rarely, as mutualists (6). They are characterized by reductive evolution and minimal genomes. MRE are the first Mollicutes to be found associated with fungi. They reside directly in the host cytoplasm and have been detected in all major lineages of Glomeromycota worldwide (4, 7). Molecular evolution studies suggest that some MRE populations exhibit substantial levels of intrahost genetic diversity (4, 7) whereas laboratory observations indicate that MRE are transmitted vertically through asexual spores of AMF (8).
To elucidate MRE biology, we sequenced three metagenomes of MRE associated with divergent AMF species. Our aim was to determine the degree of adaptation of MRE to the fungal host. In particular, we were interested in identifying genomic footprints of the MRE endosymbiotic lifestyle, including metabolic capabilities, mechanisms of genome evolution, and genetic underpinnings of MRE interaction with AMF. Although the reduced gene content of MRE suggests that they are metabolically dependent on the AMF hosts, their genomes are highly plastic, with genome rearrangements facilitated by recombination machinery and transposable elements. The MRE genomes encode proteins with domains involved in protein–protein interactions with eukaryotic hosts. They also contain products of interdomain horizontal transfer from fungi. Overall, the MRE genomes show a high degree of adaptation to the fungal host, indicating the significance of these endobacteria in the biology of Glomeromycota.
Results
MRE Diversity, Metagenome Sequencing, and Analysis.
Previous surveys revealed that MRE are abundant in all major lineages of Glomeromycota worldwide (4). To explore MRE genomes across AMF host diversity, we selected three distantly related Glomeromycota species: Claroideoglomus etunicatum (CE, family Claroideoglomeraceae), Racocetra verrucosa (RV, Gigasporaceae), and Rhizophagus clarus (RC, Glomeraceae). Unlike other vertically transmitted endosymbionts, which display genetic homogeneity facilitating assembly of their genomes (9), MRE are diverse within host individuals (4, 7). To better understand the population structure of MRE within each focal host, we conducted a phylogenetic analysis of MRE 16S rRNA gene sequences obtained from individual AMF spores (Fig. 1). As expected based on the substantial phylogenetic divergence among the three AMF hosts, our target MRE populations provided a good representation of MRE diversity detected thus far (4). MRE populations of the RC and RV hosts formed distinct clades, with 16S rRNA gene sequence similarity ranging from 99.9% in RC to 97% in RV. In contrast, sequence similarity in the MRE population from CE was only 88.3%, with individual sequences dispersed across the diversity of MRE surveyed in the past (4).
Fig. 1.

16S rRNA phylogeny reveals inter- and intrahost diversity of MRE populations. Cloned 16S rRNA gene sequences were obtained from three individual spores (A, B, or C) representing populations MRE-RC, MRE-RV, and MRE-CE (bolded). Numbers following spore designations indicate individual sequences. Reference MRE represent operational taxonomic units defined at a 97% sequence similarity level. Bayesian posterior probabilities greater than 0.90 are shown above branches. Branches with maximum likelihood bootstrap support over 70% are thickened.
Because MRE are uncultivable (8), their cells were extracted directly from multiple spores of their respective AMF hosts for sequencing. The metagenomes were sequenced using the Illumina HiSeq platform with 2 × 100 paired-end reads, followed by quality control, assembly, and annotation. The estimated sequencing coverage ranged from 4,100× to 12,000× across the three metagenomes (SI Appendix, Table S1). The statistics of the metagenomic assemblies for each MRE population are summarized in Table 1. Although two of the three metagenomes (MRE-RC and MRE-RV) were expected to be of low complexity based on the low 16S rRNA gene sequence diversity (Fig. 1), MRE contigs in all three hosts revealed high genomic intricacy, with chromosomal rearrangements apparent through alterations of gene synteny (Fig. 2). This genetic property of MRE prevented assembly of a single genome from any of the focal MRE populations. Nevertheless, we were able to assess the biological capabilities of the MRE population as a whole within each fungal host. In particular, the metagenomic assemblies show that the MRE genomes have a low guanine-cytosine (GC) content (Table 1), comparable with that of other mycoplasmas (10). Furthermore, based on the number of contigs containing the same gene content, we established that the MRE genomes are fairly small, with the size ranging from 0.7 to 1.2 Mb.
Table 1.
Metagenome assembly statistics for MRE associated with three AMF hosts
| Feature | MRE-RC | MRE-RV | MRE-CE |
| No. of chromosomal contigs | 34 | 99 | 67 |
| Combined size of contigs, bp | 739,936 (125,736) | 1,227,948 (70,737) | 662,952 (55,942) |
| GC content, % | 32.0 | 33.6 | 34.3 |
| No. of CDSs | 1,032 (180) | 1,842 (111) | 1,078 (88) |
| No. of orphan genes | 640 | 1,303 | 732 |
| tRNA | 24 | 29 | 34 |
Numbers in parentheses represent contigs sharing duplicate regions.
