

Significance
Epidemiological studies show that individuals exposed to repeated stress, a major trigger of depression, increase their caffeine intake, which correlates inversely with the incidence of depression. However, the mechanism underlying this protective effect is unknown. We used an animal model of chronic unpredictable stress (CUS) to show that caffeine prevents the maladaptive changes caused by CUS in a manner mimicked by the selective blockade of adenosine A2A receptors (A2AR). CUS enhanced A2AR in synapses, and the selective elimination of neuronal A2AR abrogated CUS modifications. Moreover, A2AR blockade also afforded a therapeutic benefit, paving the way to consider A2AR blockers as a strategy to manage the negative impact of chronic stress on mood and memory.
Keywords: chronic stress, adenosine A2A receptor, caffeine, synaptic dysfunction, mood dysfunction
Abstract
The consumption of caffeine (an adenosine receptor antagonist) correlates inversely with depression and memory deterioration, and adenosine A2A receptor (A2AR) antagonists emerge as candidate therapeutic targets because they control aberrant synaptic plasticity and afford neuroprotection. Therefore we tested the ability of A2AR to control the behavioral, electrophysiological, and neurochemical modifications caused by chronic unpredictable stress (CUS), which alters hippocampal circuits, dampens mood and memory performance, and enhances susceptibility to depression. CUS for 3 wk in adult mice induced anxiogenic and helpless-like behavior and decreased memory performance. These behavioral changes were accompanied by synaptic alterations, typified by a decrease in synaptic plasticity and a reduced density of synaptic proteins (synaptosomal-associated protein 25, syntaxin, and vesicular glutamate transporter type 1), together with an increased density of A2AR in glutamatergic terminals in the hippocampus. Except for anxiety, for which results were mixed, CUS-induced behavioral and synaptic alterations were prevented by (i) caffeine (1 g/L in the drinking water, starting 3 wk before and continued throughout CUS); (ii) the selective A2AR antagonist KW6002 (3 mg/kg, p.o.); (iii) global A2AR deletion; and (iv) selective A2AR deletion in forebrain neurons. Notably, A2AR blockade was not only prophylactic but also therapeutically efficacious, because a 3-wk treatment with the A2AR antagonist SCH58261 (0.1 mg/kg, i.p.) reversed the mood and synaptic dysfunction caused by CUS. These results herald a key role for synaptic A2AR in the control of chronic stress-induced modifications and suggest A2AR as candidate targets to alleviate the consequences of chronic stress on brain function.
Repeated stress elicits neurochemical and morphological changes that negatively affect brain functioning (1, 2). Thus, repeated stress is a trigger or a risk factor for neuropsychiatric disorders, namely depression, in both humans and animal models (2, 3). Given the absence of effective therapeutic tools, novel strategies to manage the impact of chronic stress are needed, and analyzing particular lifestyles can provide important leads. Notably, caffeine consumption increases in stressful conditions (4) and correlates inversely with the incidence of depression (5, 6) and the risk of suicide (7, 8). However, the molecular targets operated by caffeine to afford these beneficial effects have not been defined.
Caffeine is the most widely consumed psychoactive drug. The only molecular targets for caffeine at nontoxic doses are the main adenosine receptors in the brain, namely the inhibitory A1 receptors (A1R) and the facilitatory A2A receptors (A2AR) (9). A2AR blockade affords robust protection against noxious brain conditions (10), an effect that might result from the ability of neuronal A2AR to control aberrant plasticity (11, 12) and synaptotoxicity (13–15) or from A2AR’s impact on astrocytes (16) or microglia (17). The protection provided by A2AR blockade prompts the hypothesis that A2AR antagonism may underlie the beneficial effects of caffeine on chronic stress, in accordance with the role of synaptic (18, 19) or glial dysfunction (20) in mood disorders. Thus, A2AR antagonists prolonged escape behavior in two screening tests for antidepressant activity (21–23) and prevented maternal separation-induced long-term cognitive impact (12). We combined pharmacological and tissue-selective A2AR transgenic mice (24, 25) to test if neuronal A2AR controlled the modifications caused by chronic unpredictable stress (CUS).
Results
Validation of the CUS Model.
Chronic stress is expected to cause decreased weight gain, increased corticosterone levels, helpless-like and anxiogenic-like behaviors, and decreased performance in memory tests (2, 3). Accordingly, compared with control (i.e., nonstressed) mice, the mice exposed to the 3-wk CUS protocol (Table S1) displayed (i) reduced weight gain (Fig. S1C); (ii) increased corticosterone plasma levels (Fig. S2A); (iii) no significant modification in the number of crossings in an open-field task (Fig. S3A), indicating the absence of altered locomotion that might have confounded analysis in other behavioral tasks; (iv) increased immobility time in the forced-swimming test (Fig. 1A) and in the tail-suspension test (Fig. 1B) indicative of a helpless-like state; (v) reduced sucrose preference (Fig. 1C), indicative of anhedonia; (vi) reduced time spent in the open arms of an elevated-plus maze (Fig. 1D), indicative of an anxiogenic state; (vii) decreased spatial reference memory, gauged by reduced time spent in the novel arm of a modified Y-maze test (Fig. 1E) and by a lower recognition index in an object-displacement test (Fig. 1F) (n = 36–39 mice per group).
Table S1.
Protocol of CUS showing the schedule of application of the different stressors during the 3-wk CUS period
| Week | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 |
| Week 1 | Damp bedding | Paired housing (1 h) | Restraint stress (2 h) | Cold bath (15 °C, 20 min) | Inescapable shock (0.7 mA) | Apparatus exposure, no footshock | Light/dark cycle inversion |
| Week 2 | Cage tilt (45°) | Food + water deprivation (24 h) | Empty bottle exposure (1 h) | Damp bedding | Paired housing (1 h) | Restraint stress (2 h) | Cold bath (15 °C, 20 min) |
| Week 3 | Inescapable shock (0.7 mA) | Apparatus exposure, no footshock | Food + water deprivation (24 h) | Empty bottle exposure (1 h) | Light/dark cycle inversion | Cage tilt (45°) | Restraint stress (2 h) |
Fig. S1.

Mice subjected to CUS do not change their liquid intake and have a decreased weight gain. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1). (A) Liquid intake is not significantly altered upon CUS or with caffeine consumption. (B) Caffeine reaches similar plasma concentrations in control mice and in mice subjected to CUS, as measured by HPLC. (C–F) Compared with control mice (ctr), CUS caused decreased weight gain, which was prevented by the regular consumption of either caffeine (1 g/L) [CUS F(1,70) = 17.95; caffeine F(1,70) = 0.16; interaction F(1,70) = 7.15] (C) or the selective A2AR antagonist KW6002 (3 mg/kg) [CUS F(1,33) = 6.64; KW6002 F(1,33) = 0.49; interaction F(1,33) = 2.27] (D) and which also was abrogated by the global deletion of A2AR (g-A2AR-KO) [CUS F(1,36) = 14.33; genotype F(1,36) = 11.69; interaction F(1,36) = 7.50] (E) or by the selective deletion of neuronal A2AR in fb-A2AR-KO mice [CUS F(1,32) = 7.92; genotype F(1,32) = 9.89; interaction F(1,32) = 11.84] (F). Data are shown as mean ± SEM. n = 19–27 mice per group in the liquid consumption experiment (A); n = 6 or 7 mice per group in the HPLC experiments (B); n = 7–19 mice per group in the weight-gain determinations (C–F). *P < 0.05 using two-way ANOVA followed by a Newman–Keuls post hoc test; ns, nonsignificant.
