

Abstract
Nucleophilic radiofluorination is an efficient synthetic route to many positron-emission tomography (PET) probes, but removal of water to activate the cyclotron-produced [18F]fluoride has to be performed prior to reaction, which significantly increases overall radiolabeling time and causes radioactivity loss. In this report, we demonstrate the possibility of 18F-radiofluorination in highly aqueous media. The method utilizes titania nanoparticles, 1:1 (v/v) acetonitrile-thexyl alcohol solvent mixture and tetra-n-butylammonium bicarbonate as a phase-transfer agent. Efficient radiolabeling is directly performed with aqueous [18F]fluoride without the need for a drying/azeotroping step to significantly reduce radiosynthesis time. High radiochemical purity of the target compound is also achieved. The substrate scope of the synthetic strategy is demonstrated with a range of aromatic, aliphatic and cycloaliphatic tosylated precursors.
1. INTRODUCTION
Positron emission tomography (PET) is an extremely effective imaging tool in clinical care, preclinical and clinical research, and drug discovery. PET enables visualization of physiological states or changes in the living body, investigation of the mechanism of disease, and quantification of biological processes such as receptor occupancy, cell proliferation, metabolic activity, apoptosis, and gene expression. Since PET allows for highly sensitive measurement of phenotypic changes associated with a malignant condition, the onset of a particular disease such as cancer can be detected, diagnosed, and treated at an early stage prior to the development of metastasis.1 Typically, 18F-labeled PET tracers are synthesized using nucleophilic fluorination of activated precursors with [18F]fluoride/[18O]H2O obtained from a cyclotron.2–5 Due to high 18F-ion solvation energy in water, aqueous fluoride is relatively unreacttive unless it is released from its aqueous surrounding. This is usually achieved by mixing aqueous fluoride with a phase transfer agent (e.g., K2CO3/Kryptofix-222 (K222), tetra-n-butylammonium bicarbonate (TBAB), or Cs2CO3) and azeotropically drying the resulting solution with acetonitrile. The dried active complex (e.g., [18F]KF/K222) is then used for radiolabelling in an anhydrous organic solvent. Depending on the technical method of fluoride activation (e.g., cartridge-based solvent exchange followed by evaporation), this procedure may take up to 20–30 minutes and some loss of initial radioactivity is observed.6–8
To reduce the need for a lengthy drying process, there has been recent interest in the development of radiofluorination methods that allow the direct use of aqueous [18F]fluoride without a separate activation step. Several novel approaches were reported, such as enzymatic9–12 and transition-metal mediated/catalyzed13–18 reactions, and the development of specific substrates and fluorinating reagents.19,20 The majority of these methods directly use cyclotron-delivered [18F]fluoride/[18O]H2O, and reactions are conducted in organic-aqueous media with water content of 0.5–20%. However, most of these methods are suited only for a narrow compound class (e.g., aromatic, benzylic nucleophilic fluorination), and the reported fluorination efficiency was typically low to moderate (4–48% of the desired fluorinated product). For enzymatic catalysis, the substrate scope is limited to only a few specific precursors.
We report herein a conceptually new method based on titania (titanium dioxide, TiO2) nanoparticles (crystalline composition: 45% rutile, 55% anatase; <200 nm size) as a catalyst. The precursor solution is first incubated with the particles, and then a mixture of phase-transfer agent and [18F]fluoride/[18O]H2O (taken directly from cyclotron-delivered vial or trapped and eluted from QMA-cartridge) is added and reacted, followed by removal of the catalyst particles by filtration and then purification and reformulation steps. The use of TiO2 obviates the laborious synthesis and purification of a metal-precursor complex prior to radiolabeling, and the method is compatible with many commercially-available or easily-synthesized precursors. We have demonstrated that this route for radiofluorination is suitable for aromatic, aliphatic, and cycloaliphatic precursors in organic-aqueous media with a tolerated water content up to 25 vol%.
Titania is widely used in a variety of chemical processes, including electro- and photochemistry.21,22 Along with broad catalytic activity, it possesses a strong ability to adsorb water from surrounding media.23–25 Numerous reports have demonstrated the mechanism of water dissociation at titania surface,26,27 and we hypothesize that this feature of TiO2 might be used for in situ desolvation of [18F]fluoride from water to catalyze nucleophilic radiofluorination reactions in organic-aqueous solutions without preliminary drying of the [18F]fluoride. We present an investigation of the influence of various reaction parameters on the radiofluorination of a model PET probe precursor and also discuss studies performed to further elucidate the mechanism of TiO2-catalyzed radiofluorination.
2. RESULTS AND DISCUSSION
2.1 Preliminary observations
As a model system for our primary studies, we have chosen the radiofluorination of tosyl-Fallypride 1a to [18F]Fallypride 2a (Scheme 1), a highly specific dopamine D2/D3 receptor radioligand used in PET imaging of the brain to study receptor occupancy and density and which has potential clinical application in relation to neuropsychiatric conditions and aging.28,29
Scheme 1.

Catalytic formation of [18F]Fallypride
The radiofluorination reaction was performed in a 1:1 v/v mixture of acetonitrile (MeCN) and 2,3-dimethyl-2-butanol (thexyl alcohol). Thexyl alcohol was included due to a report by Lee et al.30 that showed the addition of alcohol to the reaction medium facilitates the radiofluorination SN2-process, and the report by Javed et al.31 that showed a substantial increase in fluorination efficiency of [18F]Fallypride when using this solvent mixture compared to pure MeCN. Reactions were performed in a sealed vial, heated to 110°C for 7 minutes using a Peltier heater. The reaction mixture was mixed by refluxing solvent; magnetic stirring was not used. After completing the reaction, methanol was added to the reaction mixture, and solid catalyst particles were removed by filtration through a 20 nm filter (Whatman Anotop 10 Plus). In general, we found that some [18F]fluoride trapped onto the catalyst and could not be extracted. Using a dose calibrator, radioactivity measurement of the filtered extract was compared to the initial radioactivity to determine the radioactivity extraction efficiency (REE): REE = [decay-corrected radioactivity recovered in the organic extract]/[initial radioactivity]. The fraction of initial radioactivity trapped onto the catalyst is calculated as 1 − REE. Analysis with radio-TLC and radio-HPLC were used to determine fluorination efficiency. This in turn enabled calculation of the radiochemical conversion (RCC), RCC = REE x [fluorination efficiency]. All results are averaged over at least n = 3 experiments. Identity of the radiofluorinated product was confirmed by co-injection with standard [19F]Fallypride (ABX GmbH, Germany).