Fig. 2.

Contig structures reveal that MRE genomes are highly recombinant, resulting in disruptions of gene synteny. Contigs are shown as black elongated ovals, and colored lines indicate sequences and gene order. Sequences that are identical and have the same gene order are depicted in the same color. The bases corresponding to identity between the contigs are indicated. (A) Example of two contigs of MRE-RV sharing identical sequences (orange) followed by completely different genetic content (green and red). (B) Example of three contigs of MRE-RC sharing regions of identity (pink) whereas flanking regions contain unrelated gene content (green, white, yellow, orange, and blue).
Plasticity of MRE Genomes.
The 16S rRNA gene phylogenies indicated that the rRNA gene sequences in the MRE-RC population were nearly monomorphic (Fig. 1), which is consistent with the presence of a single 16S rRNA locus in the MRE-RC genome assembly and may suggest genetic homogeneity of the MRE-RC population. However, the MRE-RC contigs revealed extensive chromosomal rearrangements (Fig. 2). In particular, we observed contigs with stretches of DNA sequences exhibiting 100% identity between the two contigs, followed by stretches of DNA that were entirely divergent. PCR amplifications and sequencing of a collection of such junctions between regions of sequence similarity and dissimilarity confirmed that their alternative versions exist in MRE populations and are not results of misassembly. Analysis of the junction regions further revealed that the main sources of the chromosomal rearrangements in the MRE population of RC were mobile element activity and genetic recombination (SI Appendix, Table S2). These two mechanisms were also important in MRE populations associated with RV and CE.
MRE genomes contain an unusually high number of transposase-encoding genes: i.e., 31 of 394 nonorphan genes encode transposases in MRE-RC, 19 of 539 in MRE-RV, and 5 of 346 in MRE-CE. Most of these genes are located at the junctions where chromosomal rearrangements are evident (SI Appendix, Table S2). Some junctions lack noticeable gene products that may have caused rearrangements, and we believe this pattern to be a consequence of recombination events that occur in the genome. Specifically, the genomes in all three MRE populations are riddled with simple sequence repeats (SSRs), genomic features common in many Mycoplasma species (11). These sequence features are known to facilitate homologous recombination events in the Mycoplasma genomes and contribute to high intraspecies variability (12). Furthermore, genes involved in DNA recombination are present in the MRE genomes (SI Appendix, Table S3), a trait also shared with other Mycoplasma genomes (12, 13). In particular, genomic assemblies of MRE revealed the presence of RecA, which is involved in homologous recombination (14). They also showed an overabundance of the XerC/D tyrosine recombinase genes (SI Appendix, Table S3). The Xer recombination system normally is used by bacteria for dimer resolution, to maintain monomeric states of chromosomes. Moreover, it is also responsible for generating inversions, as well as for integration and excision of gene cassettes (15). The Xer system has been implicated as the source of DNA rearrangements in other Mycoplasma species (16). The unusually high copy number of the XerC/D genes, combined with the AT richness of MRE genomes, increases the likelihood of Xer recombinase target sites and suggests that Xer recombinase is important for the chromosome structure and plasticity of MRE genomes.
In addition to mobile elements and recombination events as factors responsible for genome plasticity (SI Appendix, Table S2), the genomes of MRE-CE and MRE-RV display evidence of a historical invasion by plectrovirus, a phage known to infect Spiroplasma species (17). Although the MRE genomes harbor only partial plectroviral genes, some genomic rearrangements may be a result of past phage activity, as seen in the Spiroplasma genomes.
The preponderance of SSRs as well as evidence of multiple recombination systems and mobile elements collectively suggest that MRE genomes are highly plastic and undergo continuous rearrangements, not unlike the genomes of other mycoplasmas (13). As a consequence, each AMF host is expected to harbor an MRE population in which individual cells contain different products of genomic rearrangements. The metagenomic assemblies provide a snapshot of this incessant genome-shuffling activity taken at the host population level.
Limited Metabolic Capabilities of MRE.
Biological functionalities of MRE are shown in SI Appendix, Fig. S1. As expected of Mollicutes with an intracellular lifestyle, the genomic capacities of MRE are very limited, and the majority of their functional capabilities are those of basic cell maintenance. Even the most essential cellular functions have been reduced to a minimum. For example, all three MRE populations have lost all but the α-subunit of the DNA polymerase III holoenzyme, and their RNA polymerase consists only of the α-, β-, and β′-subunits. MRE are incapable of amino acid and nucleic acid biosynthesis, and so these metabolites must be obtained from the AMF host cytoplasm. As with other Mycoplasma species, the MRE genomes do not encode enzymes catalyzing the TCA cycle and oxidative phosphorylation. In some mycoplasmas, ATP energy is generated through the arginine dihydrolase pathway, specified by the arcABC genes (18). However, this system is also lacking in the MRE populations. Consequently, the mechanism of energy production in MRE remains unclear, as it is in other nonfermentative Mycoplasma species (19).