Fig. S2.

CUS increased the plasma levels of corticosterone, and caffeine, and A2AR blockade prevented this increase. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1). Three days later, animals were gently immobilized, and blood was collected from the tail vein. Animals were killed at 9:00 AM on the day of blood collection. Compared with control mice (ctr), CUS increased the plasma levels of corticosterone. This increase was prevented by the regular consumption of either caffeine (1 g/L) [CUS F(1,70) = 7.95; caffeine F(1,70) = 2.34; interaction F(1,70) = 4.36] (A) or the selective A2AR antagonist KW6002 (3 mg/kg) [CUS F(1,33) = 15.30; KW6002 F(1,33) = 6.78; interaction F(1,33) = 4.99] (B) and also was abrogated by the global deletion of A2AR (g-A2AR-KO) [CUS F(1,36) = 14.91; genotype F(1,36) = 1.50; interaction F(1,36) = 6.41] (C) or by the selective deletion of neuronal A2AR in fb-A2AR-KO mice [CUS F(1,32) = 16.79; genotype F(1,32) = 20.55; interaction F(1,32) = 4.56] (D). Data are shown as mean ± SEM of 7–19 mice per group. *P < 0.05 using two-way ANOVA followed by a Newman–Keuls post hoc test; ns, nonsignificant.
Fig. S3.

CUS does not significantly affect spontaneous locomotion. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were evaluated 24 h after the last stressor in an open-field arena. Both the number of crossings and the number of rearing events were globally similar in nonstressed control mice (ctr) and in mice subjected to CUS. Furthermore, neither the regular consumption of caffeine (1 g/L) (A) or of the selective A2AR antagonist KW6002 (3 mg/kg) (B) nor the global deletion of A2AR (g-A2AR-KO) (C) or the selective deletion of neuronal A2AR in fb-A2AR-KO mice (D) affected spontaneous locomotion. (E) Likewise, the pattern of spontaneous locomotion was similar in control mice and mice subjected to CUS that subsequently were treated for 3 wk with the A2AR antagonist SCH58261. Data are shown as mean ± SEM of 7–19 mice per group. *P < 0.05 using two-way ANOVA followed by a Newman–Keuls post hoc test; ns, nonsignificant.
Fig. 1.

Mice subjected to CUS display the expected features of depressed mice, which are largely prevented by the regular consumption of caffeine. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were evaluated behaviorally 24 h after the last stressor. Compared with nonstressed control mice (ctr, open bars), CUS-mice (checkered bars) displayed helpless-like behavior as evaluated by the forced-swimming (A) and tail-suspension (B) tests, anhedonia as evaluated by a sucrose preference test (C), anxiety-like behavior as evaluated by the elevated-plus maze test (D), and impaired memory performance as evaluated by a modified Y maze test (E) and an object-displacement test (Student’s t test comparing displaced vs. nondisplaced object) (F). After mice were killed, the CA3 area of hippocampi from CUS-subjected mice did not display overt neuronal damage, as gauged by the preservation of cresyl violet staining (G, Top Row) and lack of FluoroJade C staining (G, Middle Row) or microgliosis as evaluated by CD11b immunoreactivity (G, Bottom Row) but did display increased GFAP (H and I) and decreased synaptophysin immunoreactivity (J and K). Similar findings were obtained in the hippocampal CA1 area. Western blot analysis of whole hippocampal membranes confirmed the increase in GFAP density with CUS (L) and the decrease of synaptic markers, namely SNAP25 (M) and syntaxin (N). The administration of caffeine (1 g/L via the drinking water) to mice beginning 3 wk before CUS and continuing until mice were killed did not modify behavior or histology, except for increased anxiety in the elevated-plus maze (D), but did prevent all CUS-induced behavioral and morphological alterations. (Scale bars, 100 μm.) Data are shown as mean ± SEM; n = 9–19 mice per group in the behavioral assays (A–F); n = 4–7 mice per group in the morphological analysis; n = 5 or 6 mice per group in the neurochemical analysis. *P < 0.05 and #P < 0.05 using a two-way ANOVA followed by a Newman–Keuls post hoc test, except when stated otherwise. ns, not significant.
CUS caused no alteration in neuronal organization (determined by cresyl violet staining), no neuronal damage (shown by the lack of FluoroJadeC staining), and no microgliosis (shown by CD11 immunoreactivity) (Fig. 1G) but did cause increased GFAP immunoreactivity (Fig. 1 H and I), which was confirmed by Western blot analysis (Fig. 1L). Notably, CUS decreased the immunoreactivity of synaptophysin, a synaptic marker, mainly in the hippocampus (Fig. 1 J and K), in accordance with the critical impact of synaptic modifications in stress-induced behavioral alterations (26, 27). This putative hippocampal synaptotoxicity caused by CUS was confirmed further by Western blot analysis (Fig. 1 M and N). Overall, these behavioral, biochemical, and morphological alterations validate our CUS protocol.
Prophylactic Effect of Caffeine on Chronic Stress.
Caffeine consumption in drinking water (1 g/L for 6 wk; n = 18–19 mice per group) did not alter behavioral, morphological, or biochemical parameters in control mice (Fig. 1 and Figs. S1–S3), with the exception of the time spent in the open arms of the elevated-plus maze (Fig. 1D), which is suggestive of an anxiogenic effect. In contrast, caffeine prevented the CUS-induced reduction in weight gain (Fig. S1C) and the increase in corticosterone plasma levels (Fig. S2A) and blunted all other behavioral alterations caused by CUS, namely the increased immobility in the forced-swimming (Fig. 1A) and tail-suspension tests (Fig. 1B), the anhedonic-like behavior (Fig. 1C), the memory impairment in the modified Y maze (Fig. 1E) and object-displacement tests (Fig. 1F), as well as the loss of synaptic markers gauged by immunohistochemistry (Fig. 1 J and K) or Western blot (Fig. 1 M and N).
Alterations of Adenosine Receptors upon Chronic Stress.
We tested if caffeine’s ability to prevent CUS-induced changes while having little effect in nonstressed mice might be explained by CUS-induced changes in A1R and A2AR density. CUS decreased the A1R antagonist [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) binding, both in total membranes and in synaptosomal membranes but not in gliosomal [i.e., astrocytic (16)] membranes of the hippocampus (Fig. 2A). In contrast, CUS enhanced the A2AR antagonist [3H]5-amino-7-2-phenylethyl.-2-2-furyl-pyrazolo[4,3-ex-1,2,4-triazolo-1,5]pyrimidine ([3H]SCH58261) binding in the synaptosomal membranes (which was 60.1 ± 8.7% larger in CUS-treated mice than in control mice; n = 6, P < 0.05) but not in total or in gliosomal membranes of the hippocampus (Fig. 2B).
Fig. 2.