We performed a series of experiments (Table 1) to confirm the importance of each species in the reaction. Entries 1 and 2 show the conventional synthesis conditions where [18F]fluoride is pre-dried and reaction takes place in anhydrous organic media. The improvement in RCC due to addition of thexyl alcohol is apparent. Entry 5 shows 68% RCC resulting from catalytic synthesis conditions with all species included as described above. Comparative runs without the phase transfer catalyst resulted in only 18% RCC. Thus, the phase transfer agent appears to be important as well, possibly due to better solubilization of in situ generated tetra-n-butylammonium fluoride ([18F]TBAF), compared to [18F]fluoride, in organic-aqueous media. As expected, experiments in the absence of catalyst did not lead to formation of desired product 2a in organic-aqueous media (Entry 3), indicating that TiO2 is essential when there is water in the reaction mixture. If the [18F]fluoride is dried (as in conventional synthesis) prior to catalytic reaction, no conversion was observed (Entry 6). In fact, the filtrate contained neither fluorinated product nor parent [18F]fluoride; all the radioactivity was found bound to the catalyst. This means that in non-aqueous media, TiO2 addition results in total [18F]fluoride trapping (REE ~0%). On the other hand, REE was found to be remarkably constant (~80%) for all of the TiO2 catalyzed conditions containing water. Even if the reaction was performed without the precursor, the REE was unchanged (80±2%).
Table 1.
Influence of reaction components on formation of 2a.a
| # | Catalyst | Phase transfer agentb | MeCN, μl | Thexyl alcohol, μl | Water, μlc | REE, % | RCC, % |
|---|---|---|---|---|---|---|---|
| 1 | none | TBAB | 40 | 0 | 0 | 100d | 31±2 |
| 2 | none | TBAB | 20 | 20 | 0 | 100d | 64±4 |
| 3 | none | None | 15 | 15 | 10 | 100d | 0 |
| 4 | TiO2 | None | 15 | 15 | 10 | 79±4 | 18±3 |
| 5 | TiO2 | TBAB | 15 | 15 | 10 | 80±3 | 68±2 |
| 6 | TiO2 | TBAB | 20 | 20 | 0 | 0 | 0 |
| 7 | TiO2 | K2CO3/K222 | 15 | 15 | 10 | 78±3 | 39±6 |
| 8 | none | K2CO3/K222 | 20 | 20 | 0 | 100d | 31±4 |
| 9 | MgSO4 | TBAB | 15 | 15 | 10 | 99±1 | 0 |
| 10 | CaCl2 | TBAB | 15 | 15 | 10 | 99±1 | 0 |
Reactions were performed with 2.3 μmol 1a, 140 μmol TiO2 in 40 μL reaction volume at 110°C for 7 min without magnetic stirring.
Amounts of phase transfer agent used, when applicable: 0.36 μmol TBAB; 0.36 μmol K2CO3 and 0.36 μmol K222.
For cases where water is 0 μL, [18F]fluoride added as dry complex with phase-transfer agent (1.5 – 4 mCi) reconstituted in 10 μl of MeCN-thexyl alcohol (1:1 v/v); for other cases, radioactivity introduced as solution of aqueous [18F]fluoride (1.5 – 4 mCi) containing phase-transfer agent;
In case of catalyst absence no extraction performed.
The influence of the type of phase-transfer agent was evaluated by carrying out the reaction with K2CO3/K222 mixture (Entry 7). This resulted in a substantial decrease in RCC suggesting that TBAB is a superior phase transfer agent for these reactions.
Finally, to assess whether the role of TiO2 was simply to sequester water to facilitate fluorination in mixed aqueous-organic media, comparative runs with particles of other common non-oxide drying agents, MgSO4 and CaCl2 (140 μmol each), were also performed (Entries 9–10). These showed zero RCC, suggesting an effect of TiO2 beyond simple water adsorption, but most probably its ability of water splitting.
2.3. Hypothesized mechanism of TiO2-catalyzed radiofluorination
Previous reports show that oxo- and oxy-containing species readily coordinate on a TiO2 surface through hydrogen bonding.32–35 Thus, we hypothesize that in addition to an interaction with water, the TiO2 catalyst could also serve to coordinate tosyl-Fallypride 1a via oxygen atoms of the sulfonyl group, facilitating reaction with desolvated [18F]fluoride in close proximity. Based on these ideas, the following mechanism of catalyzed fluorination is suggested (Figure 1):
Figure 1.

Proposed mechanism for TiO2-catalyzed radiofluorination. SN2 substitution is improved with alcohol as a co-solvent, perhaps by inclusion in intermediate complex formation as determined by Oh et al.30 (not shown on scheme).
Hydrogen bonding occurs between oxygen atoms of the sulfonyl moiety and TiO2, which coordinates tosylated precursor to the surface of the catalyst; when aqueous [18F]fluoride/TBAB solution is added, solvated [18F]fluoride is adsorbed at active sites of TiO2 where the aqueous shell is split resulting in [18F]fluoride release (i.e., activation); TBAB phase transfer catalyst then serves as a [18F]fluoride-trapping agent and intercepts activated [18F]fluoride, subsequently conducting the phase-transfer of [18F]fluoride to the surface-coordinated precursor to faciliate the SN2-type reaction. Because the coordination with the precursor is at the leaving group, the resulting radiofluorinated product is released upon formation from the TiO2.
