

Significance
In nature, cells sense and fine-tune their metabolism in response to nutrient availability. Protein synthesis is one of the most energy-demanding metabolic processes and as such is subject to a tight regulation. A key open question, however, is how the components of the translation machinery, which are among the most abundant cellular proteins, can be regulated quickly and robustly in response to acute nutrient deprivation. We show that starved cells down-regulate protein synthesis by phosphorylation of essential and universally conserved translational GTPase Elongation factor tu (EF-Tu). Importantly, phosphorylated EF-Tu has a dominant-negative effect in elongation, resulting in the overall inhibition of protein synthesis. Thus, this novel regulatory mechanism allows for the quick and efficient regulation of protein synthesis.
Keywords: dormancy, sporulation, Ser/Thr kinase, GTPase, EF-Tu
Abstract
In nature, most organisms experience conditions that are suboptimal for growth. To survive, cells must fine-tune energy-demanding metabolic processes in response to nutrient availability. Here, we describe a novel mechanism by which protein synthesis in starved cells is down-regulated by phosphorylation of the universally conserved elongation factor Tu (EF-Tu). Phosphorylation impairs the essential GTPase activity of EF-Tu, thereby preventing its release from the ribosome. As a consequence, phosphorylated EF-Tu has a dominant-negative effect in elongation, resulting in the overall inhibition of protein synthesis. Importantly, this mechanism allows a quick and robust regulation of one of the most abundant cellular proteins. Given that the threonine that serves as the primary site of phosphorylation is conserved in all translational GTPases from bacteria to humans, this mechanism may have important implications for growth-rate control in phylogenetically diverse organisms.
Adaptation to nutrient availability is a fundamental cellular process. From unicellular prokaryotes to complex multicellular organisms, cells sense and adjust their metabolism to respond to variations in nutrient levels. Protein synthesis is one of the most energy-intensive cellular processes, and both the initiation and elongation stages of translation are down-regulated in response to nutrient limitation (1). Proteins that mediate translation, such as the essential and universally conserved GTPase Elongation factor Tu (EF-Tu), are observed in the phosphoproteomes of diverse organisms (2), suggesting that they are subject to regulatory phosphorylation. However, EF-Tu is the most abundant protein in bacteria, present at ∼100,000 copies per cell of growing Escherichia coli (3), but less than 10% of the EF-Tu molecules are phosphorylated (4). Thus, a key question is how this relatively small fraction of phosphorylated EF-Tu can cause a down-regulation of protein synthesis.
GTP-bound EF-Tu binds and delivers an aminoacyl-tRNA (aa-tRNA) molecule to the translating ribosome (3). Upon formation of a correctly base-paired mRNA codon–aa-tRNA anticodon interaction, the ribosome activates the GTPase activity of EF-Tu, followed by accommodation of the aa-tRNA into the ribosomal aa-tRNA binding (A) site, and release of the inactive GDP-bound EF-Tu from the ribosome. EF-Tu belongs to the GTPase superfamily which comprises molecular switches that share a core catalytic domain and mechanism but have evolved to perform diverse roles in many cellular processes (5). Central to their function is the hydrolysis of GTP, which controls the switch between the ON, GTP-bound, and the OFF, GDP-bound, states. Hydrolysis of GTP is followed by large conformational changes in two flexible regions known as “switch I” and “switch II.” These regions are composed of highly conserved motifs that surround and contact the nucleotide and are involved in interactions with both exchange factors and effectors that regulate GTPase function.
GTPases also can be regulated by direct inhibition of their GTPase activity. For example, Clostridium difficile toxin B glycosylation of the small GTPases RhoA, Rac1, and Cdc42 reduces their intrinsic GTPase activity (6). In addition, during infection by the intracellular pathogen Toxoplasma gondii, a secreted serine/threonine (Ser/Thr) kinase phosphorylates the host immunity-related GTPases Irgb6 and Irga6 on conserved Thr residues located in the switch I region of the GTP-binding domain, abolishing GTP hydrolysis and inhibiting GTP-dependent oligomerization (7, 8). GTP hydrolysis also is necessary for the delivery of the aa-tRNA and the subsequent release of EF-Tu from the ribosome. Compounds that inhibit hydrolysis, such as nonhydrolysable GTP analogs, stabilize the association of EF-Tu with the ribosome (9). A likely consequence of this stabilization is that subsequent interaction of the ribosome with a new EF-Tu molecule is prevented, and elongation stalls. Thus, if the ability of phosphorylated EF-Tu to hydrolyze GTP were impaired, this phosphorylation could act as a regulatory switch to inhibit translation.
To investigate this question in a physiological context, we took advantage of the Gram-positive bacterium Bacillus subtilis that responds to nutrient limitation by forming a metabolically quiescent, highly resistant spore (10). In the onset of this process, called “sporulation,” cells undergo an asymmetric division that generates two compartments with very distinct fates (Fig. 1A). The smaller compartment, called the “forespore,” differentiates into a dormant cell (endospore), and the larger compartment, designated the “mother cell,” remains metabolically active. Ultimately, the mother cell lyses and releases the mature dormant spore into the environment. Thus, B. subtilis sporulation allows the study of the differential regulation of protein synthesis in two adjacent cells that are genetically identical but experience different metabolic fates.
Fig. 1.

Expression of the YabT kinase during sporulation. (A) The B. subtilis cell cycle. Upon nutrient limitation, B. subtilis ceases vegetative growth and initiates sporulation. This process starts with an asymmetric division at one pole and produces two cell compartments: the mother cell and the forespore. The mother cell engulfs the forespore and remains metabolically active throughout sporulation, synthesizing the protective layers that surround the forespore. During this process, the forespore becomes metabolically inactive, eventually becoming a fully dormant spore that is released into the environment following lysis of the mother cell. The mature spore can endure extreme stresses and will exit from dormancy and resume vegetative growth in favorable conditions. (B) Mother cell/forespore fractionation strategy. Sporulating cells were treated with lysozyme resulting in the formation of protoplasts, which then were lysed, releasing the forespore. Free forespores were separated from mother cell lysates and lysed separately. The fractionation process was monitored by fluorescent microscopy using fluorescent reporters specifically expressed in the mother cell (YFP) and forespore (CFP). (C) Expression of YabT in vivo. Mother cell (MC) and forespore (FS) fractions isolated from a sporulating B. subtilis strain expressing a FLAG-tagged YabT were analyzed by immunoblotting with a FLAG antibody.
Here, we describe a novel posttranscriptional regulatory mechanism by which protein synthesis is down-regulated in dormancy. Underlying this mechanism is the reversible phosphorylation of EF-Tu on an absolutely conserved Thr that lies within the switch I region of the GTP-binding domain (11). This modification is accomplished by a eukaryotic-like Ser/Thr kinase that is expressed selectively during sporulation in the compartment that will become metabolically quiescent. We demonstrate that this phosphorylation stabilizes the interaction between the ribosome and EF-Tu and has a dominant-negative effect in elongation leading to the down-regulation of protein synthesis both in vitro and in vivo.
Results
YabT Ser/Thr Kinase Is Expressed in the Cell Compartment That Becomes Dormant.
The B. subtilis Ser/Thr kinase YabT is expressed in sporulation (Fig. S1 and ref. 12) under the control of σF, a sigma factor that is active in the forespore but not in the mother cell (13). We confirmed that YabT is present only in the forespore by developing a technique to separate the two cells biochemically (Fig. 1B). This technique relies on the higher sensitivity to the cell wall hydrolase lysozyme of the mother cell compared with the forespore. Sporulating B. subtilis cells were protoplasted by lysozyme treatment and lysed to release the forespore. The mother cell fraction was collected, and free endospores were isolated and subsequently lysed. We monitored the fractionation process using fluorescent reporters specifically expressed in the mother cell (YFP) and forespore (CFP) (Fig. 1B), and, to detect YabT, we generated a C-terminal FLAG-tagged YabT fusion under its native promoter (Fig. 1C). Lysates prepared from both mother cells and forespores were analyzed by Western blotting with a FLAG antibody, and YabT expression was detected only in the forespore fraction (Fig. 1C). This observation is consistent with the involvement of YabT in spore development (14) and suggests a possible regulatory role for this kinase in the progressive metabolic quiescence of the forespore.
Fig. S1.

YabT expression during sporulation. The transcriptional level of yabT during the B. subtilis cell cycle, including vegetative growth and sporulation, is shown in red (12). To determine YabT protein levels, samples of a B. subtilis strain expressing FLAG-tagged YabT were collected at exponential and stationary phases in vegetative growth or at different time points after the induction of sporulation and were analyzed by SDS/PAGE and immunoblotting using a FLAG antibody (1:1,000; Sigma). The levels of CFP (expressed from a sporulation-specific promoter) and EF-Tu also were determined as loading controls using GFP and EF-Tu antibodies (1:3,000).
Phosphorylation of EF-Tu.
B. subtilis EF-Tu is phosphorylated in vivo on multiple Ser/Thr residues by an as-yet-unidentified kinase (15, 16). We examined whether B. subtilis EF-Tu is an in vitro substrate of YabT by incubating the two proteins in the presence of [32P]ATP and monitoring transphosphorylation using autoradiography. A band with mobility similar to that expected from YabT autophosphorylation (∼33 kDa) and a closely spaced doublet with mobility similar to that expected from EF-Tu (∼44 kDa) can be seen (Fig. 2A). Phosphorylation often causes the production of a doublet, presumably resulting from partial phosphorylation of a multiply-phosphorylated substrate. To confirm the identity of the doublet, the two bands were isolated and analyzed by mass spectrometry. This analysis indicated that both bands corresponded to phosphorylated EF-Tu and identified Thr-63, an absolutely conserved Thr residue within switch I of the GTP-binding domain (11), as the main phosphorylated residue (Fig. S2 A–C). We confirmed this observation by generating and purifying an EF-Tu–mutant protein in which Thr-63 was replaced by an alanine (Ala) residue (T63A). This protein was still transphosphorylated by YabT, but the upper band of the doublet was no longer observed (Fig. 2A). In addition, the mass spectrometry analysis identified three other phosphorylated residues: Ser-11, Thr-300, and Thr-385 (Fig. S2D). However, phosphorylation on these residues was identified only in the absence of Thr-63 (i.e., using the T63A EF-Tu mutant), as is consistent with Thr-63 being the primary phosphorylation site.