CUS alters the adenosine neuromodulation system in the hippocampus. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were killed for preparation of total, synaptosomal (i.e., from synapses), and gliosomal (i.e., from astrocytes) membranes from the hippocampus. (A) The binding density of A1R [evaluated with the A1R antagonist 3H-DPCPX (10 nM)] was decreased in total and synaptosomal membranes and was unaltered in gliosomal membranes of CUS-subjected mice (checkered bars) compared with nonstressed mice (control; open bars). (B) In contrast, there was a selective increase in the binding density of the A2AR antagonist 3H-SCH58261 in synaptosomal membranes from CUS-subjected mice, without changes in its binding density in total or gliosomal membranes. (C and D) Double-labeling immunocytochemical analysis of plated purified nerve terminals confirmed that A2AR are located mostly in glutamatergic (immunopositive for vGluT1) rather than GABAergic (immunopositive for vGAT) nerve terminals and showed that in CUS-exposed mice the number of glutamatergic terminals was enhanced selectively (C), rather than GABAergic terminals endowed with A2AR (D). Note that this immunocytochemistry approach allows only the relative colocalization of epitopes to be quantified, irrespective of their absolute staining, which varies among groups of plated nerve terminals. We previously validated the selectivity of 3H-SCH58261 and of the anti-A2AR antibodies used, which do not yield any signal in tissue from A2AR-KO mice (47). Data are shown as mean ± SEM of 5 or 6 mice per group; *P < 0.05 using an unpaired Student’s t test.
Double-labeling immunocytochemistry of individual nerve terminals further revealed that CUS selectively increased the number of hippocampal glutamatergic [vesicular glutamate transporter 1 (vGluT1)-positive] terminals (Fig. 2C) rather than the number of GABAergic [vesicular GABA transporter (vGAT)-positive] terminals (Fig. 2D) endowed with A2AR.
Neuronal A2AR Control the Burden of Chronic Stress.
In accordance with this CUS-induced enhancement of A2AR, the selective A2AR antagonist KW6002 (3 mg/kg) mimicked the protection observed with caffeine against CUS-induced alterations (n = 8–10 mice per group) (Fig. 3). Thus, although KW6002 was devoid of effects in nonstressed mice (Fig. 3), it prevented the CUS-induced reduction in weight gain (Fig. S1D), the increase in corticosterone plasma levels (Fig. S2B), and all other measured behavioral, morphological, and neurochemical alterations caused by CUS (Fig. 3 A–E and Fig. S4). KW6002 also prevented the CUS-induced decrease in the density of a presynaptic glutamatergic marker, vGluT1 (Fig. 3F); this finding is compatible with the synaptic atrophy known to occur in chronic stress (18, 19).
Fig. 3.

The pharmacological or genetic blockade of A2AR prevents CUS-induced behavioral, neurochemical, and electrophysiological alterations in the hippocampus. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) before behavioral evaluation 24 h after the last stressor. In CUS-subjected mice (checkered bars), as compared with vehicle-treated mice (A–F) (open bars) or wild-type mice (G–M) (open bars and symbols), the consumption of the A2AR antagonist KW6002 (3 mg/kg, through the drinking water, starting 3 d before CUS until mice were killed) (A–F), or the genetic elimination of A2AR in global A2AR-KO mice (G–M), prevented the CUS-induced helpless-like behavior evaluated in the forced-swimming test (A and G), the anxiety-like behavior evaluated in the elevated-plus maze test (B and H), the impaired memory performance evaluated in a modified Y maze test (C and I), and the decreases in synaptic markers such as syntaxin (D and J), SNAP-25 (E and K), and markers of glutamatergic terminals (vGluT1) (F) in hippocampal nerve terminals. Additionally, A2AR blockade with the antagonist SCH58261 (SCH, 50 nM) prevented the CUS-induced depression of LTP [triggered by a high-frequency stimulation train at time 0 in Schaffer fibers (collateral synapses of CA1 pyramidal cells)] of hippocampal slices from wild-type mice (L) and CUS failed to modify LTP in A2AR-KO mice (M) (Student’s t test). Data are shown as mean ± SEM; n = 8–10 mice per group in the behavioral assays (A–C and G–I); n = 5 or 6 mice per group in the neurochemical analyses (D–F, J, and K); and n = 5 or 6 mice per group in the electrophysiological analyses (L and M). *P < 0.05 using a two-way ANOVA followed by a Newman–Keuls post hoc test, except when stated otherwise; ns, nonsignificant.
Fig. S4.
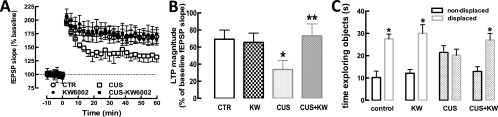
The pharmacological inhibition of A2AR blunts behavioral and electrophysiological alterations caused by CUS. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were evaluated 24 h after the last stressor in an open-field arena. Mice were killed 8 d after the end of the CUS period. (A and B) Synaptic plasticity (induced by a high-frequency stimulation train at time 0) recorded in Schaffer fibers (CA1 pyramid synapses) was decreased in hippocampal slices from mice subjected to CUS, and this decrease was prevented in mice consuming the A2AR antagonist KW6002 (KW) (3 mg⋅kg−1⋅d−1) from 3 d before the beginning of the protocol until the end of the CUS protocol. n = 5–6 mice per group. *P < 0.05 vs. control; **P < 0.05 vs. CUS; two-way ANOVA [CUS F(1,11) = 5.24; KW6002 F(1,11) = 8.71; interaction F(1,11) = 12.60] followed by a Newman–Keuls post hoc test. CTR, control. (C) In an object-displacement test, mice were exposed to two identical objects for 3 min and 90 min later were exposed for 3 min to the same objects, but with one of the objects in a different location. Control mice spent more time sniffing/exploring the displaced object than the nondisplaced object. This spatial recognition memory was blunted in mice subjected to CUS but was present in mice consuming KW6002. n = 8–10 mice per group. *P < 0.05 vs. time exploring the nondisplaced object; Student’s t test.
This prophylactic effect on pharmacological A2AR blockade was mimicked by global genetic deletion of A2AR (n = 9–10 mice per group). Thus, CUS did not change the behavior of global A2AR-KO mice in the forced-swimming (Fig. 3G), elevated-plus maze (Fig. 3H), or modified Y-maze tests (Fig. 3I) or the density of synaptic proteins such as syntaxin (Fig. 3J) or synaptosomal-associated protein 25 (SNAP-25) (Fig. 3K) in the hippocampus, in contrast to the alterations found in wild-type littermates (Fig. 3). In contrast to wild-type mice, A2AR-KO mice also failed to display behavioral alterations in the tail suspension, splash, social recognition, and object-displacement tests and in the levels of vGluT1 (Fig. S5). This CUS-induced loss of synaptic markers translated into a synaptic dysfunction, typified by a reduction of long-term potentiation (LTP) amplitude in CUS (16.2 ± 2.8% over baseline, n = 5) compared with nonstressed wild-type (control) mice (63.5 ± 6.3% over baseline, n = 5) (Fig. 3L). As previously reported (11), the acute blockade of A2AR with SCH58261 (50 nM) during LTP induction decreased LTP amplitude in wild-type control mice (by 40.7 ± 4.4% over baseline, n = 5; P < 0.05 vs. control) but increased LTP amplitude in wild-type mice subjected to CUS (by 36.5 ± 3.1% over baseline, n = 5 with SCH58261; P < 0.05 compared with 16.2 ± 2.8% without SCH58261) (Fig. 3L). Notably, LTP amplitude was unchanged by CUS in global A2AR-KO mice (Fig. 3M) and in mice drinking KW6002 (Fig. S4).
Fig. S5.