Electron micrographs of MRE in the cytoplasm of various AMF specimens indicate that the MRE cells are surrounded by an electron-dense wall-like layer not found in other wall-less bacteria of the Mollicutes class (4, 7). However, genes involved in the biosynthesis of a Gram-positive cell wall were not found in the genomes of any of the MRE populations. In other mycoplasmas with reduced biosynthetic capabilities, it is not uncommon to find exogenous lipids incorporated into membranes (20). Similarly, heritable mutualists with reduced genomes commonly lose genes for cell envelope biosynthesis and thus are dependent on host-derived membranes (21). Therefore, we speculate that the unusual wall-like layer apparent in the micrographs of MRE may be a product of the AMF host rather than of a novel biosynthetic gene cluster acquired by MRE.
Finally, it should be noted that, although MRE have reduced genomes and seemingly limited metabolic capabilities, more than half of their genomes encode orphan genes (Table 1) with no known functional orthologous groups. Thus, MRE may harbor novel metabolic pathways not yet characterized.
Host-Interactive MRE Proteins.
We found that the MRE genomes from all three AMF species encode proteins with domains predominantly used for protein–protein interactions in eukaryotes. They include ankyrin repeats, ANK, and leucine-rich repeats (LRRs) (SI Appendix, Table S4). The functions of these genes are unknown; annotations based on amino acid sequence similarity to proteins in the National Center for Biotechnology Information (NCBI) databases (recovered by BLASTp) revealed similarities to hypothetical proteins. ANK proteins in bacteria generally are found in endosymbionts, where they interact with host proteins to fulfill a variety of functions, including control of differential transcription of host genes, changes in host cytoskeleton, and degradation of host proteins (22, 23). LRR proteins in bacteria are also relatively rare although they have been found in pathogens of eukaryotic hosts, where they interact with host proteins in diverse ways, including adhesion, internalization, and virulence (24–26).
MRE Genes of Fungal Origin.
The MRE genomes contain many genes that seem to have been horizontally transferred from their AMF host. These genes represent 5% of total coding DNA sequences (CDSs) in the MRE-RC metagenome, 4% in MRE-CE, and 3% in MRE-RV (SI Appendix, Table S5). They initially were identified based on amino acid sequence similarity to proteins in the NCBI databases (recovered by BLASTp), with the majority of high-similarity genes found in an AMF species Rhizophagus irregularis. R. irregularis is the only AMF species with a sequenced genome (27), and it is one of the few AMF that do not harbor MRE (4). The genes of putative fungal origin were confirmed through PCR to be true components of the MRE metagenomic contigs. Many of these MRE genes differ from their R. irregularis homologs in GC content, a pattern consistent with MRE genomic assemblies being on average more GC-rich than those of R. irregularis, with 32–34% GC in MRE versus 28% in R. irregularis (27) (SI Appendix, Table S5). These differences in GC content suggest that some of the gene transfer events may be ancient enough to allow sufficient time for acquired genes to evolve toward a GC content consistent with MRE codon use. Similarly, some of these putative fungus-derived genes seem to have lost introns present in their fungal homologs (Fig. 3 and SI Appendix, Figs. S2–S4).
Fig. 3.
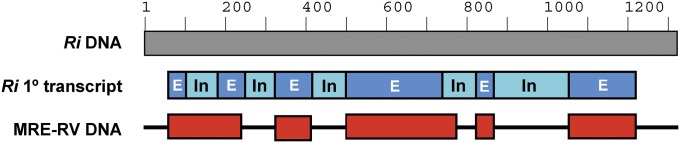
Horizontal gene transfer of the SUMO protease gene from AMF to MRE. Homologs of the AMF SUMO protease-encoding gene (ESA14994/KI282387) are present in the MRE-RV and MRE-CE metagenomes. The gene from MRE-RV (GenBank locus tag MRERV_24c013) is shown here as a representation of the horizontal gene transfer and subsequent evolution of the gene. Note that the AMF SUMO protease sequence used is that of R. irregularis (Ri) because it is the only AMF sequence available; this sequence may not be the original one that was transferred. The full DNA sequence of the SUMO protease gene of AMF (Ri) is shown as a gray bar. The corresponding primary transcript sequence is shown below, with exons (E) represented by blue bars, and introns (In) shown as pale blue bars. The DNA sequence of the gene from MRE-RV is displayed as red bars, with the horizontal line indicating loss of sequences/deletions in the MRE genome. The MRE gene still maintains traces of the introns.