Fig. 2.

Reversible phosphorylation in vitro and in vivo of EF-Tu Thr-63. (A) Purified wild-type B. subtilis EF-Tu or T63A point mutant (T63A) incubated in vitro with YabT kinase in the presence of [γ-32P]ATP. Phosphorylation was assayed by phosphorimager analysis. (B, Left) SDS/PAGE and Coomassie blue staining of whole-cell lysates of E. coli coexpressing B. subtilis wild-type EF-Tu or the T63A point mutant (T63A) and YabT kinase. (Right) Phosphorylation of EF-Tu was assayed by transferring lysates to a PVDF membrane and staining with Pro-Q Diamond. (C) EF-Tu was phosphorylated in the presence of YabT in vitro, the reaction was split, and one half was incubated with the B. subtilis Ser/Thr phosphatase PrpC. Phosphorylation was assayed by Pro-Q Diamond staining. Positions of EF-Tu (closed arrowheads) and YabT (open arrowhead) in the gels are indicated.
Fig. S2.

EF-Tu phosphorylation sites. (A) Representative MS/MS spectra of the phosphopeptide obtained after trypsin digestion of wild-type B. subtilis EF-Tu. The phosphorylation site was unambiguously assigned to Thr-63 (represented as a red-labeled pT). (B) Amino acid sequence alignment of the switch I region in EF-Tu for B. subtilis (B.su; P33166), E. coli (E.co; NP_417798), M. tuberculosis (M.tb; P0A558), and mitochondrial EF-Tu for Saccharomyces cerevisiae (Yeast; P02992), Mus musculus (Mouse; NP_766333), and Homo sapiens (Human; NP_003312). Dark and light shading represent identities and similarities, respectively. The absolutely conserved residues are shown in bold in the consensus sequence. The Thr residue in the GITI motif of the switch I region that is the primary site of phosphorylation in B. subtilis EF-Tu is shown in red. The sequence logo was generated using GENIO/logo (www.biogenio.com/logo). (C) GTP-binding domain of E. coli EF-Tu (Protein Data Bank ID code 2XQD). The structure shows the interactions between the GTP analog GDPCP and EF-Tu p-loop (red), Thr-61 in the switch I region (yellow), and the catalytic His-84 residue in the switch II region (orange). The Mg2+ ion and the catalytic water molecule are shown as green and red spheres, respectively. (D) Representative MS/MS spectra of phosphopeptides from the B. subtilis EF-Tu T63A mutant showing phosphorylation at Ser-11, Thr-300, and Thr-385. Phosphopeptides were obtained after digestion with endoprotease Asp-N (Ser-11 peptide) or trypsin (remaining peptides). The red-labeled pS and pT represent phospho-Ser and phospho-Thr, respectively.
We further confirmed Thr-63 phosphorylation by constructing an E. coli strain that coexpressed B. subtilis YabT and EF-Tu. Lysates of this strain were subjected to SDS/PAGE, and phosphorylation was assayed by Pro-Q Diamond phosphoprotein staining. The double-band pattern was observed when YabT was coexpressed with wild-type EF-Tu, but the upper band was no longer present when YabT was coexpressed with the T63A mutant (Fig. 2B). Thus, EF-Tu is a bona fide YabT substrate, and Thr-63 is the primary site of phosphorylation.
EF-Tu is an in vitro substrate of Ser/Thr phosphatases in Listeria monocytogenes and in B. subtilis (17, 18). In the latter study, EF-Tu phosphorylated in vitro by the PrkC kinase was dephosphorylated by its cognate phosphatase PrpC. We determined that EF-Tu phosphorylated by YabT (P∼EF-Tu) also was a substrate for PrpC by incubating P∼EF-Tu with PrpC and assaying phosphorylation by Pro-Q Diamond staining (Fig. 2C). In the presence of PrpC, only the lower EF-Tu band was detected, and phosphorylation levels were close to background (EF-Tu in the absence of YabT). Thus, in vitro, YabT and PrpC can reversibly regulate the phosphorylation of EF-Tu Thr-63.
We investigated if phosphorylation of EF-Tu Thr-63 is restricted to spore-forming bacteria. The Gram-positive bacterium Mycobacterium tuberculosis also enters a state of metabolic dormancy, and in vitro phosphorylation of M. tuberculosis EF-Tu affects its interaction with GTP (19). Consistently, incubation of M. tuberculosis PknA, a homolog of YabT, with EF-Tu resulted in the phosphorylation of the homologous Thr residue (Thr64) (Figs. S2B and S3). The homologous Thr residue also is phosphorylated in vivo in E. coli (20), further indicating that this modification is phylogenetically conserved.
Fig. S3.
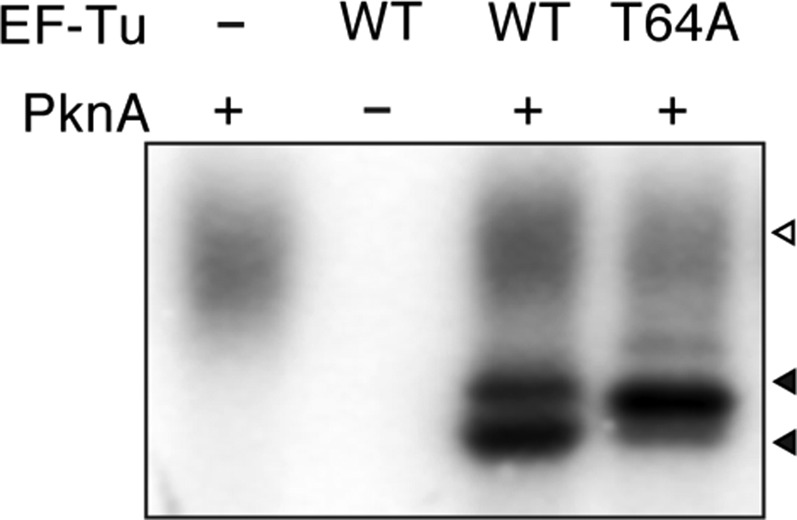
Phosphorylation of M. tuberculosis EF-Tu. Purified wild-type M. tuberculosis EF-Tu or the T64A point mutant (T64A) incubated with the Ser/Thr kinase PknA in the presence of [γ-32P]ATP. Phosphorylation was assayed by phosphorimaging. Bands containing EF-Tu and kinase are indicated by closed and open arrowheads, respectively.
Phosphorylation of EF-Tu Inhibits GTP Hydrolysis and Translation Elongation.
Crystal structures of Thermus aquaticus EF-Tu (21) and Thermus thermophilus EF-Tu (22) indicate that B. subtilis EF-Tu Thr-63 is located within the switch I region of the GTP-binding domain, in close proximity to the nucleotide (Fig. S2C). Consistently, substitution of the homologous Thr with an Ala residue results in a dramatic reduction in GTP hydrolysis activity of both T. thermophilus and E. coli EF-Tu (23, 24). Thus, phosphorylation could affect binding and/or nucleotide hydrolysis. To examine the effect of phosphorylation on GTP binding, we used the differential radial capillary action of ligand assay (DRaCALA), which is based on the ability of dry nitrocellulose to bind and sequester proteins at the application site (25). Protein–ligand complexes bind to nitrocellulose and are separated from unbound free ligand, which diffuses away. We incubated increasing concentrations of EF-Tu or P∼EF-Tu with radiolabeled GTP and spotted the reactions on nitrocellulose (Fig. S4). As expected, EF-Tu significantly retarded 32P-GTP diffusion compared with a GTP-binding–deficient (K138E) mutant of EF-Tu (26). Interestingly, incubation of 32P-GTP with P∼EF-Tu led to a slight, but not significant, decrease in 32P-GTP migration, indicating that EF-Tu phosphorylation did not substantially affect GTP binding in this assay.
Fig. S4.

EF-Tu GTP-binding kinetics. Increasing concentrations of wild-type EF-Tu, K138E GTP-binding–deficient mutant (K138E), and phosphorylated EF-Tu (P∼EF-Tu) were incubated with α-[32P]GTP. The reactions were spotted on nitrocellulose, and the diffusion patterns of the radiolabeled nucleotide were visualized by phosphorimaging and are shown in representative autoradiograms. The fraction of bound nucleotide was calculated as described in Materials and Methods and is plotted as a function of the EF-Tu concentration. Error bars indicate the SD for at least three spots.
We then determined whether EF-Tu phosphorylation affects GTP hydrolysis using thin-layer chromatography (TLC) to separate radiolabeled GDP and GTP (Fig. 3A). Consistent with previous observations (27), ribosomes are required to stimulate EF-Tu–mediated GTP hydrolysis, and virtually no hydrolysis of GTP was detected in their absence. EF-Tu hydrolyzed GTP in the presence of ribosomes in a time-dependent manner, and by 30 min ∼60% of the GTP was converted into GDP. The relatively modest levels of GTP hydrolysis observed likely are caused by the absence of mRNA and aa-tRNA in the reactions, which are required for full activation of EF-Tu GTPase activity (27). However, under the same conditions, GTP hydrolysis was reduced significantly in the presence of P∼EF-Tu to levels similar to those observed in the absence of ribosomes. In addition, dephosphorylation of P∼EF-Tu by PrpC restored GTP hydrolysis to wild-type levels. Taken together, these results indicate that phosphorylation of EF-Tu severely attenuates its GTPase activity.
Fig. 3.