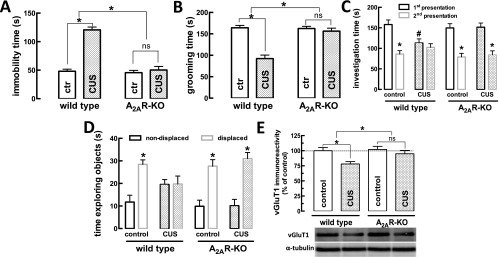
The genetic deletion of A2AR blunts behavioral and neurochemical alterations caused by CUS. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were evaluated in an open-field arena 24 h after the last stressor. Mice were killed 8 d after the end of the CUS period. In contrast with the effect of CUS (patterned bars) in wild-type mice, in global A2AR-KO mice CUS failed to trigger helpless-like behavior in the tail-suspension test (A) [CUS F(1,36) = 65.33; genotype F(1,36) = 58.16; interaction F(1,36) = 50.48], anhedonia-like behavior in the splash test (B) [CUS F(1,36) = 35.61; genotype F(1,36) = 22.87; interaction F(1,36) = 25.51], or impaired social interaction (C) [first presentation, CUS F(1,36) = 4.32; genotype F(1,36) = 2.09; interaction F(1,36) = 5.04; social interaction memory, Student’s t test comparing first vs. second presentation of the foreign mouse] or impaired short-term spatial memory in the object-displacement test (D) (n = 10; *P < 0.05 vs. time exploring the nondisplaced object, Student’s t test). (E) The density of the vGluT1, a marker of glutamatergic terminals, was reduced in the hippocampus of wild-type mice subjected to CUS. This reduction was not observed in A2AR-KO mice subjected to CUS. n = 5–6 mice per group. *P < 0.05, two-way ANOVA [CUS F(1,20) = 11.42; genotype F(1,20) = 7.53; interaction F(1,20) = 5.29] followed by a Newman–Keuls post hoc test. n = 5–6 mice per group; nonsignificant (ns) differences at P > 0.05, two-way ANOVA [CUS F(1,20) = 0.19; genotype F(1,20) = 0.01; interaction F(1,20) = 0.02] followed by a Newman–Keuls post hoc test.
Given the prominent changes in synaptic proteins and synaptic function paralleling the CUS-induced behavioral changes, together with the changes in the A2AR density mainly in glutamatergic terminals, we hypothesized that neuronal A2AR plays a pivotal role in the emergence of CUS-induced changes. Thus, we tested the impact of CUS on calcium/calmodulin-dependent protein kinase II-α (CaMKII-α) gene promoter-driven forebrain A2AR knockout (hereafter, fb-A2AR-KO) mice (n = 7–9 mice per group), in which we previously had shown neuronal A2AR to be eliminated in the forebrain and A2AR-mediated control of glutamatergic synapses to be blunted (24, 25). The behavior of fb-A2AR-KO mice was similar to that of wild-type mice under control conditions (Fig. 4); however, they did not display behavioral alterations in mood (Fig. 4 A and B) and memory tests (Fig. 4 D–F and Fig. S6) after CUS. CUS in these mice also failed to modify the density of synaptic proteins such as syntaxin (Fig. S6), SNAP-25 (Fig. 4G), vGluT1 (Fig. 4H), in contrast to the effect of CUS in wild-type mice (Fig. 4 G and H). Accordingly, hippocampal LTP amplitude in fb-A2AR-KO mice (50.9 ± 5.0% over baseline, n = 6) was not affected by either SCH58261 (41.4 ± 4.3% over baseline, n = 5) or CUS (Fig. 4I).
Fig. 4.

The selective deletion of neuronal A2AR prevents CUS-induced behavioral, neurochemical, and electrophysiological alterations in the hippocampus. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) before behavioral evaluation 24 h after the last stressor. In contrast with the impact of CUS in wild-type mice (checkered bars and filled symbols; control: open bars and symbols), in fb-A2AR-KO mice (in which neuronal A2AR is eliminated selectively in the forebrain and A2AR-mediated control of glutamatergic synapses is blunted) CUS failed to trigger helpless-like behavior in the forced-swimming test (A), anhedonia-like behavior in the splash test (B), anxiety-like behavior in the elevated-plus maze test (C), impaired social interaction and social interaction memory (Student’s t test comparing first vs. second presentation of the foreign mouse) (D), or impaired memory performance in a modified Y maze (E). (F) Additionally, in a spatial reference memory version of the Morris water maze test, both the acquisition (days 1–5) and the retention of the location of the hidden platform (day 6) were diminished by CUS in wild-type mice but not in fb-A2AR-KO mice. Also in contrast with the deleterious effects of CUS, which decreased the density of synaptic markers such as syntaxin (G) and vGluT1 (H) in hippocampal nerve terminals in wild-type mice, CUS did not affect the density of either of these synaptic markers (G and H) or the amplitude of LTP (I) in fbA2AR-KO mice. Data are shown as mean ± SEM; n = 7–9 mice per group in the behavioral assays (A–F); n = 5 mice per group in the neurochemical analysis (G and H) and in the electrophysiological analysis (I). *P < 0.05 using a two-way ANOVA followed by a Newman–Keuls post hoc test, with repeated measures for the Morris water maze test. ns, nonsignificant.
Fig. S6.

The selective deletion of neuronal A2AR blunts behavioral and neurochemical alterations caused by CUS. Male mice (10 wk old) were subjected to a 3-wk period of CUS (Table S1) and were evaluated 24 h after the last stressor in an open-field arena. Mice were killed 8 d after the end of the CUS period. In contrast with the impact of CUS in wild-type mice (patterned bars), in fb-A2AR-KO mice (in which neuronal A2AR is eliminated selectively in the forebrain and A2AR-mediated control of glutamatergic synapses is blunted), CUS failed to trigger helpless-like behavior in the tail-suspension test [CUS F(1,32) = 61.10; genotype F(1,32) = 86.09; interaction F(1,32) = 91.89] (A) or impaired short-term spatial memory in the object-displacement test (n = 9; *P < 0.05 vs. time exploring the nondisplaced object; Student’s t test) (B). (C) The density of the presynaptic marker syntaxin was reduced in the hippocampus of wild-type subjected to CUS. This reduction was not observed in fb-A2AR-KO mice subjected to CUS. n = 5; *P < 0.05, two-way ANOVA [CUS F(1,16) = 19.15; genotype F(1,16) = 10.13; interaction F(1,16) = 4.16] followed by a Newman–Keuls post hoc test.
A2AR manipulations did not affect ambulation in the open-field test (Fig. S3).
Therapeutic Effect of A2AR Antagonists in Reversing Chronic Stress-Induced Deficits.
The impact of CUS on the behavioral, neurochemical, and electrophysiological measures was stable for at least 3 wk after completion of the CUS protocol (Fig. 5), allowing us to test whether treatment with an A2AR antagonist after the CUS protocol reversed the CUS-induced modifications (n = 9–10 mice per group). After the 3-wk CUS protocol, mice showed increased immobility in the forced-swimming test (Fig. 5A), decreased time spent in the open arm of an elevated-plus maze (Fig. 5B), and decreased time spent in the novel arm of a modified Y maze (Fig. 5C). At 6 wk, these alterations were maintained in mice subjected to CUS that were injected daily with vehicle (Fig. 5 A–C). In contrast, in mice subjected to CUS and then treated with SCH58261 (0.1 mg⋅kg−1⋅d−1) for 3 wk, the altered behavior in the forced-swimming (Fig. 5A), elevated-plus maze (Fig. 5B), and modified Y maze (Fig. 5C) tests reverted to values similar to nonstressed mice. At the end of this 6-wk protocol, mice subjected to CUS displayed reduced hippocampal density of syntaxin (Fig. 5D), SNAP-25 (−21.15 ± 4.67% compared with nonstressed mice; n = 7–9), and vGluT1 (Fig. 5E) and lower amplitude of hippocampal LTP (Fig. 5F). Notably, in stressed mice that were treated for 3 wk with SCH58261 (Fig. 5 D–F), these changes also reverted to values similar to those in nonstressed mice. SCH58261 treatment per se did not cause behavioral or neurochemical alterations in nonstressed mice (Fig. 5).