Although most of the products of putative fungal origin genes are described only as “hypothetical” and their functions are unknown, the vast majority of these genes contain domains involved in protein–protein interactions (Pfam domains ANK and LRR) or signal transduction, such as tyrosine protein kinases (Pfam domain Pkinase_Tyr). Interestingly, close to half of the ANK and LRR proteins described in the previous section seem to be derived from AMF. For example, our phylogeny reconstructions revealed that the various LRR proteins in all three MRE populations cluster within the same clade as LRR proteins from the AMF R. irregularis (SI Appendix, Fig. S5).
In all three MRE populations, hypothetical proteins with the AIG1 domain are also common (SI Appendix, Fig. S6). The AIG1 domain is associated with proteins of the family IAN (immune-associated nucleotide-binding protein) or GIMAP (GTPase of immunity-associated proteins), which are conserved functionally uncharacterized GTP-binding proteins involved in plant immunity, development, and responses to biotic and abiotic stresses, as well as in immunity of vertebrates (28). The role of these GTPase proteins in AMF and MRE is unknown.
Exclusively in the MRE-RV population, four genes contain the HET (heterokaryon incompatibility) domain (SI Appendix, Fig. S7). This domain is conserved in Ascomycota fungi, where it is involved in the programmed death of fungal cells when genetically incompatible hyphae fuse, a phenomenon known as vegetative incompatibility (29). However, vegetative incompatibility involving HET proteins has not been reported in AMF, and thus the role of these proteins in MRE has yet to be resolved. For example, these proteins may facilitate competition against rhizospheric ascomycetes by disrupting their hyphae, similar to the HET-C domain protein in Pseudomonas syringae B728a, which induces fungal cell death (30).
Both MRE-RV and MRE-CE contain sentrin/small ubiquitin-like modifier (SUMO) proteases that seem to have been acquired from AMF hosts (SI Appendix, Fig. S8). SUMO proteins are members of the ubiquitin-like family involved in the process named SUMOylation. They covalently conjugate to other proteins to affect their stability, localization, or interacting partners (31). SUMO proteases function by deconjugating SUMO proteins from their substrates. Because SUMOylation is a strictly eukaryotic process, the SUMO proteases encoded in the MRE genomes are likely used to alter the SUMOylation state of their AMF hosts. When the SUMOfi (SUMO motif finder) tool was used to scan MRE proteins for possible SUMOylation targets, none were detected. Genes encoding MRE SUMO proteases are one of the examples of transfer events that originally included the full gene sequence containing both introns and exons, followed by selective degradation of introns to maintain gene functionality (Fig. 3 and SI Appendix, Fig. S2).
Differences Among MRE Populations.
Despite the common ancestry of MRE (Fig. 1) and their specialized niche within the cytoplasm of Glomeromycota hosts, the three MRE populations display differences in gene content. Although the MRE genomes are not closed, the sequence coverage of our data is well over 1,000× (SI Appendix, Table S1), assuming a genome size of ∼1 Mb. Given that a single species genome can be accurately assembled from a complex metagenome of at least 20× coverage (32), we propose that the patterns of gene presence/absence between genomic assemblies of individual MRE populations are reflective of genes gained and lost throughout independent evolution rather than the result of insufficient sequence coverage.
Maintenance of distinct genes in each separate MRE population likely depends on their importance in interaction between MRE and specific AMF host species. For example, the increased size of the MRE-RV genome relative to the MRE-CE and MRE-RC genomes suggests that different host environments select for distinct genomic features in the associated MRE populations. This hypothesis is supported by SUMO proteases, present only in the MRE-CE and MRE-RV populations, and HET domain proteins, found only in the MRE-RV population.
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas1/2 proteins are another example of divergent gene content among MRE populations because genes encoding them are found only in MRE-RC and MRE-RV. CRISPR-Cas are bacterial defense systems against foreign DNA, such as bacteriophages, plasmids, and non-self foreign chromosomal DNA (33). They may be also involved in DNA repair and genome evolution (34). For example, the loss of CRISPR from MRE-CE may be a source of the increased genetic diversity seen in this population, similar to the accelerated evolution in the bird pathogen Mycoplasma gallisepticum, also associated with the loss of the CRISPR-Cas system (35).
Taxonomic Position of MRE.