Phosphorylation inhibits EF-Tu GTP hydrolysis activity and translation elongation in vitro. (A) EF-Tu GTP hydrolysis assay. Nonphosphorylated, phosphorylated, or dephosphorylated EF-Tu was incubated with [α-32P]GTP in the presence of 70S ribosomes. Samples were collected at different time points, and GTP was separated from GDP by TLC. GTP hydrolysis was measured by phosphorimager analysis and is shown below a representative autoradiogram. Control reactions in which [α-32P]GTP was incubated by itself or in the presence of either ribosomes or EF-Tu were carried out for 30 min. (B) Protein elongation assay. Nonphosphorylated or phosphorylated EF-Tu was incubated with EF-Ts and Lys-tRNALys or Phe-tRNAPhe in the presence of GTP. The resulting ternary complexes then were incubated with 70S ribosomes preloaded with a RNA message and f-[35S]Met-tRNAfMet in the presence of EF-G. Samples were collected at different time points, and TLC was used to separate the different peptide species. The percentage of radiolabeled Met incorporated into a dipeptide or a tripeptide was determined by phosphorimager analysis as above. Error bars indicate the SD for at least three independent experiments.
To characterize further the effect of this modification on EF-Tu activity, we performed in vitro translation reactions using a fully purified, recombinant in vitro translation system (Fig. 3B, Top) (28). GTP hydrolysis is key to the catalytic cycle of EF-Tu, and when EF-Tu is incubated with 70S ribosomes in the presence of a nonhydrolysable GTP analog GDPNP, elongation is blocked (9). Similarly, the inhibition of GTP hydrolysis by phosphorylation also should block elongation. As expected, a polypeptide is synthesized in the presence of EF-Tu, and after 5-min incubation ∼60% of the radiolabeled methionine was found as part of a tripeptide (Fig. 3B, Bottom). In contrast, P∼EF-Tu exhibited a significant decrease in the rate of elongation, particularly noticeable in the levels of tripeptide, which corresponded to only 20% of the radiolabeled Met in the same incubation period (Fig. 3B, Bottom). We note that, in contrast to the GTP hydrolysis assay reported in Fig. 3A, technical challenges associated with phosphoenriching P∼EF-Tu in the quantities required for this experiment prevented us from testing whether dephosphorylation of P∼EF-Tu by PrpC could reverse the inhibitory effect that phosphorylation of EF-Tu has on the rate of elongation. Nonetheless, the observation that dephosphorylation of P∼EF-Tu by PrpC reverses the inhibitory effect on GTP hydrolysis (Fig. 3A) strongly suggests that it likewise would reverse the inhibitory effect of phosphorylating EF-Tu on the rate of translation elongation.
Phosphorylation Stabilizes the Interaction Between EF-Tu and the Ribosome.
The antibiotic kirromycin prevents the conformational changes in EF-Tu that follow GTP hydrolysis, thereby blocking EF-Tu release from the ribosome (29). To determine if the inhibition of GTP hydrolysis by phosphorylation has a similar effect, we assayed the interaction between EF-Tu (nonphosphorylated, phosphorylated, or dephosphorylated) and 70S ribosomes in the presence of GTP. Free EF-Tu was separated from ribosome-bound EF-Tu by ultracentrifugation through a sucrose cushion, and both supernatant and pellet fractions were analyzed by Western blotting with antibodies against EF-Tu and the ribosomal protein S3. As expected, EF-Tu was found mostly in the supernatant fraction, independently of the presence of ribosomes (Fig. 4A). In the presence of P∼EF-Tu, however, we observed a significant enrichment of EF-Tu in the pellet fraction with the ribosomes, indicating that phosphorylation of EF-Tu stabilizes its interaction with the ribosome, presumably by inhibiting GTP hydrolysis (Fig. 4A). Dephosphorylated EF-Tu was found almost exclusively in the supernatant, as is consistent with this modification having an inhibitory effect on EF-Tu release.
Fig. 4.

EF-Tu–ribosome interaction is stabilized by phosphorylation in vitro. (A) EF-Tu/ribosome cosedimentation assay. Nonphosphorylated (WT), phosphorylated (P∼), or dephosphorylated (dePhos) EF-Tu was incubated with 70S ribosomes in the presence of GTP. A reaction carried out in the absence of ribosomes was used as control. The binding reactions were layered into a sucrose cushion, and the ribosome-bound EF-Tu was separated from the free EF-Tu by ultracentrifugation. The supernatant (S) and pellet (P) fractions were probed with antibodies to EF-Tu and the ribosomal protein S3, and the relative amount of EF-Tu in the fractions was determined by quantitative densitometry and is shown below a representative Western blot. (B) Phosphorylated EF-Tu was incubated with 70S ribosomes in the presence of GTP. The reaction was split, and PrpC phosphatase was added to one half. Both reactions were incubated further for 30 min at 37 °C. Reactions then were pelleted in a sucrose cushion and analyzed as above. Dashed lines separate noncontiguous lanes. Error bars indicate the SD for at least three independent experiments.
We next determined whether dephosphorylating P∼EF-Tu that was preincubated with ribosomes reversed this effect. P∼EF-Tu was incubated with ribosomes as before, but in this case the reaction was split, and one part of the reaction was incubated with PrpC (Fig. 4B). As in the previous assay, EF-Tu was significantly present in the pellet with ribosomes only when phosphorylated. In contrast, in the presence of the phosphatase, EF-Tu levels in the pellet fraction reverted to those observed in the nonphosphorylated EF-Tu. This observation is consistent with phosphatase being able to dephosphorylate P∼EF-Tu that presumably is bound to the ribosome.
Phosphorylation of EF-Tu Occurs During B. subtilis Sporulation.
The presence of YabT in the forespore (Fig. 1C) suggested that it could phosphorylate EF-Tu in this compartment. Because P∼EF-Tu is stably associated with ribosomes (Fig. 4A), we fractionated sporulating B. subtilis into mother cell and forespore fractions (as in Fig. 1B), generated lysates, and isolated ribosomes by ultracentrifugation through sucrose cushions. The amount of ribosome-bound EF-Tu then was determined by analyzing the pellets by Western blotting with an antibody against EF-Tu. There were equivalent amounts of EF-Tu in the mother cell fraction from both wild-type and ∆yabT strains (Fig. 5A, Left). In contrast, there was an approximately fourfold enrichment in the forespore fraction of the wild-type strain compared with a ∆yabT strain (Fig. 5A, Center). These results are consistent with the in vitro effect of EF-Tu phosphorylation and support the role of YabT as the kinase that phosphorylates EF-Tu in vivo in the developing spore. Next, we analyzed ribosomes isolated from mature spores. As in the forespore, we detected an enrichment of EF-Tu in the ribosome pellet only when the kinase was present (Fig. 5A, Right), confirming the role of this kinase during dormancy. Finally, we probed the ribosomal fraction isolated from forespores (Fig. 5A, Center) with a phospho-Thr antibody and observed that the antibody cross-reacted with EF-Tu from lysates derived from the wild-type strain but not from a strain lacking the YabT kinase (Fig. 5B). Thus, YabT is responsible for phosphorylating the EF-Tu molecules associated with ribosomes in the forespore.
Fig. 5.

Enrichment of EF-Tu in ribosomes isolated from spores. (A) Isolation of ribosomes from sporulating cells. Intact ribosomes from wild-type and ∆yabT strains were isolated from mother cells (Left), forespores (Center), and mature spores (Right) by ultracentrifugation through a sucrose cushion and were probed with an EF-Tu antibody. The relative amount of EF-Tu present in the ribosomal fraction (pellet) was determined by quantitative densitometry and is expressed as a fraction of WT EF-Tu. Quantification is shown below a representative Western blot. ***P < 0.001, unpaired t test with Welch’s correction. Error bars indicate the SD for at least three independent experiments. (B) Forespore fractions shown in A, Center were probed with a phospho-Thr antibody.
Phosphorylation of EF-Tu Inhibits Protein Synthesis.
EF-Tu in its active, GTP-bound state binds and delivers the aa-tRNA to the translating ribosome. Upon delivery, EF-Tu exits in its inactive, GDP-bound state, and the ribosome undergoes translocation, moving one codon forward and allowing a new elongation round to occur. Phosphorylated EF-Tu could have a dominant-negative effect on translation by halting ribosome progression. We tested this hypothesis using an in vitro transcription/translation system consisting of reconstituted E. coli components including EF-Tu. The yield of the reporter protein (CotE) is not affected significantly by the presence of nonphosphorylated B. subtilis EF-Tu (Fig. 6A). However, the addition of phosphorylated B. subtilis EF-Tu to the reaction reduced CotE levels threefold, suggesting that P∼EF-Tu inhibits translation even in the presence of the native E. coli EF-Tu. Unfortunately, technical challenges identical to those described for the translation elongation assay reported in Fig. 3B prevented us from testing directly whether dephosphorylation of P∼EF-Tu by PrpC could reverse the inhibitory effect that phosphorylation of EF-Tu has on the yield of CotE. Nonetheless, the observations that dephosphorylation of P∼EF-Tu reverses the inhibitory effect on GTP hydrolysis (Fig. 3A) and the stabilizing effect of EF-Tu phosphorylation on the interactions between EF-Tu and the ribosome (Fig. 4) strongly suggest that it likewise would reverse the inhibitory effects of phosphorylating EF-Tu on the yield of CotE.
Fig. 6.