2.4. Evidence for coordination
To explore the role of coordination in the reaction mechanism, we performed radiolabeling after first incubating the tosylated substrate with catalyst. If coordination indeed occurs, prolonged exposure of the precursor to the catalyst should increase the amount of precursor bound to the surface of the catalyst and thus promote increased SN2-reactions (i.e., radiolabeling efficiency). Thus, a solution of tosyl-Fallypride 1a in 1:1 (v/v) MeCN-thexyl alcohol was added to the catalyst and incubated at room temperature for various durations. After incubation, aqueous [18F]fluoride/TBAB mixture was added, and radiolabeling was performed by heating at 110 °C for 12 min using an oil bath. The reaction mixture was mixed by refluxing solvent; no magnetic stirring was used. We observed a modest enhancement in RCC starting after 20–30 min of incubation and reaching a maximum improvement after ~1 hr (Figure 2).
Figure 2.

Effect of substrate-catalyst pre-incubation time on radiofluorination efficiency of 1a.
Analytical samples of organic solution after incubation with catalyst contained significant amount of precursor 1a; thus, it is likely that the observed saturation is due to occupation of all accessible active sites of TiO2 capable of binding the oxygen-atoms of the leaving group. Attempts to perform pre-incubation at elevated temperatures (30, 45 and 60°C) showed similar results, i.e., the timescale was not shortened. It should be noted that the pre-incubation step was performed before the introduction of [18F]fluoride; therefore, incorporation of pre-incubation into the synthesis protocol does not introduce any delays that would affect the yield due to decay.
Studies were performed by running the radiofluorination reaction in the presence of various amounts of a non-reactive SO-containing compound (dimethylsulfoxide, DMSO) to assess whether the presence of another co-coordinating species could block the binding of the precursor and lower the RCC (Figure 3).
Figure 3.

Influence of DMSO on TiO2 catalyzed radiochemical conversion of 1a.
It was revealed that even slight addition of DMSO dramatically affected the RCC; 2-fold decrease in RCC was registered at 5 vol% DMSO with nearly complete inhibition at 37.5 vol% DMSO, suggesting that DMSO molecules may indeed bind to active sites of TiO2, thus reducing sites available for precursor coordination. Surprisingly, the REE was also significantly affected, and the amount of [18F]fluoride trapped onto the catalyst increased with increasing DMSO content. Almost total [18F]fluoride trapping (90%) was registered at 75 vol% DMSO. This suggests that instead of merely affecting precursor coordination, DMSO may interact with the catalyst in additional ways leading to trapping of fluoride, such as DMSO-water cluster formation,36–39 intercalation of DMSO-water clathrate inside the oxide structure,40 or DMSO-induced creation of positively-charged sites.41
To investigate further the need for leaving group coordination at the TiO2 surface, experiments were performed comparing tosylated compounds 1b, 1c and 1s with their brominated versions 3–5 (not suspected to coordinate with surface) in TiO2-catalyzed [18F]fluorination (Table 2). No product was observed in case of bromo-derivatives, while high RCCs of ~80% were determined for tosylated precursors, which agrees with the necessity for the proposed oxygen-coordinating mechanism at the leaving group. The REE in case of bromo-substituted substrates remained close to 80%, i.e., similar to that for tosylated reactants.
Table 2.
TiO2 catalyzed radiofluorination of tosylated vs. brominated substratesa.
| Entry | Structure | |
|---|---|---|
| 1 |
 1b (78 ± 3% RCC) |
 3 (0 % RCC) |
| 2 |
 1c (80 ± 3% RCC) |
 4 (0 % RCC) |
| 3 |
 1s (70 ± 7% RCC) |
 5 (0 % RCC) |
1 h pre-incubation time; 2.3 μmol of precursor; 140 μmol of TiO2; 130°C, 5 min; 40 μl total reaction volume; no magnetic stirring; radioactivity introduced as 10 μl solution of aqueous [18F]fluoride (1.5 – 4 mCi) containing 0.36 μmol TBAB. ~80% of radioactivity extraction efficiency (REE) observed for every entry.
2.5 Optimization of catalyst loading
It was found that the amount of catalyst added had a substantial effect on RCC. Studies with different catalyst amounts (see Supporting Information) revealed that the optimal loading of TiO2 is 140 μmol for 40 μl reaction volume with 25 vol% water content for the catalyst particle size used (<200 nm). This translates to ~60:1 ratio of catalyst to precursor 1a and enables production of target compound 2a with the highest RCC (78%). The RCC drops for both increasing and decreasing amounts of catalyst.
Looking at fluorination efficiency, we observed catalyst amounts lower than the optimum to result in decreasing fluorination efficiency, perhaps due to reduced desolvation of fluoride. Surprisingly, increasing the amount of catalyst also decreased the fluorination efficiency. Perhaps as the catalyst amount is increased, one of the reaction components becomes depleted, reducing the interactions necessary for fluorination.
On the other hand, looking at extraction efficiency, we observed that REE decreases (i.e., trapping increases) with increasing amount of catalyst. Due to this linear relation, we suspect there are specific trapping sites for fluoride on the TiO2, the number of which depends on the amount of catalyst present. Perhaps trapping occurs via exchange of fluoride with terminal hydroxyl groups.42–44 The consistent ~20% trapping for a fixed amount of catalyst may represent the equilibrium exchange between the surface and the reaction solution containing both fluoride and hydroxide ions. The case of 100% trapping under dry conditions (Table 1, entry 6), where the fluoride concentration dominates could represent a shift in this equilibrium.45
To determine if the optimal amount of catalyst is universal or should be adjusted for every precursor, similar experiments were performed with substrates 1i, 1q and 1t. The loading of 140 μmol of TiO2 remained optimal for these substrates as well (Figure 4).