Reconstructions of the mollicute phylogenetic history based on 16S rRNA gene sequences are known to be problematic compared with other groups of bacteria (10). Therefore, to resolve the relationship between MRE and other mollicute lineages, we used concatenated amino acid sequences of 19 genes, selected based on the Genomic Encyclopedia of Bacteria and Archaea project (36). We excluded MRE-CE from this analysis because the high genetic diversity in its population was likely to confound the results (Fig. 1). The multigene phylogenies clustered MRE with members of the family Mycoplasmataceae (Fig. 4), suggesting that MRE share a closer relationship with the Mycoplasma species than the 16S rRNA phylogeny would imply (4).
Fig. 4.

Multigene phylogeny reveals shared ancestry of MRE and the members of the Mycoplasmataceae family. Phylogenetic reconstructions were performed on the concatenated amino acid sequences of the following genes: dnaG, infC, nusA, rplA, rplB, rplC, rplE, rplF, rplM, rplN, rplP, rplT, rpmA, rpsB, rpsC, rpsE, rpsJ, rpsS, and smpB. Bayesian posterior probabilities greater that 0.90 are shown above branches. Branches with maximum likelihood bootstrap support over 70% are thickened. Data from MRE-CE were not included due to high diversity of 16S rRNA gene sequences in this population (Fig. 1).
Discussion
This study provides fundamental insights into the biology of a group of endobacteria widely associated with Glomeromycota, the most common and oldest symbionts of plants (1). Because of the role of AMF in mineral nutrient and carbon cycles, the tripartite association between plants, AMF, and MRE is one of the most important symbioses on the planet. Our genomic data indicate that MRE are highly adapted to AMF and that the association between the partners is evolutionarily stable. Moreover, MRE genomic features imply extensive interdomain horizontal gene transfer and maintenance of genes involved in creating genomic diversity.
Evolution of MRE Genomes.
Genome reduction is one of the hallmarks of bacterial reproductive dependence on the host (21). In heritable mutualists, genome reduction is a degenerative process related to the small effective size of endosymbiont populations that is an outcome of population bottlenecks punctuating transmission between host generations, population subdivision, and clonality (37). In populations of small effective size, genetic drift is magnified relative to natural selection, which leads to accumulation of slightly deleterious mutations, followed by genome erosion and contraction. Only endobacterial genes important for the provision of goods and services essential to the host are retained, owing to host-level selection (38).
The minimal genomes of vertically transmitted MRE represent a stark departure from the small genomes typical for heritable mutualists. The phylogenetic position of MRE suggests that they are derived from animal-associated mycoplasmas of the Mycoplasmataceae family. Consistent with this origin, MRE genomes have retained traits shared by members of Mycoplasmataceae, including propensity for mutation accumulation and gene loss, as well as features contributing to genome plasticity (13). Rapid mutation accumulation in mycoplasmas is related to the loss of proofreading ability of DNA polymerase. This functionality is conferred by the polymerase subunits ε (dnaQ/mutD) and θ (holE) (39). Genes encoding these proteins are missing in the genomes of mycoplasmas and also from the MRE genomes. The features responsible for genome plasticity include the presence of recombination genes, as well as mobile DNA elements that facilitate recombination (13). In line with the existence of these features in the MRE genomes is evidence of recombination detected in MRE populations using population genetics tools (7, 8). The role of recombination in the restoration of high fitness genotypes (40) and facilitation of adaptation (41) is well established. It is, therefore, clear that the minimal genomes of MRE are not exclusively products of degenerative evolution. Instead, genome degradation from rapid mutation accumulation seems to be countervailed by recombination, which restores high fitness MRE genotypes. Moreover, the patterns of gene presence/absence differentiating MRE populations suggest that MRE are very effective in responding to distinct selective pressures from their fungal hosts.
Horizontal Gene Transfer as a Source of Host-Interactive MRE Proteins.
MRE genomes contain a substantial proportion of genes likely involved in the interaction with the regulatory networks of the AMF host. Many of these genes were acquired horizontally from AMF. Horizontal gene transfer (HGT) is a common and important factor in prokaryotic evolution. Anywhere from 1% to 15% of genes in a bacterial genome are estimated to be derived from HGT (42). Interdomain HGT events from eukaryotes to bacteria, such as the ones evidenced in the MRE–Glomeromycota system, although not as common, are well documented between organisms that live in close association. For instance, Legionella possess many genes acquired via HGT from aquatic protozoa (43). These genes are known to allow the bacteria to replicate within eukaryotic hosts.