EF-Tu phosphorylation inhibits protein translation. (A) In vitro synthesis of CotE-FLAG in the presence of B. subtilis EF-Tu. PURExpress reactions in the absence (−) and presence (+) of nonphosphorylated B. subtilis EF-Tu or phosphorylated B. subtilis EF-Tu (P∼). The amount of CotE-FLAG was determined by quantitative densitometry and is expressed relative to CotE-FLAG synthesized in the absence of B. subtilis EF-Tu. Quantification is shown below a representative Western blot. **P < 0.005, unpaired t test with Welch’s correction. Error bars indicate the SD for at least three independent experiments. (B) The expression of the Pspac-yfp reporter was induced in wild-type and ∆kinase (JDB3566) cells at hour 4 of sporulation. The magnitude of the YFP signal was measured across the spore 40 min following reporter induction. The distribution of fluorescence values observed in both strains is represented as the relative frequency of cells within each range of fluorescence values. Randomly selected fields were analyzed in at least three independent experiments. (C) yfp mRNA levels in forespores. Samples in B were processed as in Fig. 1B to isolate forespores, and total mRNA was extracted. yfp mRNA levels were determined by quantitative RT-PCR and are expressed relative to tuf. Error bars indicate the SD for at least three independent experiments. (D) Spores prepared from wild-type and ∆kinase (JDB3566) were incubated in 10 mM Tris⋅HCl, pH 7.5, for 48 h at room temperature and visualized by phase-contrast microscopy. Randomly selected fields were analyzed in at least three independent experiments. Error bars indicate the SD. Quantification is shown next to representative fields.
The phosphorylation of EF-Tu in the forespore of sporulating cells (Fig. 5) suggests that this in vitro inhibition of translation (Fig. 6A) could occur in vivo as well. We investigated this possibility by constructing a strain expressing a yfp reporter gene under the control of an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible promoter (Fig. 6B, Left). Thus, YFP levels following induction of the reporter should correspond to the ability of cells to translate YFP. Our protein expression data indicated that YabT is detected 2 h after sporulation initiation (T2; Fig. S1) and reaches maximum levels around 3–4 h (T3–T4) after sporulation initiation, so we induced YFP expression at T4 and 40 min later measured the fluorescence in the forespore. We observed higher fluorescent levels in the ∆kinase cells than in wild-type cells (Fig. 6B, Right), but the yfp mRNA levels in the two strains were not significantly different (Fig. 6C), suggesting that the rate of protein synthesis is increased in the absence of the kinase. These results are consistent with an inhibitory effect of EF-Tu phosphorylation on translation in the cell that is becoming dormant.
Finally, we asked if the absence of this regulatory mechanism had phenotypic consequences for the spore. Dormant spores contain significant amounts of mRNA molecules (30–32), suggesting that, in principle, spores are capable of undergoing translation. We therefore reasoned that spores would have an increased predisposition to translate these mRNAs and perhaps initiate germination spontaneously if protein synthesis was not inhibited. That is, the absence of this metabolic “brake” would make spores less stable. Approximately half the spores from the ∆kinase strain transitioned from phase-bright to phase-dark (a hallmark of germination) without being stimulated (Fig. 6D). Thus, consistent with the hypothesis, spores spontaneously initiate exit from a fully dormant state in the absence of the kinase.
Discussion
Here, we describe a novel mechanism for the regulation of protein synthesis involving phosphorylation of EF-Tu and demonstrate that it occurs during the process of sporulation when B. subtilis produces a metabolically dormant spore. EF-Tu supplies ribosomes with aa-tRNAs in each elongation cycle during growth (Fig. 7A), but EF-Tu is phosphorylated as the cell initiates dormancy (Fig. 7B). Because P∼EF-Tu is unable to hydrolyze GTP, it remains bound to translating ribosomes and stalls protein synthesis. When nutrients become available, the stalling could be reverted by the action of a phosphatase, which dephosphorylates EF-Tu, thereby releasing it from the ribosome and consequently allowing elongation to resume (Fig. 7C). This model predicts that a single phosphorylated EF-Tu would be sufficient to stall a ribosome on an mRNA. Thus, the ability of a single P∼EF-Tu to act as a dominant negative provides a mechanistic basis for the quick and robust regulation of the highly abundant components of the translation machinery observed in cells responding to nutrient limitation.
Fig. 7.

Regulation of protein synthesis by reversible phosphorylation of EF-Tu. (A) During growth, the translation GTPase EF-Tu cycles rapidly between its active (GTP-bound) and inactive (GDP-bound) state. In the active state, EF-Tu binds and delivers the aa-tRNA to the ribosome. If there is a codon/anticodon match, GTP hydrolysis is activated, and EF-Tu releases the aa-tRNA and exits the ribosome. (B) In cells entering dormancy (e.g., in the forespore of sporulating bacteria), an Ser/Thr kinase (STK) is expressed and phosphorylates EF-Tu. This phosphorylation inhibits GTP hydrolysis; as a result, EF-Tu remains stably bound to the ribosome, stalling protein translation. Ribosomes are kept in this quiescent state throughout dormancy. (C) In cells exiting dormancy, EF-Tu can be dephosphorylated by an Ser/Thr phosphatase (STP). GTP hydrolysis can occur, and protein synthesis can resume.
The low intrinsic GTPase activity of EF-Tu is stimulated by interaction with the ribosome. Specifically, a cognate codon–anticodon interaction in the ribosomal A site induces conformational changes in the ribosome and the aa-tRNA that are transmitted to the switch I and II regions of EF-Tu (3). The catalytic mechanism underlying the activation of the GTPase activity of EF-Tu is the subject of much recent debate (33–35). Nevertheless, it is clear that the absolutely conserved Thr residue (Fig. S2B and ref. 11) within switch I of the GTP-binding domain of EF-Tu plays a key role by mediating the coordination of the Mg2+ ion, the catalytic water molecule, and the γ-phosphate of GTP and is directly involved in the activation of the GTPase activity of EF-Tu by the ribosome (36). Our in vitro kinase assays and mass spectrometry data indicate that this Thr residue, Thr-63 in B. subtilis, is the primary site of phosphorylation of EF-Tu by the YabT kinase (Fig. 2 and Fig. S2). However, because this residue is essential for EF-Tu function (23, 24), it is difficult or impossible to interpret experiments involving mutations at this site unambiguously. Therefore we cannot irrefutably ascribe the reduced GTPase activity of the phosphorylated protein to phosphorylation of this Thr residue. Nonetheless, the reduced GTPase activity that we observe for EF-Tu that has been primarily phosphorylated at Thr-63 (Fig. 3A) is consistent with previous reports that replacing the homologous Thr residue with an Ala residue is sufficient to reduce the GTPase activity very significantly in both T. thermophilus and E. coli EF-Tu (23, 24).
EF-Tu species from the nonsporulating Gram-positive bacterium M. tuberculosis (Fig. S3) and the Gram-negative bacterium E. coli (20) also are phosphorylated on this Thr residue. Thus, the mechanism that we describe here is very likely to be phylogenetically conserved. In addition, because this Thr residue is absolutely conserved among translational GTPases (Fig. S2B and ref. 11), our findings may have general implications for the mechanism of GTP hydrolysis in this family of proteins (37).
The inhibition of GTP hydrolysis by phosphorylation prevents EF-Tu from dissociating from the ribosome (Figs. 4 and 5). This effect is reminiscent of nonhydrolysable GTP analogs, which stabilize EF-Tu association with the ribosome (9). A similar inhibitory mechanism also has been observed previously for the GTPase eIF5B, the eukaryotic homolog of the translation initiation factor IF2. A mutation of a single Thr residue, which blocks GTP hydrolysis, prevents release of eIF5B from the ribosome and thereby inhibits translation (38). Thus, this mechanism of inhibition appears to be conserved in both eukaryotic and prokaryotic translational GTPases.
An unresolved aspect of the phosphorylation of the Thr residue located within the nucleotide-binding pocket is how the kinase gains access to this non–surface-exposed residue. In the case of isocitrate dehydrogenase (IDH), which is regulated by direct phosphorylation of an Ser residue in the active site, the flexible nature of the IDH catalytic center likely facilitates this modification (39). Interestingly, the switch I region of EF-Tu assumes an alpha-helical conformation in the closed form of EF-Tu but changes to a beta-hairpin conformation in the open form of EF-Tu (40, 41). Consistent with this flexibility, this region of EF-Tu is disordered in most EF-Tu structures, whether alone or in complex with the ribosome (36). Thus, the dynamics of switch I may facilitate access to Thr-63 by both the kinase and the phosphatase.
Translation elongation in eukaryotes also is subject to regulatory phosphorylation. For example, the eukaryotic EF-Tu homolog eEF1α is phosphorylated in glial cells in response to glutamate, and this modification was correlated with a reduction in the rate of polypeptide chain elongation (42). Also, phosphorylation of Thr-56 inhibits the activity of the eukaryotic EF-G homolog eEF2 by reducing its affinity for the ribosome (43). The eEF2K kinase responsible for eEF2 phosphorylation is itself subject to control by the AMP-activated kinase (44), suggesting a direct link between nutritional availability and the inhibition of protein synthesis. This mechanism protects tumor cells during acute nutrient deprivation, suggesting that it is a key switch in the fate of these cells (45).
Many organisms respond to nutrient limitation by entering a metabolically dormant or quiescent state. This response allows them to persist through unfavorable conditions until the environment becomes conducive for growth, whereupon they exit dormancy and reinitiate growth. Although dormancy traditionally has been associated with a relatively small number of bacterial species that generate specialized forms such as spores or cysts, the majority of the microbial biomass is quiescent (46). The ability to exist in such a state becomes advantageous when cells are faced with unfavorable environmental conditions. For example, metabolically quiescent pathogens show high tolerance to chemotherapy and host defenses and thus commonly are associated with recurrent and chronic infections. However, the advantage provided by this strategy relies on the ability of dormant cells to reverse this state under the appropriate conditions; if they cannot, they will be outcompeted by growing cells.
Our understanding of the mechanisms underlying entry into and exit from dormancy is still incomplete. An appealing hypothesis is that these two processes are mechanistically related. Hence, when cells reduce protein synthesis in response to nutrient limitation, they need to ensure that they can reverse this state efficiently to respond to changes in the environment. The observations presented in this paper suggest that reversible phosphorylation of the translation factor EF-Tu could function as part of this mechanism. Thus, the phosphorylation of EF-Tu that results in a stable association with the ribosome and thereby inhibits translation also enables the ribosome to resume translation quickly by ensuring that EF-Tu already is present as part of what can be termed the “ribosome holoenzyme.”