Fig. 5.

Blockade of adenosine A2AR reverses CUS-induced alterations. Male mice (10 wk old) were behaviorally evaluated (baseline). Then mice were randomized into two groups. One group (checkered bars) was subjected to a 3-wk period of CUS (Table S1). The mice in the control group (ctr) were handled daily. All mice were behaviorally evaluated after 3 wk. Finally, half of the mice in each group were i.p. injected daily with saline (bars filled with white), and the other half were injected with the A2AR antagonist SCH58261 (SCH, 0.1 mg⋅kg−1⋅d−1, gray-outlined bars). All mice were behaviorally evaluated again at 6 wk and then were killed for neurochemical and electrophysiological analysis. Compared with nonstressed (control) mice (noncheckered bars), the mice subjected to CUS (checkered bars) displayed increased immobility in the forced-swimming test [F(2,36) = 182.0, P < 0.0001] (A), decreased time in the open arms of the elevated-plus maze test [F(1,36) = 77.14, P < 0.0001] (B), and decreased time spent in the novel arm of a modified Y maze test [F(1,36) = 77.14, P < 0.0001] (C), both at the end of the CUS protocol (3 wk) and 3 wk later (6 wk). As shown in A–C, SCH58261 treatment did not affect the behavior of control mice (noncheckered bars) but reversed the CUS-induced alterations in helpless behavior [F(1,36) = 77.14, P < 0.0001] (A), anxiety [F(2,36) = 21.35, P < 0.0001] (B), and spatial reference memory [F(2,36) = 14.88, P = 0.0005] (C) to the level of control (noncheckered bars). SCH58261 treatment also reversed the CUS-induced reduction in the density of the synaptic markers syntaxin [CUS F(1,16) = 4.62; SCH58261 F(1,16) = 10.54; interaction F(1,16) = 18.30] (D) and vGluT1 [CUS F(1,16) = 11.60; SCH58261 F(1,16) = 6.25; interaction F(1,16) = 4.54] (E) in hippocampal nerve terminal membranes and reversed the CUS-induced decrease in the amplitude of LTP in hippocampal slices [CUS F(1,16) = 49.89; SCH58261 F(1,16) = 16.74; interaction F(1,16) = 29.56] (F). Data are shown as mean ± SEM. n = 9 or 10 mice per group in the behavioral assays (A–C); n = 5 mice per group in the neurochemical analysis (D and E) and in the electrophysiological analysis (F). *P < 0.05, #P < 0.05 using a repeated ANOVA followed by a Newman–Keuls post hoc test.
Discussion
This study shows that caffeine prevents the behavioral, neurochemical, and electrophysiological alterations caused by chronic stress in a manner mimicked by the pharmacological or genetic blockade of adenosine A2AR. We also show that neuronal A2AR plays a critical role in controlling the burden of chronic stress in adult mice and also has the ability to reverse the maladaptive changes caused by repeated stress. These findings in a model of CUS that caused a constellation of behavioral changes comparable to these observed in stressed or depressed individuals (28, 29) confirm the previously reported inverse relationship between caffeine consumption and the incidence of depression (5, 6) or suicide (7, 8) and suggest that A2AR has a pivotal role in controlling mood disorders. However, the role of A2AR in anxiety still remains unclear (30), as shown by the inconsistent effects of the different A2AR manipulations.
A major advance provided by this study is the finding that neuronal A2AR controls behavioral dysfunction upon CUS, as demonstrated by the elimination of CUS-induced changes in fb-A2AR-KO mice (24, 25). This result excludes a major participation by peripheral adenosine receptors (22) and does not support a prominent role for glial A2AR in controlling CUS-induced alterations, as occurs in animal models of Parkinson’s disease (25) or upon treatment with lipopolysaccharide (17). However, the data are compatible with A2AR having a key role in glutamatergic terminals defining the synaptic dysfunction underlying the behavioral alterations associated with repeated stress (26, 27). In accordance with the synaptic atrophy observed in different models of chronic stress (18, 19), CUS led to alterations in synaptic markers and synaptic function in the hippocampus, a brain region known to play a pivotal role in the maladaptive changes upon chronic stress (2, 3). Furthermore, A2AR are located most abundantly in hippocampal nerve endings, and A2AR are selectively engaged to control synaptic plasticity (11). Additionally, noxious brain conditions trigger A2AR up-regulation, which is most evident in synapses (14, 31, 32), particularly in glutamatergic synapses (11, 31), as now shown also under CUS. This A2AR up-regulation is accompanied by an A2AR gain of function (reviewed in ref. 33) that leads to synaptic dysfunction, as demonstrated by the ability of A2AR antagonists to prevent synaptic plasticity dysfunction with aging (11) and maternal separation (12) and by the loss of synaptic markers in different noxious brain conditions (14, 15, 31), namely upon repeated restraint stress (32). How enhanced A2AR function triggers synaptic dysfunction remains to be determined, given our current ignorance of the transducing systems operated by these pleiotropic A2AR (34, 35). This unanswered question has significant importance, because the mechanism seems to be common to the A2AR-mediated control of memory and of mood dysfunction.
Overall, this robust ability of A2AR to control CUS-induced alterations provides a rationale explaining the ability of caffeine to attenuate the burden of CUS. The prevention of different alterations caused by repeated stress by regular (not acute) caffeine consumption had been noted previously by others (36–38) and is in tight agreement with the inverse correlation between caffeine intake and the incidence of depression (5, 6). Given that caffeine intake increases in stressed individuals (4), it is tempting to speculate that this increased intake may be a prophylactic antistress measure to normalize mood-related behavioral changes by normalizing synaptic functions via A2AR blockade. The present results also show that the selective antagonism of A2AR offers a therapeutic benefit in stressed rats similar to the reversion of memory impairment in aged rodents (39). Thus we prompt the suggestion that the up-regulated A2AR might be an effective target to correct brain disorders that involve a synaptic dysfunction, as now observed for the maladaptive responses to chronic stress.
Materials and Methods
Animals.
Male C57BL/6 mice (10–12 wk old) were obtained from Charles River. Global A2AR-KO and fb-A2AR-KO mice, both in the C57BL/6 background (24), were raised based on mating of heterozygotes. Mice were handled according to European Union guidelines, as approved by the CNC Ethical Committee for Animal Research (ORBEA-78/2013).
CUS and Administration of Drugs.
Prophylactic studies with caffeine or KW6002 were done using previously validated doses (14, 40). Mice were divided into two groups: a drug-free group drinking water or vehicle and a treated group drinking either 1 g/L caffeine (Sigma), starting 3 wk before CUS, or KW6002 (istradefylline; 3 mg/kg, dissolved in 0.5% nitrocellulose), synthesized as described previously (41), starting 4 d before CUS. At the end of treatments, blood samples were collected at 9:00 AM, and the plasma concentrations of caffeine and corticosterone were determined by HPLC (31) and RIA (MP Biomedicals), respectively. Half the animals in each group were subjected to a CUS protocol (42) for 21 d (Table S1). Control (i.e., nonstressed) and stressed mice were housed individually and were submitted to behavioral tests 24 h after the last stressor.