A notable example of a putative host manipulation mechanism used by MRE involves SUMO proteases, which seem to be horizontally acquired from the fungal host. These proteases may allow the endobacteria to alter host protein SUMOylation levels. Although the role of the MRE SUMO proteases in the host biology requires empirical exploration, these proteases retain the conserved Peptidase C48 domain, which is essential for the SUMO deconjugation function. Although rare, the manipulation of the host SUMOylation system by bacteria has been found in several pathogens. For example, the XopD protein of Xanthomonas campestris, a pathogen of tomato and pepper plants, is delivered into the plant’s cytosol where it translocates to the nucleus and deSUMOylates host nuclear proteins (44). The direct effect of XopD deSUMOylation is unknown, but it is presumed to aid in the pathogen’s virulence. Similarly, Listeria monocytogenes, a human pathogen capable of intracellular invasion, uses one of its virulence factors to decrease host SUMOylation levels, which, in turn, allows for increased bacterial invasion and replication (45). We hypothesize that the acquisition of the fungal SUMO protease genes permits MRE for direct manipulation of host cytoplasmic conditions to benefit endobacterial fitness. It is intriguing, therefore, that not all MRE populations harbor them.
The Nature and Evolutionary Stability of the MRE–Glomeromycota Association.
The phylogenetic position of MRE as a sister clade of the Mycoplasma pneumoniae group in the family Mycoplasmataceae suggests that the origin of the MRE lineage was likely associated with a host switch event that enabled ancestral MRE to spread from animals to fungi. The host switch by MRE from animals to fungi is entirely plausible because the estimated origin of the Mycoplasmataceae at 410 mya (46) and fossil record of Glomeromycota from 460 mya (47) predate the putative origin of MRE. The ancient origin of the MRE–Glomeromycota association and its extant ubiquity suggest that this symbiosis is evolutionarily and ecologically stable despite the complete metabolic dependence of MRE on their AMF hosts and the ability of some AMF fungal species to exist without MRE infection.
MRE share with other Mycoplasma species a collection of genomic features, including metabolic dependence on the host as well as recombination machinery and transposable elements that ensure genomic diversity (13). However, unlike present-day mycoplasmas that rely primarily on horizontal dispersal, MRE are transmitted vertically from one host generation to the next, at least under laboratory conditions (8). A shift from horizontal to vertical transmission could be expected to eliminate selective pressures that maintain mechanisms promoting genomic diversity. With few notable exceptions (48), improved genome stability related to losses of recombination machinery is common in bacteria that are essential heritable mutualists (37). The fact that MRE retain the mechanisms responsible for genomic diversity suggests that, despite vertical transmission and in contrast to heritable mutualists, these mechanisms may be selected for. In interspecific interactions, antagonism is a powerful selective force that favors genetic exchanges and recombination over clonal proliferation (49, 50). Consequently, we hypothesize that retention of the mechanisms underlying genomic diversity in MRE is the result of antagonistic interactions with the AMF hosts. Under the scenario of MRE–Glomeromycota antagonism, stability and longevity of this ancient association are expected to depend on the mechanisms that generate MRE genomic diversity. Testing of these hypotheses awaits empirical analyses of the functional effects of MRE on AMF fitness.
Conclusion
This study for the first time, to our knowledge, explores the biological capabilities and mechanisms of adaptation of MRE to their Glomeromycota hosts. Although MRE existence has been known for close to half a century (3), the ubiquity of the MRE–Glomeromycota associations and their biological importance are only now being discovered. Analyses of three MRE metagenomes indicate that MRE are highly adapted to a host-dependent lifestyle and play an important role in the biology of their Glomeromycota hosts. As microbiome studies have revealed the importance of microbial communities in higher eukaryotes, this work has uncovered a tightly knit network of interactions between fungi and their own microbial communities.
Materials and Methods
Detailed descriptions of materials and methods are provided in SI Appendix. In brief, cells of MRE from C. etunicatum CA-OT135 (CE), R. clarus NB112A (RC), and R. verrucosa VA103A (RV) were separated from the fungal host cell content, and their metagenomes were sequenced using the Illumina HiSeq platform. The genomes were assembled and annotated using standard computational tools. Standard approaches were also used for phylogeny reconstructions.
Supplementary Material
Acknowledgments
We thank S. Purin for the in vitro culture of R. clarus and D. Douds for root-inducing T-DNA–transformed carrot roots (clone DC2). This work was supported by National Science Foundation Grants DEB-0918880 and IOS-1261004 (to T.E.P.) and CSBR-1349308 (to J.B.M.) and by the Cornell University Agricultural Experiment Station federal formula funds, Project NYC-153434, received from the National Institute of Food and Agriculture, US Department of Agriculture.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. JPXG00000000, JPXH00000000, and JQIB00000000).
See Commentary on page 7622.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1501676112/-/DCSupplemental.