Finally, the mechanism described here may have direct implications for the regulation of translation in eukaryotic organelles of bacterial origin. For example, mitochondrial protein translation is responsible for the synthesis of components of the respiratory chain complexes that are encoded in the mitochondrial genome and that are necessary for oxidative phosphorylation. The translation components of this organelle, including the elongation factor mtEF-Tu, are closely related to the bacteria homologs (47). Because the target of regulatory phosphorylation in B. subtilis EF-Tu (Thr-63) is conserved in both yeast and mammalian mtEF-Tu (Fig. S2B), a question to be addressed in future work is whether mtEF-Tu is subject to similar reversible regulation.
Materials and Methods
See SI Materials and Methods for descriptions of growth conditions and strain construction and detailed descriptions of experimental conditions and buffers. Strains, plasmids, and primers used in this study are listed in Tables S1–S3.
Table S1.
Bacterial strains
| Strain | Relevant genotype | Description | Source or reference |
| E. coli strains | |||
| DH5a | Invitrogen | ||
| BL21(DE3) | Stratagene | ||
| JDE1715 | pSFP65 amp | BL21(DE3) expressing PrpC-Strep | This study |
| JDE1741 | pSFP75 amp | BL21(DE3) expressing His-YabT (amino acids 1–277) | This study |
| JDE1788 | pSFP80 amp | BL21(DE3) expressing His-EF-Tu | This study |
| JDE1824 | pSFP80T63A amp | BL21(DE3) expressing His-EF-TuT63A | This study |
| JDE1840 | pSFP83 amp | BL21(DE3) expressing His-EF-TuT63A | This study |
| JDE1890 | pSFP89 amp | BL21(DE3) coexpressing YabT (amino acids 1–277) and His-EF-Tu | This study |
| JDE2386 | pSFP180 amp | BL21(DE3) expressing His-EF-Tu from M. tuberculosis | This study |
| JDE2424 | pSFP187 kan | BL21(DE3) expressing His-PknA (amino acids 1–338) | This study |
| JDE2517 | pSFP180T64A amp | BL21(DE3) expressing His-EF-Tu T64A from M. tuberculosis | This study |
| B. subtilis strains | |||
| 168 | TrpC2 | B. subtilis wild-type strain | (57) |
| JDB3485 | ∆cotE::tet ∆gerE::cat | Coat-deficient mutant | This study |
| JDB3516 | amyE::PspoIIQ-cfp spec | Reporter strain used in mother cell/endospore fractionation | This study |
| sacA::PspoIID-yfp kan | |||
| ∆cotE::tet ∆gerE::cat | |||
| JDB3557 | amyE::Phyperspanc-yfp cat | YFP reporter strain used to assess protein synthesis in sporulating cells | This study |
| JDB3566 | ∆prkC ∆yabT::erm ∆ybdM::spec | YFP reporter in a ∆kinase background used to assess protein synthesis in sporulating cells | This study |
| amyE::Phyperspanc-yfp cat | |||
| JDB3564 | ∆yabT::erm | ∆yabT in JDB3557 | This study |
| amyE::Phyperspanc-yfp cat | |||
| JDB3598 | ∆yabT::erm ∆cotE::tet ∆gerE::cat | ∆yabT in JDB3485 | This study |
| JDB3709 | ∆yabT::erm ∆cotE::tet ∆gerE::cat | ∆yabT in JDB3516 | This study |
| amyE::PspoIIQ-cfp spec | |||
| sacA::PspoIID-yfp kan | |||
| JDB3969 | ∆cotE::tet ∆gerE::cat | YabT-FLAG translation fusion expressed from yabT native locus | This study |
| amyE::PspoIIQ-cfp spec | |||
| yabT-FLAG erm | |||
| M. tuberculosis strain | |||
| Erdman | M. tuberculosis wild-type strain | (58) | |
Table S3.
Primers
| Oligo | Sequence | Characteristics | Origin |
| SFP237 | GGAATTCCATATGttgttaacagccttaaaaacag | NdeI; ATG | This study |
| SFP238 | TGAGGATCCTTATTTTTCGAACTGCGGGTGGCTCCAAGCGCCgcactgatcttcaccctc | BamHI; STOP Strep-tag | This study |
| SFP239 | GGAATTCCATatgatgaacgacgctttgacgag | NdeI | This study |
| SFP255 | GAAGATCTTTAaggctgttttctttgtgctgc | BglII; STOP | This study |
| SFP271 | GGAATTCCATatggctaaagaaaaattcg | NdeI | This study |
| SFP272 | GTCGGATccatactattactcagtg | BamHI | This study |
| SFP273 | cgagcgcggtatcGcaatctctactg | T63A | This study |
| SFP274 | cagtagagattgCgataccgcgctcg | T63A | This study |
| SFP286 | CATGCCATGGGCatgatgaacgacgctttgacgag | NcoI | This study |
| SFP296 | CTCGCTCGAGccatactattactcagtg | XhoI | This study |
| SFP311 | GGAAGATCTACATCATCATCATCATCATGGTatggctaaagaaaaattcg | BglII; His-tag | This study |
| SFP494 | ATTCCATATGgtggcgaaggcgaagttcc | NdeI | This study |
| SFP495 | CGCGGATCCcctacttgatgatcttgg | BamHI | This study |
| SFP506 | TGAGGATCCatgagcccccgagttggcgt | BamHI | This study |
| SFP507 | TAGTGTCGACTTAacgctgaccggacgaaaacgtgc | SalI; STOP | This study |
| SFP527 | cagcgcggtatcGccatcaacatcg | T64A | This study |
| SFP528 | cgatgttgatggCgataccgcgctg | T64A | This study |
| SFP656 | ggccctgtccttttaccaga | yfp qPCR F | This study |
| SFP657 | atgccatgtgtaatcccagca | yfp qPCR R | This study |
| SFP658 | gcgacatggtagacgacgaa | tuf qPCR F | This study |
| SFP659 | tcagcgtctccttcaagagc | tuf qPCR R | This study |
Homology sequences are shown in lowercase. Nonhomolog sequences are shown in uppercase and include restriction sites (in italic), STOP codons (underlined), tags (bold), and point mutations.
Table S2.
Plasmids
| Plasmid | Description | Source or reference |
| pET11a | Cloning vector, ampR | Novagen |
| pETPhos | Cloning vector, ampR | (59) |
| pETDuet-1 | Cloning vector, ampR | EMD Biosciences |
| pSMT3 | Cloning vector, kanR | (60) |
| pSFP65 | pET11a with prpC amplified from TrpC2 genomic DNA with oligos SFP237 and SFP238 (NdeI/BamHI) | This study |
| pSFP75 | pETPhos with yabT amplified from TrpC2 genomic DNA with oligos SFP239 and SFP255 (NdeI/BamHI) | This study |
| pSFP80 | pETPhos with tuf amplified from TrpC2 genomic DNA with oligos SFP271 and SFP272 (NdeI/BamHI) | This study |
| pSFP80T63A | pSFP80 with tufT63A generated by site-directed mutagenesis with oligos SFP273 and SFP274 | This study |
| pSFP83 | pETDuet-1 with yabT amplified from TrpC2 genomic DNA with oligos SFP286 and SFP255 (NcoI/BamHI) | This study |
| pSFP89 | pSFP83 with tuf amplified from TrpC2 genomic DNA with oligos SFP311 and SFP296 (BglII/XhoI) | This study |
| pSFP180 | pETPhos with tuf amplified from M. tuberculosis genomic DNA with oligos SFP494 and SFP495 (NdeI/BamHI) | This study |
| pSFP187 | pSMT3 with pknA amplified from M. tuberculosis genomic DNA with oligos SFP506 and SFP507 (BamHI/XhoI) | This study |
| pSFP180T64A | pSFP180 with tufT64A generated by site-directed mutagenesis with oligos SFP527 and SFP528 | This study |
Restriction sites used are shown in parentheses.
Fractionation of Mother Cells and Forespores.
Cells sporulated for 5 h were protoplasted by incubation with 1 mg/mL of lysozyme for 10 min at 37 °C in protoplasting buffer. Protoplasts were lysed by vigorous vortexing in lysis buffer, and supernatant (lysed mother cells) and pellet (forespores) were separated by centrifugation at 13,000 × g for 3 min at 4 °C. Mother cell lysate was cleared at 20,000 × g for 20 min at 4 °C. Forespores were isolated from the pellet and lysed in the presence 4 mg/mL of lysozyme for 5 min at 37 °C, processed in a FastPrep homogenizer, and incubated with 1% Nonidet P-40. Forespore lysate then was cleared by centrifugation at 20,000 × g for 20 min at 4 °C. The strains used in this experiment were coat-deficient to facilitate lysis and contained fluorescent reporters specifically expressed in the mother cell (PspoIID-yfp) or forespore (PspoIIQ-cfp) compartments to monitor fractionation by fluorescence microscopy.
EF-Tu Phosphorylation.
EF-Tu was phosphorylated in vitro by incubating kinases (0.5 µM) and substrates (2 µM) with 0.1 M unlabeled ATP and 1 µCi of [-32P]ATP in kinase buffer for 30 min at 37 °C. B. subtilis EF-Tu was phosphorylated in vivo by coexpression with a tagless YabT kinase domain from a pETDuet-derived plasmid in E. coli and was phosphoenriched using the Pro-Q Diamond phosphoprotein enrichment kit (Molecular Probes).
GTP Hydrolysis and Translation Elongation.
GTP hydrolysis and translation elongation assays were adapted from ref. 28.
Cosedimentation Assay.
The EF-Tu ribosome-binding assay was adapted from ref. 48.
In Vitro Translation of CotE-FLAG.
The PURExpress system (New England Biolabs) was used to assay translation of CotE-FLAG in the presence of B. subtilis EF-Tu.
Isolation of Intact Ribosomes from B. subtilis.