In therapeutic studies, mice first were characterized behaviorally, then were subjected to the 3-wk CUS protocol, and were characterized behaviorally again. They then were randomized into two groups. One group was treated daily with saline, and the other group was treated with a validated selective dose of the A2AR antagonist SCH58261 (0.1 mg/kg, i.p.) for 3 wk (15) before the final behavioral characterization. Then all mice were killed by decapitation after halothane-induced deep anesthesia.
Behavioral Tests.
Locomotor and exploratory behavior was monitored using an open-field apparatus (14). Evaluation of anxiety was carried out using the elevated-plus maze, and the helpless-like behavior was evaluated in the tail-suspension and forced-swimming tests (43). Anhedonic-like behavior was evaluated with the sucrose (1.2%) preference test (44) by measuring the intake of sucrose solution versus water intake at the end of a 16-h test period (12-h dark phase plus 4-h light phase) or with the splash test by measuring grooming bouts (head washing and nose/face and body grooming) over 5 min after a 10% sucrose solution was squirted on the dorsal coat (45). Social recognition memory was evaluated as previously described (39), and spatial memory was evaluated using an object-displacement test (40), a reference memory version of the Morris water maze (46), and a two-trials Y-maze test in which mice first explored the maze for 8 min while one arm was blocked and explored it again 2 h later for 8 min with all three arms accessible (14). Behavioral experiments were conducted between 10:00 AM and 5:00 PM in a sound-attenuated room under low-intensity light and were monitored by two researchers who were unaware of phenotypes or drug treatments.
Western Blot Analysis in Hippocampal Membranes.
Western blot analyses of total membranes [to evaluate the astrocytic marker GFAP (1:10,000; Sigma)] or Percoll-purified synaptosomal membranes [to probe synaptic markers, using antibodies against syntaxin (1:5,000; Sigma), SNAP-25 (1:5,000; Sigma), and vGluT1 (1:5,000; Chemicon)] were performed as described previously (14, 31). Membranes then were reprobed with α-tubulin (1:10,000; Sigma) as a loading control.
Membrane-Binding Assays.
The density of A1R and A2AR was estimated by radioligand-binding assays using supramaximal concentrations of the A1R antagonist [3H]DPCPX (10 nM; DuPont NEN) or the A2AR antagonist [3H]SCH58261 (6 nM; provided by E. Ongini, Schering-Plough, Milan, Italy), as described previously (31, 32). Specific binding was determined by the subtraction of nonspecific binding measured in the presence of 3 μM XAC (Tocris), a mixed A1R/A2R antagonist.
Immunocytochemistry of Purified Nerve Terminals.
Immunocytochemistry of purified nerve terminals was performed as previously described (31) by double labeling with goat anti-A2AR (1:200; Santa Cruz Biotechnology) together with either guinea pig anti-vGAT (1:1,000; Calbiochem) or guinea pig anti-vGluT1 (1:1,000; Chemicon) followed by incubation with Alexa Fluor-labeled secondary antibodies (1:2,000; Molecular Probes). The preparations were examined under a Zeiss Z2 microscope, and each coverslip was analyzed by counting an average of 500 elements (31).
Mouse Brain Histochemistry.
Neuronal morphology was assessed using cresyl violet staining of Nissl bodies, and neuronal degeneration was evaluated by FluoroJade-C staining in 20-μm brain sections (14, 32). Immunohistochemical analysis of microglia (CD11-b staining; 1:500; Serotec), astrocytes (GFAP staining; 1:1,000; Sigma), or nerve terminals (synaptophysin staining; 1:200; Sigma) was done as previously described (17, 31).
Electrophysiological Analysis of Synaptic Plasticity.
Electrophysiological recordings of synaptic plasticity were performed in 400-μm hippocampal slices, as described previously (11). Briefly, a bipolar electrode was placed onto Schaffer fibers, and the evoked field excitatory postsynaptic potentials (fEPSP) were recorded through an extracellular microelectrode (4 M NaCl; 1–2 MΩ resistance) placed in the CA1 stratum radiatum. LTP was induced with a high-frequency stimulation train (100 pulses at 100 Hz, over a 0.066-Hz basal stimulation) and was quantified as the percentage change between the fEPSP slopes 60 min after and 10 min before the train.
Statistics.
Results are given as mean ± SEM of n animals, and significance was considered at P < 0.05 using Student’s t test for comparison between two groups or two-way ANOVA followed by a Newman–Keuls post hoc test for comparison of multiple groups (Table S2).
Table S2.
Statistical values resulting from the two-way ANOVA analysis of the different groups of mice subjected to the 3-wk CUS protocol and treated with different drugs or having different genotypes
| Group | Assay | Effect of CUS | Effect of drug or genotype | Interaction |
| Caffeine | Weight gain | F(1,70) = 17.95; P < 0.0001 | F(1,70) = 0.1585; P = 0.6918 | F(1,70) = 7.145; P = 0.0093 |
| Cortisol | F(1,70) = 7.952; P = 0062 | F(1,70) = 2.336; P = 0.1309 | F(1,70) = 4.361; P = 0.0404 | |
| Forced swim | F(1,43) = 12.25; P = 0.0011 | F(1,43) = 7.098; P = 0.0108 | F(1,43) = 9.033; P = 0.0044 | |