References
- 1.Smith SE, Read DJ. Mycorrhizal Symbiosis. 3rd Ed Academic; New York: 2008. [Google Scholar]
- 2.Gianinazzi S, et al. Agroecology: The key role of arbuscular mycorrhizas in ecosystem services. Mycorrhiza. 2010;20(8):519–530. doi: 10.1007/s00572-010-0333-3. [DOI] [PubMed] [Google Scholar]
- 3.Mosse B. Honey-coloured, sessile Endogone spores: II. Changes in fine structure during spore development. Arch Microbiol. 1970;74(2):129–145. [Google Scholar]
- 4.Naumann M, Schüssler A, Bonfante P. The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J. 2010;4(7):862–871. doi: 10.1038/ismej.2010.21. [DOI] [PubMed] [Google Scholar]
- 5.Sirand-Pugnet P, Citti C, Barré A, Blanchard A. Evolution of mollicutes: Down a bumpy road with twists and turns. Res Microbiol. 2007;158(10):754–766. doi: 10.1016/j.resmic.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Jaenike J, Unckless R, Cockburn SN, Boelio LM, Perlman SJ. Adaptation via symbiosis: Recent spread of a Drosophila defensive symbiont. Science. 2010;329(5988):212–215. doi: 10.1126/science.1188235. [DOI] [PubMed] [Google Scholar]
- 7.Desirò A, et al. Detection of a novel intracellular microbiome hosted in arbuscular mycorrhizal fungi. ISME J. 2014;8(2):257–270. doi: 10.1038/ismej.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naito M. 2014. The biology and evolution of the Mollicutes/Mycoplasma-related endobacteria of arbuscular mycorrhizal fungi. PhD dissertation (Cornell University, Ithaca, NY)
- 9.Tamas I, et al. 50 million years of genomic stasis in endosymbiotic bacteria. Science. 2002;296(5577):2376–2379. doi: 10.1126/science.1071278. [DOI] [PubMed] [Google Scholar]
- 10.Thompson CC, Vieira NM, Vicente AC, Thompson FL. Towards a genome based taxonomy of Mycoplasmas. Infect Genet Evol. 2011;11(7):1798–1804. doi: 10.1016/j.meegid.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 11.Mrázek J. Analysis of distribution indicates diverse functions of simple sequence repeats in Mycoplasma genomes. Mol Biol Evol. 2006;23(7):1370–1385. doi: 10.1093/molbev/msk023. [DOI] [PubMed] [Google Scholar]
- 12.Sirand-Pugnet P, et al. Being pathogenic, plastic, and sexual while living with a nearly minimal bacterial genome. PLoS Genet. 2007;3(5):e75. doi: 10.1371/journal.pgen.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marenda MS. Genomic mosaics. In: Browning GF, Citti C, editors. Mollicutes: Molecular Biology and Pathogenesis. Caister Academic; Norfolk, UK: 2014. pp. 15–54. [Google Scholar]
- 14.Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453(7194):489–4. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 15.Das B, Martínez E, Midonet C, Barre FX. Integrative mobile elements exploiting Xer recombination. Trends Microbiol. 2013;21(1):23–30. doi: 10.1016/j.tim.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Ron Y, Flitman-Tene R, Dybvig K, Yogev D. Identification and characterization of a site-specific tyrosine recombinase within the variable loci of Mycoplasma bovis, Mycoplasma pulmonis and Mycoplasma agalactiae. Gene. 2002;292(1-2):205–211. doi: 10.1016/s0378-1119(02)00679-0. [DOI] [PubMed] [Google Scholar]
- 17.Carle P, et al. Partial chromosome sequence of Spiroplasma citri reveals extensive viral invasion and important gene decay. Appl Environ Microbiol. 2010;76(11):3420–3426. doi: 10.1128/AEM.02954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereyre S, et al. Life on arginine for Mycoplasma hominis: Clues from its minimal genome and comparison with other human urogenital mycoplasmas. PLoS Genet. 2009;5(10):e1000677. doi: 10.1371/journal.pgen.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Razin S, Hayflick L. Highlights of mycoplasma research: An historical perspective. Biologicals. 2010;38(2):183–190. doi: 10.1016/j.biologicals.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Kornspan JD, Rottem S. The phospholipid profile of mycoplasmas. J Lipids. 2012;2012:640762. doi: 10.1155/2012/640762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10(1):13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- 22.Al-Khodor S, Price CT, Kalia A, Abu Kwaik Y. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 2010;18(3):132–139. doi: 10.1016/j.tim.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards AM, Von Dwingelo JE, Price CT, Abu Kwaik Y. Cellular microbiology and molecular ecology of Legionella-amoeba interaction. Virulence. 2013;4(4):307–314. doi: 10.4161/viru.24290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bierne H, Sabet C, Personnic N, Cossart P. Internalins: A complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 2007;9(10):1156–1166. doi: 10.1016/j.micinf.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Brinster S, et al. Enterococcal leucine-rich repeat-containing protein involved in virulence and host inflammatory response. Infect Immun. 