Ribosomes were isolated from lysed mother cells and forespores or mature spores by ultracentrifugation in sucrose cushions and were analyzed by immunoblotting.
Translation in Cells Entering Dormancy.
Protein synthesis in endospores was measured by fluorescence microscopy by inducing the expression of an yfp reporter 4 h after initiation of sporulation. Quantitative RT-PCR was used to quantify expression of yfp under the same conditions.
Spontaneous Germination.
Purified spores were incubated in 10 mM Tris⋅HCl, pH 7.5, for 48 h at room temperature.
SI Materials and Methods
Bacterial Strains, Plasmids, and General Growth Conditions.
Strains, plasmids, and primers used in this study are listed in Tables S1–S3. All plasmids were constructed in E. coli strain DH5. Overproduction of recombinant proteins was carried out in E. coli strain BL21 (DE3). E. coli strains were routinely cultured at 37 °C in LB medium supplemented with ampicillin (100 µg/mL) or kanamycin (35 µg/mL) and 1 mM IPTG when indicated. The B. subtilis strains used in this study were derivatives of 168 Marburg strain PB2. All general methods were carried out as described (49). B. subtilis strains were routinely grown at 37 °C in LB. Sporulation was carried out by resuspension in Difco sporulation medium (DSM) (50) or in Sterlini–Mandelstam medium (51) as indicated in the text. When appropriate, medium was supplemented with chloramphenicol (5 µg/mL), MLS (1 µg/mL erythromycin and 25 µg/mL lincomycin), kanamycin (10 µg/mL), spectinomycin (100 µg/mL), or tetracycline (10 µg/mL) and IPTG (1 mM). All chemicals were obtained from Sigma-Aldrich unless otherwise specified. Deletions on the Bacillus genome were produced by PCR using long, flanking homology regions (52).
Fractionation and Lysis of Mother Cells and Forespores.
Strains were grown in CH medium at 37 °C, and sporulation was induced by resuspension in A+B exhaustion medium (51). Five hours after the initiation of sporulation (T5), cells were collected, washed in TE buffer [10 mM Tris⋅HCl (pH 7.5) and 0.5 mM EDTA] and were resuspended in protoplasting buffer [100 mM Tris⋅HCl (pH 7.5), 20 mM MgCl2, 0.6 mM KCl, 1 mM NaCl, 0.9 mM Na2SO4, 18.7 mM NH4Cl, and 5% (vol/vol) sucrose]. Lysozyme was added to 1 mg/mL, and samples were incubated for 10 min at 37 °C. Protoplasts then were pelleted, flash frozen in liquid N2, and lysed by vigorous vortexing in lysis buffer [50 mM Tris⋅HCl (pH 7.5), 250 mM NaCl, and 1 mM EDTA] with 1 mM PMSF, followed by a 30-min incubation on ice with 3 mM MgCl2 and 10 µg/mL DNase. The resulting supernatant (lysed mother cells) and pellet (forespores) were separated by centrifugation at 13,000 × g for 3 min at 4 °C and were treated separately. Mother cell lysate was cleared by a second centrifugation step at 20,000 × g for 20 min at 4 °C. The pellet was processed further to isolate forespores from cell debris by three washes with lysis buffer. Forespores then were treated with 4 mg/mL of lysozyme in lysis buffer with 1 mM PMSF, 3 mM MgCl2, and 10 µg/mL DNase for 5 min at 37 °C, followed by 20 min on ice. Lysis was performed by mixing samples with 1 volume of 100 µM Zirconia/Silica beads (BioSpec) and processing three times in a FastPrep homogenizer (MP Biomedicals) at settings of 6.5 m/s, for 45 s each time, with 5 min of ice cooling between cycles. Finally, forespores were incubated with 1% Nonidet P-40 for 30 min at 4 °C and were cleared in two centrifugation steps: (i) 1 min at 20,000 × g at 4 °C to remove the beads, and (ii) 20 min at 20,000 × g at 4 °C. To facilitate forespore lysis, the strains used in this experiment contained ∆cotE ∆gerE mutations that lead to a defective spore coat and therefore are lysed more easily (53). In addition, these strains contained fluorescent reporters that are specifically expressed in mother cell (PspoIID-yfp) or forespore (PspoIIQ-cfp) compartments to monitor fractionation and lysis by fluorescence microscopy.
Protein Expression and Purification.
The kinase domain of YabT from B. subtilis (amino acids 1–277) was expressed as an N-terminal 6xHis-tag fusion and purified as described (54). The kinase domain of PknA from M. tuberculosis (amino acids 1–338) was expressed with an N-terminal 6xHis-SUMO tag and purified essentially as described (55), but using Ni2+-NTA affinity purification. PrpC from B. subtilis was expressed as a C-terminal Strep-tag II tag recombinant protein and was purified by Strep-Tactin affinity purification (IBA GmbH) in 100 mM Tris⋅HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 1 mM TCEP supplemented with 2.5 mM desthiobiotin. Purified PrpC then was dialyzed into 20 mM Tris⋅HCl (pH 7.5), 50 mM NaCl, 10 mM MnCl2, 10% (vol/vol) glycerol, and 1 mM TCEP. EF-Tu from B. subtilis and M. tuberculosis were expressed with a cleavable N-terminal 6xHis-tag and were purified as described (28). EF-Tu point mutants were generated by site-directed mutagenesis (see Table S3 for primers used).
In Vitro Kinase Assays.
Purified kinases (0.5 µM) and substrates (2 µM) were incubated with 0.1 M of unlabeled ATP and 1 µCi of [-32P]ATP (Perkin-Elmer) in kinase buffer [50 mM Tris⋅HCl (pH 7.5), 50 mM KCl, 10 mM MgCl2, 10 mM MnCl2, and 0.5 mM TCEP] for 30 min at 37 °C. Reactions were stopped by adding 5× SDS/PAGE loading buffer and were resolved by SDS/PAGE (12%). Gels were dried, exposed to a Storage Phospho Screen (Molecular Dynamics), and imaged using a Molecular Dynamics Typhoon Trio phosphorimager.
EF-Tu Phosphorylation in Vivo.
B. subtilis EF-Tu was phosphorylated in E. coli by coexpression with a tagless YabT kinase domain from a pETDuet-derived plasmid. Purified EF-Tu was resolved by SDS/PAGE (12%) and stained by Coomassie Brilliant Blue or transferred to an Immobilon-P PVDF membrane and stained with the phosphoprotein dye Pro-Q Diamond (Molecular Probes) following the manufacturer’s instructions.
Phosphoenrichment of EF-Tu.
All biochemical assays were performed with EF-Tu phosphorylated by YabT in E. coli and phosphoenriched using the Pro-Q Diamond phosphoprotein enrichment kit (Molecular Probes) following the manufacturer’s instructions.
Dephosphorylation of EF-Tu.
EF-Tu phosphorylated by YabT in vivo or in vitro was incubated with PrpC at a 1:3 molar ratio in the kinase buffer for 1 h at 37 °C, unless otherwise indicated. Dephosphorylation was assayed by Pro-Q Diamond following the manufacturer’s instructions.
GTP-Binding Assay.
The GTP-binding assay used was adapted from ref. 25 with the following modifications: Increasing concentrations of EF-Tu samples (0, 0.125, 0.25, 0.5, 1, 1.5, 2, 3, 5, and 10 µM) were incubated with 7 nM of [-32P]ATP (Perkin-Elmer) in Tris-Polymix buffer [50 mM Tris⋅acetate (pH 7.5) at 25 °C, 100 mM KCl, 5 mM NH4OAc, 0.5 mM Ca(OAc)2, 6 mM b-mercaptoethanol (BME), 5 mM putrescine–HCl, and 1 mM spermidine-free base] with 5 mM Mg(OAc)2 for 10 min at 37 °C. The reactions were spotted on nitrocellulose, which was dried and phosphorimaged as described above. The intensity of the spots was quantified using ImageJ software (56), and the GTP-bound fraction was calculated according to ref. 25.
GTP Hydrolysis Assay.
Ribosome-dependent multiple-turnover GTP hydrolysis was performed according to ref. 28, with modifications. EF-Tu samples at 0.8 µM were incubated with 0.4 µM 70S ribosomes in the presence of 1 mM unlabeled GTP and 10 nM of [-32P]GTP in Tris-polymix buffer with 5 mM Mg(OAc)2 in a final volume of 15 µL. Reactions were carried at 37 °C, and 2-mL samples were taken at 5, 15, and 30 min and quenched in the same volume of 100-mM EDTA, pH 9.5. Samples then were boiled for 1 min at 95 °C and centrifuged for 5 min at 18,000 × g. The supernatant (1.5 µL) was spotted onto a PEI-F cellulose plate (EMD Chemicals), and separation of GTP and GDP was done by TLC using 0.9 M guanidine HCl as solvent. The TLC plates were dried and phosphorimaged as above. Bands were quantified using ImageJ (56). Control reactions in the absence of 70S ribosomes, EF-Tu, or both were carried out for 30 min. The 70S ribosomes used in this and the following assays were purified from E. coli as described (28).
Elongation Assay.