Figure 4.

Determination of optimal catalyst amount for radiolabeling of 1a, 1i, 1q and 1t.
2.6 Evaluation of optimal reaction conditions
To maximize fluorination efficiency of the precursor 1a, further evaluation of reaction conditions has been performed to determine optimal reaction parameters. The range of water content that provided maximum RCC was found to be up to 25 vol% (see Supporting Information). In this aqueous range, the RCC and fluoride trapping remained remarkably constant. With higher water content, the trapping remained constant, but a strong decrease in [18F]fluorination was observed. We hypothesize that higher water content exceeds the capacity of the catalyst to adsorb and split water, and thus the [18F]fluoride is not as effectively desolvated, reducing the fluorination efficiency. If true, this suggests that increased catalyst amount may enable improved water tolerance if desired, but in light of results in the previous section, it would also be necessary to increase the precursor amount in the same proportion as the catalyst.
We also studied RCC as a function of reaction time and temperature (see Supporting Information), to determine the optimal reaction time (5 min) and temperature (130°C). The influence of the specific type of alcohol co-solvent was evaluated as well. Several alcohols were tested in place of thexyl alcohol, but relatively little difference in RCC was observed (see Supporting Information). Thexyl alcohol showed the highest RCC of alcohols tested.
2.7. Production and quality control of clinically relevant PET probe
To demonstrate the overall radiochemical yield of isolated [18F]Fallypride 2a, we performed full production runs (radiofluorination, HPLC purification, and formulation). The final formulated product was obtained as a sterile, injectable solution. During production, radioactivity measurements were recorded at key steps to assess efficiency of each process and identify potential areas for optimization (Table 3). The biggest loss is during extraction from the catalyst (20% trapped) and an additional ~10% is lost during our purification and formulation processes. Total production times, isolated yields and specific activities are compared to those previously reported in literature (Table 4). Generally, the isolated yield of titania-catalyzed reaction tends to be higher than reported for macroscale automated production, while it is comparable to microfluidic procedures, such as syntheses on a digital microfluidic chip7 or using the Advion Nanotek capillary reactor.46 By avoiding the need for fluoride drying/azeotroping, the synthesis process is simplified.
Table 3.
Titania-catalyzed production of [18F]Fallypride.a
| Parameter | Run 1 | Run 2 | Run 3 | Average |
|---|---|---|---|---|
| Initial activity, mCi | 5.20 | 5.19 | 5.23 | 5.20 ± 0.02 |
| REE, % | 79.1 | 81.2 | 81.9 | 80.7 ± 1.5 |
| RCC, % (non-isolated) | 79.1 | 81.2 | 81.5 | 80.6 ± 1.3 |
| Activity remaining after synthesis and extraction, %b | 79.1 | 81.2 | 81.9 | 80.7 ± 1.5 |
| Activity remaining after HPLC purification, %b | 75.7 | 76.5 | 77.6 | 76.6 ± 1.0 |
| Activity remaining after reformulation, %b | 71.2 | 68.6 | 73.2 | 71.0 ± 2.3 |
| Total loss, % | 28.8 | 31.4 | 26.8 | 28.9 ± 2.3 |
| Isolated RCY, % | 71.2 | 68.6 | 73.2 | 71.0 ± 2.3 |
Optimized reaction conditions: 1 h pre-incubation time; 2.3 μmol of precursor; 140 μmol of TiO2; 40 μl total reaction volume; no magnetic stirring; radioactivity introduced as 10 μl solution of aqueous [18F]fluoride (~2.6 mCi) containing 0.36 μmol of TBAB; heated to 130° for 5 min using an oil bath. For each run, 2 vials are pooled after extraction to improve accuracy of measurements.
Fractions of remaining radioactivity determined by measuring the radioactivity after the relevant step, correcting for decay, and dividing by the initial radioactivity.
Table 4.
Comparison of catalytic production of 2a to known procedures.
| Entry | Reactor type | Radiolabeling conditions | Mean RCY, % | Total time, min |
|---|---|---|---|---|
| Moon et al., 201048 | Macroscale, automated. TracerLab FX | 100 °C, 30 min | 68 | 74 |
| Pike et al.46 | Microscale, automated. Nanotek Advion. | 150–190 °C, 4–23 min | 16–88 | 50–218 |
| Javed et al.7 | Microscale, automated. EWOD chip | 105°C, 7 min | 83 | 70 |
| Lazari et al.49 | Macroscale automated. Elixys™ | 105°C, 7 min | 66 | 56a |
| Current report | Small-volume vial, manual | 130°C, 5 min | 71 | 50 |
Total time reported without reformulation step
It is noteworthy to mention the high radiochemical purity of target compound 2a that formed during reaction. Only two radioactive peaks were detected by analytical HPLC, which consisted of unreacted [18F]fluoride and the desired [18F]Fallypride (see Supporting Information). Regarding HPLC analysis of the non-radioactive side products, the hydroxylated compound, resulting from hydrolysis, is clearly observed while no byproduct from β-elimination is apparent. With HPLC purification, the [18F]fluoride and non-radioactive side products were effectively removed, and the reformulated solution was then examined by standard quality control (QC) tests47 to evaluation its compliance with United States Food and Drug Administration requirements for injectable PET tracers (Table 5).
Table 5.