2007;75(9):4463–4471. doi: 10.1128/IAI.00279-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida K, et al. Amoebal endosymbiont Neochlamydia genome sequence illuminates the bacterial role in the defense of the host amoebae against Legionella pneumophila. PLoS ONE. 2014;9(4):e95166. doi: 10.1371/journal.pone.0095166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tisserant E, et al. Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc Natl Acad Sci USA. 2013;110(50):20117–20122. doi: 10.1073/pnas.1313452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Li X. IAN/GIMAPs are conserved and novel regulators in vertebrates and angiosperm plants. Plant Signal Behav. 2009;4(3):165–167. doi: 10.4161/psb.4.3.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutchison EA, Glass NL. Programmed cell death and heterokaryon incompatibility in filamentous fungi. In: Witzany G, editor. Biocommunication of Fungi. Springer; Dordrecht, The Netherlands: 2012. pp. 115–138. [Google Scholar]
- 30.Wichmann G, Sun J, Dementhon K, Glass NL, Lindow SE. A novel gene, phcA from Pseudomonas syringae induces programmed cell death in the filamentous fungus Neurospora crassa. Mol Microbiol. 2008;68(3):672–689. doi: 10.1111/j.1365-2958.2008.06175.x. [DOI] [PubMed] [Google Scholar]
- 31.Everett RD, Boutell C, Hale BG. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol. 2013;11(6):400–411. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- 32.Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 2012;6(4):898–901. doi: 10.1038/ismej.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasiunas G, Sinkunas T, Siksnys V. Molecular mechanisms of CRISPR-mediated microbial immunity. Cell Mol Life Sci. 2014;71(3):449–465. doi: 10.1007/s00018-013-1438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westra ER, Buckling A, Fineran PC. CRISPR-Cas systems: Beyond adaptive immunity. Nat Rev Microbiol. 2014;12(5):317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 35.Delaney NF, et al. Ultrafast evolution and loss of CRISPRs following a host shift in a novel wildlife pathogen, Mycoplasma gallisepticum. PLoS Genet. 2012;8(2):e1002511. doi: 10.1371/journal.pgen.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu D, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462(7276):1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- 38.Canbäck B, Tamas I, Andersson SGE. A phylogenomic study of endosymbiotic bacteria. Mol Biol Evol. 2004;21(6):1110–1122. doi: 10.1093/molbev/msh122. [DOI] [PubMed] [Google Scholar]
- 39.Kelman Z, O’Donnell M. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu Rev Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 40.Muller HJ. The relation of recombination to mutational advance. Mutat Res. 1964;106(1):2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 41.Fisher RA. The Genetical Theory of Natural Selection. Clarendon; Oxford: 1930. [Google Scholar]
- 42.Garcia-Vallvé S, Romeu A, Palau J. Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 2000;10(11):1719–1725. doi: 10.1101/gr.130000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez-Valero L, Bonora MN, Gribaldo S, Buchrieser C. Interdomain horizontal gene transfer shaped the genomes of Legionella pneumophila and Legionella longbeachae. In: Gophna U, editor. Lateral Gene Transfer in Evolution. Springer; New York: 2013. pp. 199–219. [Google Scholar]
- 44.Hotson A, Chosed R, Shu H, Orth K, Mudgett MB. Xanthomonas type III effector XopD targets SUMO-conjugated proteins in planta. Mol Microbiol. 2003;50(2):377–389. doi: 10.1046/j.1365-2958.2003.03730.x. [DOI] [PubMed] [Google Scholar]
- 45.Ribet D, et al. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature. 2010;464(7292):1192–1195. doi: 10.1038/nature08963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maniloff J. Phylogeny and evolution. In: Razin S, Herrmann R, editors. Molecular Biology and Pathogenicity of Mycoplasmas. Kluwer Academic/Plenum; New York: 2002. [Google Scholar]
- 47.Redecker D, Kodner R, Graham LE. Glomalean fungi from the Ordovician. Science. 2000;289(5486):1920–1921. doi: 10.1126/science.289.5486.1920. [DOI] [PubMed] [Google Scholar]
- 48.Sloan DB, Moran NA. The evolution of genomic instability in the obligate endosymbionts of whiteflies. Genome Biol Evol. 2013;5(5):783–793. doi: 10.1093/gbe/evt044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaenike J. A hypothesis to account for the maintenance of sex within populations. J Evol Theory. 1978;3:191–194. [Google Scholar]
- 50.Morran LT, Schmidt OG, Gelarden IA, Parrish RC, 2nd, Lively CM. Running with the Red Queen: Host-parasite coevolution selects for biparental sex. Science. 2011;333(6039):216–218. doi: 10.1126/science.1206360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.