B. subtilis EF-Tu samples were tested in a tripeptide synthesis assay using highly purified E. coli components as described (28). Elongation reactions were carried out in three main steps. First, an initiation reaction was started by incubating the initiation factors IF1-3 at 1.6 µM each, 1.5 µM of 70S ribosomes, and 1 mM GTP in Tris-polymix buffer with 3.5 mM Mg(OAc)2, for 10 min at 37 °C. Then, 2.1 µM of T4gp1-20 mRNA (mRNA variant encoding the first 20 amino acids of bacteriophage T4 gene product 32) was added, and the reaction was incubated for 10 min at 37 °C, followed by the addition of 0.5 µM of f-[35S]Met-tRNAfMet and another incubation for 10 min at 37 °C. Second, EF-Tu(GTP)aa-tRNA ternary complexes were prepared by incubating EF-Tu samples at 3.3 µM with 2.5 µM EF-Ts, 800 µM GTP, 2.5 mM phosphoenolpyruvate, and 0.001 U/µL pyruvate kinase in Tris-polymix buffer for 1 min at 37 °C; then 2.5 µM of Lys-tRNALys and Phe-tRNAPhe were added, and the mixture was incubated for an additional minute at 37 °C. Third, a mixture of 21 µM EF-G, 1 mM GTP, 3 µM phosphoenolpyruvate, and 0.001 U/µL pyruvate kinase was prepared in Tris-polymix buffer. Then the EF-G mixture and the initiation and ternary complex reactions were combined and incubated at 37°, allowing peptide synthesis to initiate. The final concentrations of components in the peptide synthesis reaction were as follows: 0.5 µM of initiation complexes, 0.25 µM of f-[35S]Met-tRNAfMet, 1.9 µM of mRNA, 1.5 µM of EF-Tu, 1.1 µM EF-Ts, 1.2 µM of each aa-tRNA, and 1.7 µM of EF-G. Samples (0.5 µL) taken at 15 s and 1 and 5 min were quenched with 1 µL of 500 mM KOH, and 0.5 µL of each quenched reaction was spotted onto cellulose TLC plates (EMD). Products were separated using electrophoretic TLC (eTLC) in pyridine acetate buffer (5% pyridine, 20% acetic acid, pH 2.8) for 30 min at 1,200 V. eTLC plates were air-dried and phosphorimaged, and bands were quantified using ImageJ (56) to determine the percentage of f-[35S]Met that is converted into a dipeptide and tripeptide.
Cosedimentation Assays.
The EF-Tu ribosome-binding assay used was adapted from ref. 48. EF-Tu samples at 0.05 µM were incubated with 0.025 µM of 70S ribosomes and 1 mM GTP in binding buffer [50 mM Tris⋅HCl (pH 7.5), 100 mM KCl, 5 mM NH4Cl2, 0.5 mM CaCl2, 10 mM MgCl2 and 6 mM BME] in a final volume of 25 µL for 3 min at 37 °C. Reactions were stopped by adding 25 µL of binding buffer (including 1 mM GTP) and by layering the reaction on 50 µL of 10% (vol/vol) sucrose cushions in buffer R [10 mM Tris⋅HCl (pH 7.2) at 4 °C, 30 mM NH4Cl, 10 mM MgCl2, 0.5 mM EDTA, and 6 mM BME] with 1 mM GTP. Reactions subsequently were centrifuged at 75,000 rpm for 5 min at 4 °C in a Beckman Optima TLX ultracentrifuge using a Beckman TLA 100 rotor. The supernatant (20 µL collected from the top) and pellet (resuspended in 20 µL) were analyzed by SDS/PAGE (12%) and immunoblotting with a rabbit-raised EF-Tu polyclonal antibody (1:3,000) or a monoclonal antibody against the ribosomal protein S3 (1:3,000; Developmental Studies Hybridoma Bank). A reaction in the absence of ribosomes was done as a control. The relative amounts of EF-Tu in the supernatant and pellet were determined by quantitative densitometry using ImageJ software (56).
Dephosphorylation of EF-Tu prebound to ribosomes was performed by first binding phosphorylated EF-Tu to 70S ribosomes as described above, then splitting the reaction and adding 0.25 µM of PrpC and 10 mM of MnCl2 to one part of the reaction. Both parts of the reaction were incubated further for 30 min at 37 °C. A reaction with unmodified EF-Tu was done as control.
In Vitro Translation of CotE-FLAG.
The PURExpress system (New England Biolabs) was used according to the manufacturer’s instructions to transcribe and translate a C-terminal FLAG-tagged CotE protein. The New England Biolabs control plasmid expressing CotE-FLAG under the T7 promoter was added to the complete reaction mixture (solution A + solution B), and 10-µL reactions were carried out for 5 min at 37 °C in the absence or presence of 0.92 µM of nonphosphorylated or phosphorylated B. subtilis EF-Tu. The reactions were stopped by the addition of SDS/PAGE loading buffer, resolved by SDS/PAGE (12%), and analyzed by immunoblotting with a FLAG-tag antibody (1:3,000; Sigma). The amount of CotE-FLAG was determined by quantitative densitometry using ImageJ (56) and is shown relative to the amount of CotE-FLAG synthesized in the absence of B. subtilis EF-Tu.
Fractionation and Lysis of Mother Cells and Forespores for Ribosome Isolation.
Mother cells and endospores were fractionated essentially as described above except that the lysis buffer was replaced by buffer R and lysis was carried out in the presence of 1× protease and phosphatase inhibitor mixtures (Pierce) and 40 U/mL of RNase inhibitor.
Lysis of Mature Spores for Ribosome Isolation.
Strains containing a ∆cotE ∆gerE double-knockout mutation were sporulated by exhaustion in DSM medium for 24 h at 37 °C (50). Cells then were collected, washed with distilled water, and incubated in decoating buffer (0.1 M NaCl, 0.1 M NaOH, 0.5% SDS, and 0.1 M DTT) for 1 h at 65 °C to remove the remaining layers of the coat. After extensive washes with distilled water, decoated spores were resuspended in buffer R with 1× protease and phosphatase inhibitor mixtures (Pierce) and 40 U/mL of RNase inhibitor and were lysed as described.
Isolation of Intact Ribosomes from B. subtilis.
Cell lysates from mother cells, endospores, or mature spores were produced as described above. Total cell lysates were quantified by the BCA protein assay kit (Pierce), and normalized samples were loaded in 1.5-mL sucrose cushions [buffer R with 37.7% (vol/vol) sucrose]. Ribosomes were isolated by ultracentrifugation at 85,000 rpm for 2 h at 4 °C in a Beckman Optima TLX ultracentrifuge using a Beckman TLA 100.3 rotor. The supernatant was discarded, and the pellet containing the ribosome complexes was resuspended overnight at 4 °C in 2× SDS/PAGE loading buffer. The EF-Tu levels in the lysate and sucrose pellet were analyzed by SDS/PAGE (12%) and immunoblotting with an EF-Tu polyclonal antibody (1:3,000) or a phospho-Thr antibody (1:1,000; Cell Signaling). The relative amount of EF-Tu present in the ribosomal fraction (pellet) was determined by quantitative densitometry using ImageJ software (56) and was expressed as a fraction of wild-type EF-Tu.
Induction of Protein Translation in Cells Entering Dormancy.
Strains harboring a fluorescent yfp reporter under the IPTG-inducible Phyper-spanc promoter were sporulated as described (51). At T4 (4 h after cells initiated sporulation), IPTG was added to the culture to a final concentration of 1 mM to induce yfp expression, and 40 min later cells were visualized by fluorescent microscopy. Fluorescence was measured across the spore as schematized in Fig. 6B.
yfp Expression in Forespores.
Total RNA was extracted from the same samples and in the same conditions used to determine induction of protein translation in cells entering dormancy (see above). Forespores were isolated as described above and were lysed by shaking with 100 µM Zirconia/Silica beads (BioSpec) in a FastPrep homogenizer (MP Biomedicals) at 6.5 m/s for 40 s. Total RNA was isolated using TRIzol (Invitrogen) and reverse transcribed by SuperScript III (Invitrogen) according to the manufacturer’s instructions. Quantitative RT-PCR was performed with SYBR Green (Quanta Biosciences) on a CFX96 (Bio-Rad).
Spontaneous Germination Assay.
Strains were sporulated by resuspension in DSM for 48 h at 37 °C (50). Remaining vegetative cells were removed by incubating the spore suspensions at 80 °C for 20 min, followed by three washes with 10 mM Tris⋅HCl, pH 7.5. Purified spores were stored in the same buffer and were visualized by phase-contrast microscopy after 48 h at room temperature.
Supplementary Material
Acknowledgments
We thank Rachel Fleisher and Wei Ning for technical assistance; Mary Ann Gawinowicz (Columbia University) for performing the mass spectrometry; and Max Gottesman for comments on the manuscript. This work was supported by the Fundação para a Ciência e Tecnologia, Portugal SFRH/BPD/65369/2009 (to S.F.F.P.); a Senior Scholar Award from the Ellison Medical Foundation (to J.D.); NIH Grants R01 GM83468 and R01 GM114213 (to J.D.) and R01 GM084288 (to R.G.); and a Research Initiatives for Science and Engineering award from Columbia University (to J.D. and R.L.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505297112/-/DCSupplemental.