| Tail suspension | F(1,42) = 45.40; P < 0.0001 | F(1,42) = 26.25; P < 0.0001 | F(1,42) = 35.18; P < 0.0001 | |
| Sucrose preference | F(1,42) = 8.368; P = 0060 | F(1,42) = 9.311; P = 0.039 | F(1,42) = 4.552; P = 0.0039 | |
| Elevated-plus maze | F(1,41) = 13.28; P = 0007 | F(1,41) = 2.183; P = 0.1472 | F(1,41) = 15.77; P = 0.0003 | |
| Modified Y maze | F(1,41) = 8.268; P = 0091 | F(1,41) = 7.490; P = 0.0091 | F(1,41) = 15.01; P = 0.0004 | |
| GFAP IHC | F(1,21) = 4.866; P = 0.0387 | F(1,21) = 20.81; P = 0.0002 | F(1,21) = 9.942; P = 0.0048 | |
| Synaptophysin IHC | F(1,21) = 9.531; P = 0.0056 | F(1,21) = 14.76; P = 0.0009 | F(1,21) = 6.814; P = 0.0163 | |
| GFAP | F(1,19) = 5.168; P = 0.0348 | F(1,19) = 0.8074; P = 0.3801 | F(1,19) = 11.34; P = 0.0032 | |
| Syntaxin | F(1,19) = 5.183; P = 0.0346 | F(1,19) = 1.041; P = 0.3204 | F(1,19) = 4.609; P = 0.0449 | |
| SNAP-25 | F(1,19) = 6.754; P = 0.0176 | F(1,19) = 2.212; P = 0.1534 | F(1,19) = 10.79; P = 0.0039 | |
| KW6002 | Weight gain | F(1,33) = 6.635; P = 0.0147 | F(1,33) = 0.4935; P = 0.4873 | F(1,33) = 2.274; P = 0.1411 |
| Cortisol | F(1,33) = 15.30; P = 0.0004 | F(1,33) = 6.784; P = 0.0137 | F(1,33) = 4.987; P = 0.0324 | |
| Forced swim | F(1,33) = 11.50; P = 0.0018 | F(1,33) = 22.08; P < 0.0001 | F(1,33) = 14.65; P = 0.0005 | |
| Tail suspension | F(1,33) = 108.9; P < 0.0001 | F(1,33) = 79.88; P < 0.0001 | F(1,33) = 66.12; P < 0.0001 | |
| Elevated-plus maze | F(1,33) = 17.65; P = 0.0002 | F(1,33) = 24.00; P < 0.0001 | F(1,33) = 13.50; P = 0.0008 | |
| Modified Y maze | F(1,33) = 9.987; P = 0.0034 | F(1,33) = 2.501; P = 0.1233 | F(1,33) = 7.112; P = 0.0118 | |
| Syntaxin | F(1,18) = 12.48; P = 0.0024 | F(1,18) = 0.9993; P = 0.3307 | F(1,18) = 12.76; P = 0.0022 | |
| SNAP-25 | F(1,19) = 13.50; P = 0.0016 | F(1,19) = 6.006; P = 0.0241 | F(1,19) = 6.541; P = 0.0192 | |
| vGluT1 | F(1,20) = 12.15; P = 0.0023 | F(1,20) = 0.4262; P = 0.5213 | F(1,20) = 4.822; P = 0.0401 | |
| LTP amplitude | F(1,11) = 5.241; P = 0.0428 | F(1,11) = 8.712; P = 0.0132 | F(1,11) = 12.60; P = 0.0046 | |
| Global A2AR-KO | Weight gain | F(1,36) = 14.33; P = 0.0006 | F(1,36) = 11.69; P = 0.0016 | F(1,36) = 7.495; P = 0.0096 |
| Cortisol | F(1,36) = 14.91; P = 0.0005 | F(1,36) = 1.497; P = 0.2291 | F((1,36) = 6.413; P = 0.0158 | |
| Forced swim | F(1,36) = 9.029; P = 0.0048 | F(1,36) = 14.98; P = 0.0004 | F(1,36) = 32.46; P < 0.0001 | |
| Tail suspension | F(1,36) = 65.33; P < 0.0001 | F(1,36) = 58.16; P < 0.0001 | F(1,36) = 50.48; P < 0.0001 | |
| Splash | F(1,36) = 35.61; P < 0.0001 | F(1,36) = 22.87; P < 0.0001 | F(1,36) = 25.51; P < 0.0001 | |
| Elevated-plus maze | F(1,36) = 18.89; P = 0.0001 | F(1,36) = 35.92; P < 0.0001 | F(1,36) = 12.32; P = 0.0012 | |
| Social interaction | F(1,36) = 4.318; P = 0.0449 | F(1,36) = 2.095; P = 0.1564 | F(1,36) = 5.041; P = 0.0310 | |
| Modified Y maze | F(1,36) = 12.22; P = 0.0013 | F(1,36) = 2.189; P = 0.1477 | F(1,36) = 4.959; P = 0.0323 | |
| Syntaxin | F(1,20) = 5.724; P = 0.0267 | F(1,20) = 5.068; P = 0.0358 | F(1,20) = 13.79; P = 0.0014 | |
| SNAP-25 | F(1,20) = 7.820; P = 0.0111 | F(1,20) = 4.318; P = 0.0508 | F(1,20) = 5.318; P = 0.0319 | |
| vGluT1 | F(1,20) = 11.42; P = 0.0030 | F(1,20) = 7.530; P = 0.0125 | F(1,20) = 5.289; P = 0.0324 | |
| Effect of SCH58261 in LTP amplitude, WT | F(1,16) = 67.12; P < 0.0001 | F(1,16) = 0.1595; P = 6949 | F(1,16) = 46.75; P < 0.0001 | |
| Forebrain A2AR-KO | Weight gain | F(1,32) = 7.924; P = 0.0083 | F(1,32) = 9.886; P = 0.0036 | F(1,32) = 11.84; P = 0.0016 |
| Cortisol | F(1,32) = 16.79; P = 0.0003 | F(1,32) = 20.55; P < 0.0001 | F(1,32) = 4.563; P = 0.0404 | |
| Forced swim | F(1,32) = 84.68; P < 0.0001 | F(1,32) = 53.24; P < 0.0001 | F(1,32) = 51.40; P < 0.0001 | |
| Tail suspension | F(1,32) = 61.10; P < 0.0001 | F(1,32) = 86.09; P < 0.0001 | F(1,32) = 91.89; P < 0.0001 | |
| Splash | F(1,32) = 27.69; P < 0.0001 | F(1,32) = 23.91; P < 0.0001 | F(1,32) = 17.14; P = 0.0002 | |
| Elevated-plus maze | F(1,32) = 21.01; P < 0.0001 | F(1,32) = 1.242; P = 0.2735 | F(1,32) = 31.35; P < 0.0001 | |
| Social interaction | F(1,32) = 8.525; P = 0.0064 | F(1,32) = 3.907; P = 0.0568 | F(1,32) = 6.147; P = 0.0186 | |
| Modified Y maze | F(1,30) = 20.43; P < 0.0001 | F(1,30) = 25.82; P < 0.0001 | F(1,30) = 6.918; P = 0.0133 | |
| Syntaxin | F(1,16) = 19.15; P = 0.0005 | F(1,16) = 3.902; P = 0.0658 | F(1,16) =1 0.13; P = 0.0058 | |
| SNAP-25 | F(1,16) = 6.482; P = 0.0216 | F(1,16) = 4.215; P = 0.0568 | F(1,16) = 7.443; P = 0.0149 | |
| vGluT1 | F(1,16) = 15.43; P = 0.0012 | F(1,16) = 10.76; P = 0.0047 | F(1,16) = 4.668; P = 0.0463 |
Supplementary Material
Acknowledgments
This work was supported by Brain and Behavior Research Foundation, Defense Advance Research Projects Agency Grant 09-68-ESR-FP-010; the Foundation for Science and Technology (FCT) Grant PTDC/SAU-NSC 122254/2010; The National Strategic Reference Framework's (QREN) Grant CENTRO-07-ST24-FEDER-002006; Coordination for the Improvement of Higher Education Personnel (CAPES)-FCT; and Brazilian National Council for Scientific and Technological Development (CNPq) Grant Ciência sem Fronteiras.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423088112/-/DCSupplemental.