Quality control tests of injectable [18F]Fallypride solution.
| Clinical QC test | Clinical acceptance criteria | Results of this study |
|---|---|---|
| Optical clarity | Clear and particle free | Clear and particle free |
| pH | 5.5–8.0 | 6.5 |
| Radiochemical purity | >95% | >99% |
| Radiochemical identity | Matches retention time of the standard | Matches retention time of the standard |
| 18F-radionuclide identity | Half-life 105–115 min | Half-life 111 min |
| Endotoxin level | < 5 EU/mL | < 1 EU/mL |
| Filter integrity | > 50 psig | > 100 psig |
| MeCN content | < 410 ppm | <2 ppm |
| Thexyl alcohol content | < 5000 ppm | <1 ppm |
| Sterility | No growth in 14 days | No growth in 14 days |
| Titanium content | None specified | 36±4 ng |
With the introduction of the TiO2 catalyst into the production procedure, an additional QC test to assess the titanium content in the reformulated solution may be needed. While the 20 nm filtration process and subsequent HPLC purification are expected to eliminate all particles, there is still the possibility of titanium ions in the final formulated solution. Using inductively-coupled plasma mass spectrometry (ICP-MS), the titanium content in representative samples was found to be 36±4 ng (n = 9) of titanium per batch. Titanium is considered very inert and there do not appear to be established limits for titanium in injectable solutions. According to medicinal reports50–52, however, normal levels of titanium in human body range from 0 to ~20 μg/L blood (~0–100 μg per whole body of an adult) in patients without titanium implants, while reaching 100–150 ug/L blood (500–750 μg per whole body) in patients with artificial titanium joints. Thus, an injection of formulated [18F]Fallypride produced by our method would have a very miniscule impact because it contains orders of magnitude less titanium than normally present in the blood. This could suggest that routine titanium testing may not be needed for every batch of injectable PET probe.53,54
We have, therefore, demonstrated the practical feasibility of the newly developed synthetic approach to the synthesis of a clinically-relevant PET probe, including compliance with FDA requirements for injectable solutions in clinical applications. By varying the precursor, other [18F]fluorine-labeled PET probes could be produced by this procedure and similar QC results would be expected.
2.8. Determination of specific activity
High specific activity (SA) of PET tracers is essential to minimize the injected quantity of the non-radioactive form of the tracer, which can saturate rare biological targets, such as neurological receptors, and subsequently lower the image quality and possibly cause pharmacologic effects.55 It has been confirmed that SA plays an important role in PET image quality,29,56,57 especially in small animals.58,59 Using standard methods7, the SA for TiO2 catalyzed synthesis of [18F]Fallypride 2a was found to be 5±2 Ci/μmol (n = 5). This is higher than typically obtained from macroscale synthesis. (0.4–3 Ci/μmol).49,60 This evidence suggests TiO2 catalysis could be successfully used for routine production of PET imaging probes that require high SA.
2.9. Attempts to scale-up synthesis
Previous experiments utilized operating volumes of 40 μl, 10 μL of which was [18F]fluoride solution. While we have been able to demonstrate the production of several millicuries of the isolated product starting from ~5 mCi of [18F]fluoride, it may be desirable in some cases to produce larger amounts of product (e.g., for clinical production), which requires a larger volume of the initial [18F]fluoride solution.
One simple way to scale up the quantity of tracer produced is to run several reactions in parallel and pool the results; however, this would require multiple reaction vessels and may not be convenient to handle or automate. Another approach is to scale up the reaction volume, enabling a larger volume of [18F]fluoride to be used while still keeping water content within the 25 vol% range, and thus enabling larger amount of starting radioactivity. To test this possibility, we explored proportionally increasing the amounts of all of the reaction components (i.e., precursor, TiO2, solvent, and aqueous [18F]fluoride/TBAB solution). As an example, the catalytic fluorination of 1a was scaled up to a factor of 3, such that the final volume of the reaction mixture comprised 120 μl. During these experiments, we found that 5 min reaction time was insufficient to efficiently fluorinate the precursor (fluorination efficiency = 50%). With increased heating time, it was possible to increase fluorination efficiency to the values seen prior to scaling (Figure 5). We suspect that longer time is needed for sufficient diffusive mixing in the larger volume. We also encountered some initial difficulties during the filtration step to remove the nanoparticles after the reaction was done. We observed clogging of the 20 nm filter due to the increased amount of TiO2. This issue was resolved by incorporating an initial pre-filtration step with a 0.22 μm filter prior to 20 nm fine filtering. Unfortunately, this had the effect of slightly reducing the extraction efficiency, from 80±2% to 71±13%.
Figure 5.

Correlation between reaction time and fluorination efficiency in scaled-up catalytic radiolabeling of 1a. Values are averaged from n = 9 experiments for 5 min runs, and n = 6 for each of 8 and 15 min experiments.
The tested scale-up factor of 3 is sufficient for production of a human-dose of [18F]Fallypride, and further increases in scale are presumably possible, if desired; with higher volumes, stirring during the pre-incubation and reaction steps may become important, requiring additional optimization of reaction time and extraction procedures. Another reason to perform volume scale-up is to potentially enable automation in commercially-available radiosynthesizers, which typically require at least several hundred microliters of solution in the reaction. Though there are advantages in performing reactions in extremely small volumes,7,8 it will be some time before automated and commercialized versions of such technologies are widely available.
As an alternative way to increase the amount of radioactivity in the reaction without impacting reaction volume, solid phase extraction procedures using microscale QMA cartridges could be used to concentrate the [18F]fluoride to obtain higher starting radioactivity in volumes of the [18F]fluoride/TBAB solution described here (i.e. 10 μL water content). Several reports have shown that an entire cyclotron target volume can be trapped and efficiently eluted in only 5–45 μL of eluent solution.61–63 This would likely be the preferred approach to scale-up the amount of radioactivity since no increase in reaction volume would be necessary.
2.10 Substrate scope
In order to explore the utility of the TiO2 catalytic approach for the synthesis of other PET tracers, we investigated the scope of applicable substrates. We first considered the use of other leaving groups with sulfonyl moieties (i.e., triflate and nosylate). Surprisingly, the radiofluorination of commercially available substrates 6 and 7 (precursors for 2′-deoxy-2′-[18F]fluoro-D-glucose ([18F]FDG), and 3′-deoxy-3′-[18F]fluoro-L-thymidine ([18F]FLT)) (Figure 6) resulted in RCC = 0%.