References
- 1.Browne GJ, Proud CG. Regulation of peptide-chain elongation in mammalian cells. Eur J Biochem. 2002;269(22):5360–5368. doi: 10.1046/j.1432-1033.2002.03290.x. [DOI] [PubMed] [Google Scholar]
- 2.Macek B, Mijakovic I. Site-specific analysis of bacterial phosphoproteomes. Proteomics. 2011;11(15):3002–3011. doi: 10.1002/pmic.201100012. [DOI] [PubMed] [Google Scholar]
- 3.Kavaliauskas D, Nissen P, Knudsen CR. The busiest of all ribosomal assistants: Elongation factor Tu. Biochemistry. 2012;51(13):2642–2651. doi: 10.1021/bi300077s. [DOI] [PubMed] [Google Scholar]
- 4.Soares NC, Spät P, Krug K, Macek B. Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J Proteome Res. 2013;12(6):2611–2621. doi: 10.1021/pr3011843. [DOI] [PubMed] [Google Scholar]
- 5.Verstraeten N, Fauvart M, Versées W, Michiels J. The universally conserved prokaryotic GTPases. Microbiol Mol Biol Rev. 2011;75(3):507–542. doi: 10.1128/MMBR.00009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sehr P, et al. Glucosylation and ADP ribosylation of rho proteins: Effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37(15):5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- 7.Fentress SJ, et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe. 2010;8(6):484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinfeldt T, et al. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 2010;8(12):e1000576. doi: 10.1371/journal.pbio.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stark H, et al. Visualization of elongation factor Tu on the Escherichia coli ribosome. Nature. 1997;389(6649):403–406. doi: 10.1038/38770. [DOI] [PubMed] [Google Scholar]
- 10.Higgins D, Dworkin J. Recent progress in Bacillus subtilis sporulation. FEMS Microbiol Rev. 2012;36(1):131–148. doi: 10.1111/j.1574-6976.2011.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprinzl M, et al. Regulation of GTPases in the bacterial translation machinery. Biol Chem. 2000;381(5-6):367–375. doi: 10.1515/BC.2000.049. [DOI] [PubMed] [Google Scholar]
- 12.Nicolas P, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335(6072):1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 13.Wang ST, et al. The forespore line of gene expression in Bacillus subtilis. J Mol Biol. 2006;358(1):16–37. doi: 10.1016/j.jmb.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 14.Bidnenko V, et al. Bacillus subtilis serine/threonine protein kinase YabT is involved in spore development via phosphorylation of a bacterial recombinase. Mol Microbiol. 2013;88(5):921–935. doi: 10.1111/mmi.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lévine A, et al. Analysis of the dynamic Bacillus subtilis Ser/Thr/Tyr phosphoproteome implicated in a wide variety of cellular processes. Proteomics. 2006;6(7):2157–2173. doi: 10.1002/pmic.200500352. [DOI] [PubMed] [Google Scholar]
- 16.Eymann C, et al. Dynamics of protein phosphorylation on Ser/Thr/Tyr in Bacillus subtilis. Proteomics. 2007;7(19):3509–3526. doi: 10.1002/pmic.200700232. [DOI] [PubMed] [Google Scholar]
- 17.Absalon C, et al. CpgA, EF-Tu and the stressosome protein YezB are substrates of the Ser/Thr kinase/phosphatase couple, PrkC/PrpC, in Bacillus subtilis. Microbiology. 2009;155(Pt 3):932–943. doi: 10.1099/mic.0.022475-0. [DOI] [PubMed] [Google Scholar]
- 18.Archambaud C, Gouin E, Pizarro-Cerda J, Cossart P, Dussurget O. Translation elongation factor EF-Tu is a target for Stp, a serine-threonine phosphatase involved in virulence of Listeria monocytogenes. Mol Microbiol. 2005;56(2):383–396. doi: 10.1111/j.1365-2958.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- 19.Sajid A, et al. Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J Bacteriol. 2011;193(19):5347–5358. doi: 10.1128/JB.05469-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macek B, et al. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol Cell Proteomics. 2008;7(2):299–307. doi: 10.1074/mcp.M700311-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Kjeldgaard M, Nissen P, Thirup S, Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1(1):35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 22.Berchtold H, et al. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature. 1993;365(6442):126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- 23.Ahmadian MR, Kreutzer R, Blechschmidt B, Sprinzl M. Site-directed mutagenesis of Thermus thermophilus EF-Tu: The substitution of threonine-62 by serine or alanine. FEBS Lett. 1995;377(2):253–257. doi: 10.1016/0014-5793(95)01354-7. [DOI] [PubMed] [Google Scholar]
- 24.Krab IM, Parmeggiani A. Mutagenesis of three residues, isoleucine-60, threonine-61, and aspartic acid-80, implicated in the GTPase activity of Escherichia coli elongation factor Tu. Biochemistry. 1999;38(40):13035–13041. doi: 10.1021/bi9909748. [DOI] [PubMed] [Google Scholar]
- 25.Roelofs KG, Wang J, Sintim HO, Lee VT. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc Natl Acad Sci USA. 2011;108(37):15528–15533. doi: 10.1073/pnas.1018949108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang YW, Sanchez A, Miller DL. Mutagenesis of bacterial elongation factor Tu at lysine 136. A conserved amino acid in GTP regulatory proteins. J Biol Chem. 1989;264(14):8304–8309. [PubMed] [Google Scholar]
- 27.Maracci C, Peske F, Dannies E, Pohl C, Rodnina MV. Ribosome-induced tuning of GTP hydrolysis by a translational GTPase. Proc Natl Acad Sci USA. 2014;111(40):14418–14423. doi: 10.1073/pnas.1412676111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fei J, et al. A highly purified, fluorescently labeled in vitro translation system for single-molecule studies of protein synthesis. Methods Enzymol. 2010;472:221–259. doi: 10.1016/S0076-6879(10)72008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf H, Chinali G, Parmeggiani A. 1977. Mechanism of the inhibition of protein synthesis by kirromycin. Role of elongation factor Tu and ribosomes. Eur J Biochem 75(1):67–75.
- 30.Chambon P, Deutscher MP, Kornberg A. Biochemical studies of bacterial sporulation and germination. X. Ribosomes and nucleic acids of vegetative cells and spores of Bacillus megaterium. J Biol Chem. 1968;243(19):5110–5116. [PubMed] [Google Scholar]
- 31.Jeng YH, Doi RH. Messenger ribonucleic acid of dormant spores of Bacillus subtilis. J Bacteriol. 1974;119(2):514–521. doi: 10.1128/jb.119.2.514-521.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segev E, Smith Y, Ben-Yehuda S. RNA dynamics in aging bacterial spores. Cell. 2012;148(1-2):139–149. doi: 10.1016/j.cell.2011.11.059. [DOI] [PubMed] [Google Scholar]
- 33.Wallin G, Kamerlin SC, Aqvist J. Energetics of activation of GTP hydrolysis on the ribosome. Nat Commun. 2013;4:1733. doi: 10.1038/ncomms2741. [DOI] [PubMed] [Google Scholar]
- 34.Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330(6005):835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liljas A, Ehrenberg M, Åqvist J. Comment on “The mechanism for activation of GTP hydrolysis on the ribosome”. Science. 2011;333(6038):37–, author reply 37. doi: 10.1126/science.1202532. [DOI] [PubMed] [Google Scholar]
- 36.Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- 37.Carvalho AT, Szeler K, Vavitsas K, Qvist J, Kamerlin SC. Modeling the mechanisms of biological GTP hydrolysis. Arch Biochem Biophys 2015 doi: 10.1016/j.abb.2015.02.027. , in press. [DOI] [PubMed] [Google Scholar]
- 38.Shin BS, et al. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell. 2002;111(7):1015–1025. doi: 10.1016/s0092-8674(02)01171-6. [DOI] [PubMed] [Google Scholar]
- 39.Finer-Moore J, et al. Access to phosphorylation in isocitrate dehydrogenase may occur by domain shifting. Biochemistry. 1997;36(45):13890–13896. doi: 10.1021/bi9711691. [DOI] [PubMed] [Google Scholar]
- 40.Polekhina G, et al. Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure. 1996;4(10):1141–1151. doi: 10.1016/s0969-2126(96)00122-0. [DOI] [PubMed] [Google Scholar]
- 41.Abel K, Yoder MD, Hilgenfeld R, Jurnak F. An alpha to beta conformational switch in EF-Tu. Structure. 1996;4(10):1153–1159. doi: 10.1016/s0969-2126(96)00123-2. [DOI] [PubMed] [Google Scholar]
- 42.Barrera I, et al. Glutamate regulates eEF1A phosphorylation and ribosomal transit time in Bergmann glial cells. Neurochem Int. 2010;57(7):795–803. doi: 10.1016/j.neuint.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Carlberg U, Nilsson A, Nygard O. 1990. Functional properties of phosphorylated elongation factor 2. Eur J Biochem 191(3):639–645.
- 44.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279(13):12220–12231. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 45.Leprivier G, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153(5):1064–1079. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rittershaus ES, Baek SH, Sassetti CM. The normalcy of dormancy: Common themes in microbial quiescence. Cell Host Microbe. 2013;13(6):643–651. doi: 10.1016/j.chom.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: Processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreno JM, Kildsgaard J, Siwanowicz I, Mortensen KK, Sperling-Petersen HU. Binding of Escherichia coli initiation factor IF2 to 30S ribosomal subunits: A functional role for the N-terminus of the factor. Biochem Biophys Res Commun. 1998;252(2):465–471. doi: 10.1006/bbrc.1998.9664. [DOI] [PubMed] [Google Scholar]
- 49.Harwood CR, Cutting SM. Molecular Biological Methods for Bacillus. Wiley; New York: 1990. [Google Scholar]
- 50.Schaeffer P, Millet J, Aubert JP. Catabolic repression of bacterial sporulation. Proc Natl Acad Sci USA. 1965;54(3):704–711. doi: 10.1073/pnas.54.3.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sterlini JM, Mandelstam J. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem J. 1969;113(1):29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12(3):259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S, et al. Characterization of spores of Bacillus subtilis that lack most coat layers. J Bacteriol. 2008;190(20):6741–6748. doi: 10.1128/JB.00896-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mieczkowski C, Iavarone AT, Alber T. Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J. 2008;27(23):3186–3197. doi: 10.1038/emboj.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arora G, et al. Understanding the role of PknJ in Mycobacterium tuberculosis: Biochemical characterization and identification of novel substrate pyruvate kinase A. PLoS ONE. 2010;5(5):e10772. doi: 10.1371/journal.pone.0010772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spizizen J (1958) Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci USA 44(10):1072–1078. [DOI] [PMC free article] [PubMed]
- 58. Trudeau Institute (1972) TMC #107 in Trudeau mycobacterial culture collection, U.S.-Japan Cooperative Medical Science Program, Geographic Medical Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD. (Trudeau Institute, Saranac Lake, NY) p. 13.
- 59.Canova MJ, Kremer L, Molle V. pETPhos: A customized expression vector designed for further characterization of Ser/Thr/Tyr protein kinases and their substrates. Plasmid. 2008;60(2):149–153. doi: 10.1016/j.plasmid.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 60.O'Gaora P. Expression of genes in mycobacteria. In: Parish T, Stoker NG, editors. Mycobacteria Protocols. Humana; Totowa: 1998. pp. 261–273. [Google Scholar]