References
- 1.de Kloet ER, Joëls M, Holsboer F. Stress and the brain: From adaptation to disease. Nat Rev Neurosci. 2005;6(6):463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 3.Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3(6):453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- 4.Harris A, Ursin H, Murison R, Eriksen HR. Coffee, stress and cortisol in nursing staff. Psychoneuroendocrinology. 2007;32(4):322–330. doi: 10.1016/j.psyneuen.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Lucas M, et al. Coffee, caffeine, and risk of depression among women. Arch Intern Med. 2011;171(17):1571–1578. doi: 10.1001/archinternmed.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AP. Caffeine, cognitive failures and health in a non-working community sample. Hum Psychopharmacol. 2009;24(1):29–34. doi: 10.1002/hup.991. [DOI] [PubMed] [Google Scholar]
- 7.Kawachi I, Willett WC, Colditz GA, Stampfer MJ, Speizer FE. A prospective study of coffee drinking and suicide in women. Arch Intern Med. 1996;156(5):521–525. [PubMed] [Google Scholar]
- 8.Lucas M, et al. Coffee, caffeine, and risk of completed suicide: Results from three prospective cohorts of American adults. World J Biol Psychiatry. 2014;15(5):377–386. doi: 10.3109/15622975.2013.795243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 10.Cunha RA. Neuroprotection by adenosine in the brain: From A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005;1(2):111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costenla AR, et al. Enhanced role of adenosine A2A receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34(1):12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- 12.Batalha VL, et al. Adenosine A2A receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol Psychiatry. 2013;18(3):320–331. doi: 10.1038/mp.2012.8. [DOI] [PubMed] [Google Scholar]
- 13.Silva CG, Porciúncula LO, Canas PM, Oliveira CR, Cunha RA. Blockade of adenosine A2A receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis. 2007;27(2):182–189. doi: 10.1016/j.nbd.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Cognato GP, et al. Caffeine and an adenosine A2A receptor antagonist prevent memory impairment and synaptotoxicity in adult rats triggered by a convulsive episode in early life. J Neurochem. 2010;112(2):453–462. doi: 10.1111/j.1471-4159.2009.06465.x. [DOI] [PubMed] [Google Scholar]
- 15.Canas PM, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci. 2009;29(47):14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matos M, et al. Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease of glutamate uptake. J Alzheimers Dis. 2012;31(3):555–567. doi: 10.3233/JAD-2012-120469. [DOI] [PubMed] [Google Scholar]
- 17.Rebola N, et al. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J Neurochem. 2011;117(1):100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- 18.Magariños AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA. 1997;94(25):14002–14008. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97(2):253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- 20.Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: Glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73(12):1172–1179. doi: 10.1016/j.biopsych.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Yacoubi M, Costentin J, Vaugeois JM. Adenosine A2A receptors and depression. Neurology. 2003;61(11) Suppl 6:S82–S87. doi: 10.1212/01.wnl.0000095220.87550.f6. [DOI] [PubMed] [Google Scholar]
- 22.Minor TR, Rowe M, Cullen PK, Furst S. Enhancing brain adenosine signaling with the nucleoside transport blocker NBTI (S-(4-nitrobenzyl)-6-theoinosine) mimics the effects of inescapable shock on later shuttle-escape performance in rats. Behav Neurosci. 2008;122(6):1236–1247. doi: 10.1037/a0013143. [DOI] [PubMed] [Google Scholar]
- 23.Yamada K, et al. Antidepressant activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002) on learned helplessness in rats. Psychopharmacology (Berl) 2014;231(14):2839–2849. doi: 10.1007/s00213-014-3454-0. [DOI] [PubMed] [Google Scholar]
- 24.Shen HY, et al. A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J Neurosci. 2008;28(12):2970–2975. doi: 10.1523/JNEUROSCI.5255-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L, et al. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol. 2008;63(3):338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- 26.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13(1):22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: Potential therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willner P. Validity, reliability and utility of the chronic mild stress model of depression: A 10-year review and evaluation. Psychopharmacology (Berl) 1997;134(4):319–329. doi: 10.1007/s002130050456. [DOI] [PubMed] [Google Scholar]
- 29.Hill MN, Hellemans KG, Verma P, Gorzalka BB, Weinberg J. Neurobiology of chronic mild stress: Parallels to major depression. Neurosci Biobehav Rev. 2012;36(9):2085–2117. doi: 10.1016/j.neubiorev.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Correa M, Font L. Is there a major role for adenosine A2A receptors in anxiety? Front Biosci. 2008;13:4058–4070. doi: 10.2741/2994. [DOI] [PubMed] [Google Scholar]
- 31.Duarte JM, Agostinho PM, Carvalho RA, Cunha RA. Caffeine consumption prevents diabetes-induced memory impairment and synaptotoxicity in the hippocampus of NONcZNO10/LTJ mice. PLoS ONE. 2012;7(4):e21899. doi: 10.1371/journal.pone.0021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunha GM, Canas PM, Oliveira CR, Cunha RA. Increased density and synapto-protective effect of adenosine A2A receptors upon sub-chronic restraint stress. Neuroscience. 2006;141(4):1775–1781. doi: 10.1016/j.neuroscience.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Cunha RA, Agostinho PM. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimers Dis. 2010;20(Suppl 1):S95–S116. doi: 10.3233/JAD-2010-1408. [DOI] [PubMed] [Google Scholar]
- 34.Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol. 2007;83(5):263–276. doi: 10.1016/j.pneurobio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Zezula J, Freissmuth M. The A2A-adenosine receptor: A GPCR with unique features? Br J Pharmacol. 2008;153(Suppl 1):S184–S190. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haskell CF, Kennedy DO, Wesnes KA, Scholey AB. Cognitive and mood improvements of caffeine in habitual consumers and habitual non-consumers of caffeine. Psychopharmacology (Berl) 2005;179(4):813–825. doi: 10.1007/s00213-004-2104-3. [DOI] [PubMed] [Google Scholar]
- 37.Pechlivanova DM, et al. Effect of long-term caffeine administration on depressive-like behavior in rats exposed to chronic unpredictable stress. Behav Pharmacol. 2012;23(4):339–347. doi: 10.1097/FBP.0b013e3283564dd9. [DOI] [PubMed] [Google Scholar]
- 38.Alzoubi KH, et al. Caffeine prevents cognitive impairment induced by chronic psychosocial stress and/or high fat-high carbohydrate diet. Behav Brain Res. 2013;237:7–14. doi: 10.1016/j.bbr.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 39.Prediger RD, Batista LC, Takahashi RN. Caffeine reverses age-related deficits in olfactory discrimination and social recognition memory in rats. Involvement of adenosine A1 and A2A receptors. Neurobiol Aging. 2005;26(6):957–964. doi: 10.1016/j.neurobiolaging.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 40.Silva CG, et al. Adenosine receptor antagonists including caffeine alter fetal brain development in mice. Sci Transl Med. 2013;5(197):ra104. doi: 10.1126/scitranslmed.3006258. [DOI] [PubMed] [Google Scholar]
- 41.Hockemeyer J, Burbiel JC, Müller CE. Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: Potential drugs for Parkinson’s disease. J Org Chem. 2004;69(10):3308–3318. doi: 10.1021/jo0358574. [DOI] [PubMed] [Google Scholar]
- 42.Mineur YS, Belzung C, Crusio WE. Effects of unpredictable chronic mild stress on anxiety and depression-like behavior in mice. Behav Brain Res. 2006;175(1):43–50. doi: 10.1016/j.bbr.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 43.Kaster MP, et al. Adenosine administration produces an antidepressant-like effect in mice: Evidence for the involvement of A1 and A2A receptors. Neurosci Lett. 2004;355(1-2):21–24. doi: 10.1016/j.neulet.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 44.Willner P, Moreau JL, Nielsen CK, Papp M, Sluzewska A. Decreased hedonic responsiveness following chronic mild stress is not secondary to loss of body weight. Physiol Behav. 1996;60(1):129–134. doi: 10.1016/0031-9384(95)02256-2. [DOI] [PubMed] [Google Scholar]
- 45.Yalcin I, Aksu F, Belzung C. Effects of desipramine and tramadol in a chronic mild stress model in mice are altered by yohimbine but not by pindolol. Eur J Pharmacol. 2005;514(2-3):165–174. doi: 10.1016/j.ejphar.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Soares E, et al. Spatial memory impairments in a prediabetic rat model. Neuroscience. 2013;250:565–577. doi: 10.1016/j.neuroscience.2013.07.055. [DOI] [PubMed] [Google Scholar]
- 47.Lopes LV, et al. Binding of the prototypical adenosine A2A receptor agonist CGS 21680 to the cerebral cortex of adenosine A1 and A2A receptor knockout mice. Br J Pharmacol. 2004;141(6):1006–1014. doi: 10.1038/sj.bjp.0705692. [DOI] [PMC free article] [PubMed] [Google Scholar]