Figure 6.

Additional substrates with sulfonyl-containing leaving groups and their corresponding hydrolysates.
In a presence of the catalyst, triflate and nosylate groups seemed to become overreactive, and immediate explosive hydrolysis was observed upon aqueous [18F]fluoride addition. When analyzed, only unreacted [18F]fluoride and hydroxylated compounds 6-OH and 7-OH were detected. This phenomenon suggests that reactivity of oxygen-containing leaving groups are increased when incubated with TiO2. Due to this additional activation, the ideal leaving group should initially possess lower reactivity (tosyloxy preferred before triflyloxy or nosyloxy), otherwise concurrent side-reaction of hydrolysis prevails over [18F]fluorination.
We next investigated the generality of TiO2-catalyzed radiofluorination of tosylated precursors. A library of aromatic, aliphatic and cycloaliphatic tosylates was tested, along with the commercially available tosylated PET probe precursors for [18F]-4-fluoroproline ([18F]-4-FP), [18F]-fluoroazomycin-arabinoside ([18F]FAZA) and [18F]-fluoroerythronitroimidazole ([18F]FETNIM) (Table 6). The methodology was highly efficient for low-molecular-weight precursors 1b-v (65–80% RCC) but resulted in low to moderate yields with bulky and sterically hindered substrates 1w-x (i.e., from the commercially available precursors for [18F]FAZA and [18F]FETNIM, respectively). These particular precursors also contain additional oxo-moieties, which potentially lower yields by coordinating the precursor at the catalyst surface instead of at the O=S=O moiety of the tosylate leaving group. Such coordination could significantly reduce the [18F]fluoride interaction with the tosylate reactive center by placing the reaction center further from the catalyst surface where fluoride desolvation occurs. We are currently looking into methods to further understand the reaction mechanism to predict the effectiveness of different substrates and perhaps enable improved substrate design.
Table 6.
Substrate scope for the TiO2 catalyzed radio-fluorination of tosylated substratesa
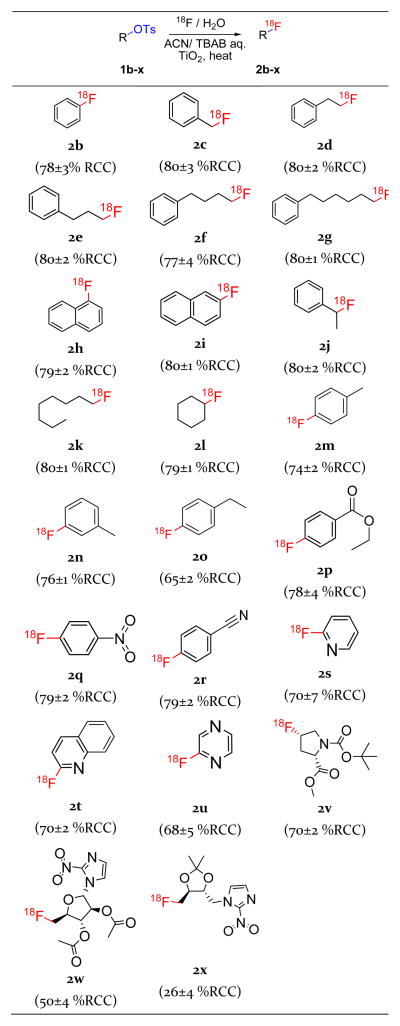
|
Optimized reaction conditions: 1 h pre-incubation time; 2.3 μmol of precursor; 140 μmol of TiO2; 130°C, 5 min; 40 μl total reaction volume; no magnetic stirring; radioactivity introduced as 10 μl solution of aqueous [18F]fluoride (1.5 – 4 mCi) containing 0.36 μmol TBAB. For all entries, REE was observed to be ~80%.
3. CONCLUSIONS
In conclusion, we have developed a novel method of TiO2-catalyzed radiofluorination of tosylated presursors and demonstrated its use for the preparation of 18F-labeled PET probes. The method avoids the need for drying of the [18F]fluoride/[18O]H2O from the cyclotron before fluorination. The wet [18F]fluoride is mixed with a phase transfer agent and added to a solution of precursor solution pre-incubated with TiO2 nanoparticles and reacted for a short time. In this fashion, nucleophilic 18F-fluorination is shown to proceed rapidly and efficiently in aqueous medium with up to 25 vol% water content, which to the best of our knowledge is the highest reported other than enzymatic methods. We have also demostrated the production of clinically-relevant amounts of [18F]Fallypride with this approach as well as shown compliance of the final formulated PET tracer with QC requirements for clinical use. The product was found to have high specific activity even with low amounts of starting radioactivity. The applicability of the reported protocol to a range of tosylated substrates was also demonstrated for organic molecules containing aromatic, aliphatic and cycloaliphatic moieties. Although extensive additional investigations are required to explore the substrate scope and further understand the mechanism, we anticipate that the facile procedure and high radiofluorination efficiency of this new method may provide a versatile tool for practitioners in the field of PET radiochemistry. Based on our hypothesized mechanism of reaction, further studies regarding the importance of the structure of the precursor and other effective catalysts are currently in progress in our group.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in part by the Department of Energy Office of Biological and Environmental Research (DE-SC00001249) and the National Institute of Biomedical Imaging and Bioengineering (NIBIB, grant number EB015540).
The authors gratefully acknowledge the invaluable services of Prof. Saman Sadeghi and the staff of the UCLA Biomedical Cyclotron for providing 18F-ion and performing QC testing of samples.
ABBREVIATIONS
- GCMS
gas-chromatography mass spectrometry
- HPLC
high-performance liquid chromatography
- ICP-MS
inductively-coupled plasma mass spectrometry
- PET
positron-emission tomography
- QC
quality control
- REE
radioactivity extraction efficiency
- RCC
radiochemical conversion
- RCY
radio-chemical yield
- SA
specific activity
- TBAB
tetra-n-butylammonium bicarbonate
- TBAF
tetra-n-butylammonium fluoride
- TLC
thin layer chromatography
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Experimental details, selected HPLC and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Schirrmacher R, Wangler C, Schirrmacher E. Mini-Rev Org Chem. 2007;4:317. [Google Scholar]
- 3.Cai L, Lu S, Pike VW. Eur J Org Chem. 2008;2008:2853. [Google Scholar]
- 4.Brooks AF, Topczewski JJ, Ichiishi N, Sanford MS, Scott PJH. Chem Sci. 2014;5:4545. doi: 10.1039/C4SC02099E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J. Tetrahedron Lett. 2014;55:4289. [Google Scholar]
- 6.Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY. Bull Korean Chem Soc. 2011;32:71. [Google Scholar]
- 7.Javed MR, Chen S, Lei J, Collins J, Sergeev M, Kim H-K, Kim C-J, Dam RM, van Keng PY. Chem Commun. 2014;50:1192. doi: 10.1039/c3cc47616b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sergeev M, Lazari M, Collins J, Morgia F, Javed MR, Keng PY, van Dam RM. Proceedings of the 6th International Symposium on Microchemistry and Microsystems (ISMM-2014); Singapore. Jul. 30–Aug. 1, 2014; pp. 77–78. [Google Scholar]
- 9.Onega M, Domarkas J, Deng H, Schweiger LF, Smith TAD, Welch AE, Plisson C, Gee AD, O’Hagan D. Chem Commun. 2010;46:139. doi: 10.1039/b919364b. [DOI] [PubMed] [Google Scholar]
- 10.Eustáquio AS, O’Hagan D, Moore BS. J Nat Prod. 2010;73:378. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sergeev ME, Morgia F, Javed MR, Doi M, Keng PY. J Mol Catal B Enzym. 2013;92:51. [Google Scholar]
- 12.Sergeev ME, Morgia F, Javed MR, Doi M, Keng PY. J Mol Catal B: Enzym. 2013;97:74. [Google Scholar]
- 13.Kamlet AS, Neumann CN, Lee E, Carlin SM, Moseley CK, Stephenson N, Hooker JM, Ritter T. PLoS ONE. 2013;8:e59187. doi: 10.1371/journal.pone.0059187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuya T, Ritter T. Org Lett. 2009;11:2860. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 15.Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Liu W, Ren H, Neelamegam R, Hooker JM, Groves JT. J Am Chem Soc. 2014;136:6842. doi: 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]
- 17.Tredwell M, Preshlock SM, Taylor NJ, Gruber S, Huiban M, Passchier J, Mercier J, Génicot C, Gouverneur V. Angew Chem. 2014;126:1. doi: 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]
- 18.Ichiishi N, Brooks AF, Topczewski JJ, Rodnick ME, Sanford MS, Scott PJH. Org Lett. 2014;16:3224. doi: 10.1021/ol501243g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun JH, Telu S, Lu S, Pike VW. Org Biomol Chem. 2013;11:5094. doi: 10.1039/c3ob40742j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McBride WJ, Sharkey RM, Goldenberg DM. EJNMMI Res. 2013;3:36. doi: 10.1186/2191-219X-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar SG, Devi LG. J Phys Chem A. 2011;115:13211. doi: 10.1021/jp204364a. [DOI] [PubMed] [Google Scholar]
- 22.Fröschl T, Hörmann U, Kubiak P, Kučerová G, Pfanzelt M, Weiss CK, Behm RJ, Hüsing N, Kaiser U, Landfester K, Wohlfahrt-Mehrens M. Chem Soc Rev. 2012;41:5313. doi: 10.1039/c2cs35013k. [DOI] [PubMed] [Google Scholar]
- 23.Zhang C, Lindan PJD. J Chem Phys. 2003;118:4620. [Google Scholar]
- 24.Diebold U. Surf Sci Rep. 2003;48:53. [Google Scholar]
- 25.Di Valentin C, Tilocca A, Selloni A, Beck TJ, Klust A, Batzill M, Losovyj Y, Diebold U. J Am Chem Soc. 2005;127:9895. doi: 10.1021/ja0511624. [DOI] [PubMed] [Google Scholar]
- 26.Hammer B, Wendt S, Besenbacher F. Top Catal. 2010;53:423. [Google Scholar]
- 27.Onal I, Soyer S, Senkan S. Surf Sci. 2006;600:2457. [Google Scholar]
- 28.Constantinescu CC, Coleman RA, Pan ML, Mukherjee J. Synapse. 2011;65:778. doi: 10.1002/syn.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchsbaum MS, Christian BT, Lehrer DS, Narayanan TK, Shi B, Mantil J, Kemether E, Oakes TR, Mukherjee J Schizophr Res. 2006;85:232. doi: 10.1016/j.schres.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 30.Oh YH, Ahn DS, Chung SY, Jeon JH, Park SW, Oh SJ, Kim DW, Kil HS, Chi DY, Lee S. J Phys Chem A. 2007;111:10152. doi: 10.1021/jp0743929. [DOI] [PubMed] [Google Scholar]
- 31.Javed MR, Chen S, Kim H-K, Wei L, Czernin J, Kim C-J, Dam RM, van Keng PY. J Nucl Med. 2014;55:321. doi: 10.2967/jnumed.113.121053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weisz AD, Regazzoni AE, Blesa MA. Croat Chem Acta. 2007;80:325. [Google Scholar]
- 33.Bahruji H, Bowker M, Brookes C, Davies PR, Wawata I. Appl Catal Gen. 2013;454:66. [Google Scholar]
- 34.Paz Y. Beilstein J Nanotechnol. 2011;2:845. doi: 10.3762/bjnano.2.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansson EMJ, Plogmaker S, Walle LE, Schölin R, Borg A, Sandell A, Rensmo H. J Phys Chem C. 2010;114:15015. [Google Scholar]
- 36.Huang A, Liu C, Ma L, Tong Z, Lin R. J Chem Thermodyn. 2012;49:95. [Google Scholar]
- 37.Kirchner B, Hutter J. Chem Phys Lett. 2002;364:497. [Google Scholar]
- 38.Kirchner B, Reiher M. J Am Chem Soc. 2002;124:6206. doi: 10.1021/ja017703g. [DOI] [PubMed] [Google Scholar]
- 39.Jerie K, Baranowski A, Rozenfeld B, Jeżowska-Trzebiatowska B, Gliński J Acta Phys Pol A. 1991;79:507. [Google Scholar]
- 40.Zhang S, Liu Q, Cheng H, Zeng F. Appl Surf Sci. 2015;331:234. [Google Scholar]
- 41.Letaief S, Leclercq J, Liu Y, Detellier C. Langmuir. 2011;27:15248. doi: 10.1021/la203492m. [DOI] [PubMed] [Google Scholar]
- 42.Minella M, Faga MG, Maurino V, Minero C, Pelizzetti E, Coluccia S, Martra G. Langmuir. 2010;26:2521. doi: 10.1021/la902807g. [DOI] [PubMed] [Google Scholar]
- 43.Minero C, Mariella G, Maurino V, Pelizzetti E. Langmuir. 2000;16:2632. [Google Scholar]
- 44.Minero C, Mariella G, Maurino V, Vione D, Pelizzetti E. Langmuir. 2000;16:8964. [Google Scholar]
- 45.Habuda-Stanić M, Ravančić M, Flanagan A. Materials. 2014;7:6317. doi: 10.3390/ma7096317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu S, Giamis AM, Pike VW. Curr Radiopharm. 2009;2:1. doi: 10.2174/1874471010902010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keng PY, Chen S, Ding H, Sadeghi S, Shah GJ, Dooraghi A, Phelps ME, Satyamurthy N, Chatziioannou AF, Kim CJ, van Dam RM. Proc Natl Acad Sci. 2012;109:690. doi: 10.1073/pnas.1117566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seok Moon B, Hyung Park J, Jin Lee H, Sun Kim J, Sup Kil H, Se Lee B, Yoon Chi D, Chul Lee B, Kyeong Kim Y, Eun Kim S. Appl Radiat Isot. 2010;68:2279. doi: 10.1016/j.apradiso.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Lazari M, Collins J, Shen B, Farhoud M, Yeh D, Maraglia B, Chin FT, Nathanson DA, Moore M, van Dam RM. J Nucl Med Technol. 2014;42:203. doi: 10.2967/jnmt.114.140392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ipach I, Schäfer R, Mittag F, Leichtle C, Wolf P, Kluba T. BMC Musculoskelet Disord. 2012;13:159. doi: 10.1186/1471-2474-13-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patton MS, Lyon TDB, Ashcroft GP. Acta Orthop. 2008;79:820. doi: 10.1080/17453670810016911. [DOI] [PubMed] [Google Scholar]
- 52.Rodushkin I, Ödman F, Branth S. Fresenius J Anal Chem. 1999;364:338. [Google Scholar]
- 53.Shao X, Schnau PL, Fawaz MV, Scott PJH. Nucl Med Biol. 2013;40:109. doi: 10.1016/j.nucmedbio.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radiopharmaceuticals for Position Emission Tomography - Compounding. Chapter 823. U.S. Pharmacopeial Convention; 2012. pp. 398–406. USP 35 - NF 50. [Google Scholar]
- 55.Liu Z, Li Y, Lozada J, Wong MQ, Greene J, Lin KS, Yapp D, Perrin DM. Nucl Med Biol. 2013;40:841. doi: 10.1016/j.nucmedbio.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Millet P, Moulin-Sallanon M, Tournier BB, Dumas N, Charnay Y, Ibáñez V, Ginovart N. NeuroImage. 2012;62:1455. doi: 10.1016/j.neuroimage.2012.05.075. [DOI] [PubMed] [Google Scholar]
- 57.Vandehey NT, Moirano JM, Converse AK, Holden JE, Mukherjee J, Murali D, Nickles RJ, Davidson RJ, Schneider ML, Christian BT. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2010;30:994. doi: 10.1038/jcbfm.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Honer M, Brühlmeier M, Missimer J, Schubiger AP, Ametamey SM. J Nucl Med. 2004;45:464. [PubMed] [Google Scholar]
- 59.Rominger A, Mille E, Zhang S, Böning G, Förster S, Nowak S, Gildehaus FJ, Wängler B, Bartenstein P, Cumming P. J Nucl Med. 2010;51:1576. doi: 10.2967/jnumed.110.078451. [DOI] [PubMed] [Google Scholar]
- 60.Mukherjee J, Yang ZY, Das MK, Brown T. Nucl Med Biol. 1995;22:283. doi: 10.1016/0969-8051(94)00117-3. [DOI] [PubMed] [Google Scholar]
- 61.Elizarov AM, van Dam RM, Shin YS, Kolb HC, Padgett HC, Stout D, Shu J, Huang J, Daridon A, Heath JR. J Nucl Med. 2010;51:282. doi: 10.2967/jnumed.109.065946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lebedev A, Miraghaie R, Kotta K, Ball CE, Zhang J, Buchsbaum MS, Kolb HC, Elizarov A. Lab Chip. 2012;13:136. doi: 10.1039/c2lc40853h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lazari M, Narayanam MK, Murphy JM, Van Dam RM. Automated concentration of 18F-fluoride in microliter volumes. Proceedings of 21st International Symposium on Radiopharmaceutical Sciences (ISRS-2015); Columbia, MO, USA. May 26–31, 2015; Oral presentation assigned. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.