

Abstract
Smad transcription factors are central to the signal transduction pathway that mediates the numerous effects of the TGF-β superfamily of cytokines in metazoan embryo development and adult tissue regeneration and homeostasis. Although Smad proteins are conserved, recent genome-sequencing projects have revealed their sequence variation in metazoan evolution, human polymorphism, and cancer. Structural studies of Smads bound to partner proteins and target DNA provided a framework to understand the significance of these evolutionary and pathologic sequence variations. Here we synthesize the extant mutational and structural data to suggest how genetic variation in Smads may affect the structure, regulation, and function of these proteins. Furthermore, we present a web-app that compares Smad sequences and displays Smad protein structures and their disease-associated variants.
Keywords: TGF-β signaling, Smad proteins, Smad Structure, Smad conservation/variation, Smad binding proteins, Smad DNA Binding, Cancer mutations
Smad proteins and the TGF-β signaling pathway
The transforming growth factor β (TGF-β) superfamily of cytokines plays key roles in metazoan organisms from the early stages of embryo development to the maintenance and regeneration of mature tissues, and in many developmental and degenerative diseases [1, 2]. The TGF-β superfamily controls this remarkable range of biologic processes by regulating the transcription of genes that control cell proliferation, differentiation, death, adhesion, movement and positioning. TGF-β itself functions as an enforcer of tissue homeostasis by regulating pluripotency and differentiation in stem and progenitor cells, by limiting the growth of epithelial, hematopoietic and neural tissues, by promoting tolerance in the immune system, and by suppressing the oncogenic progression of premalignant cells, among other effects. Bone morphogenetic proteins (BMPs) in general counterbalance TGF-β action in stem cell differentiation, and promote skeletal tissue development and regeneration. In contrast to the multifunctional nature of TGF-β and the BMPs, other family members have highly specialized functions. For example, myostatin (previously known as Growth and Differentiation Factor-8, GDF8) specifically suppresses skeletal muscle growth, GDF11 suppresses cardiac hypertrophy, and anti-Müllerian hormone (AMH) induces the regression of the Müllerian duct in male mammalian embryos.
A common pathway, with Smad transcription factors as the centerpiece, mediates the remarkably diverse effects of the TGF-β superfamily (Figure 1A). In a nutshell, TGF-β cytokines bind to receptor serine kinases that phosphorylate Smads, which then form transcriptional complexes that regulate specific genes. The choice of target genes is strongly influenced by contextual determinants including receptor and Smad regulatory inputs, Smad transcriptional cofactors and DNA binding partners, and mediators of Smad interaction with the chromatin. Many of these determinants are specific to a particular cell lineage, differentiation stage, or cell metabolic condition. Under their influence, the TGF-β/Smad signal transduction pathway functions as a context-dependent multifunctional signaling device for cell regulation throughout the lifespan of the organism [2].
Figure 1. Main components of the TGF-beta pathway.
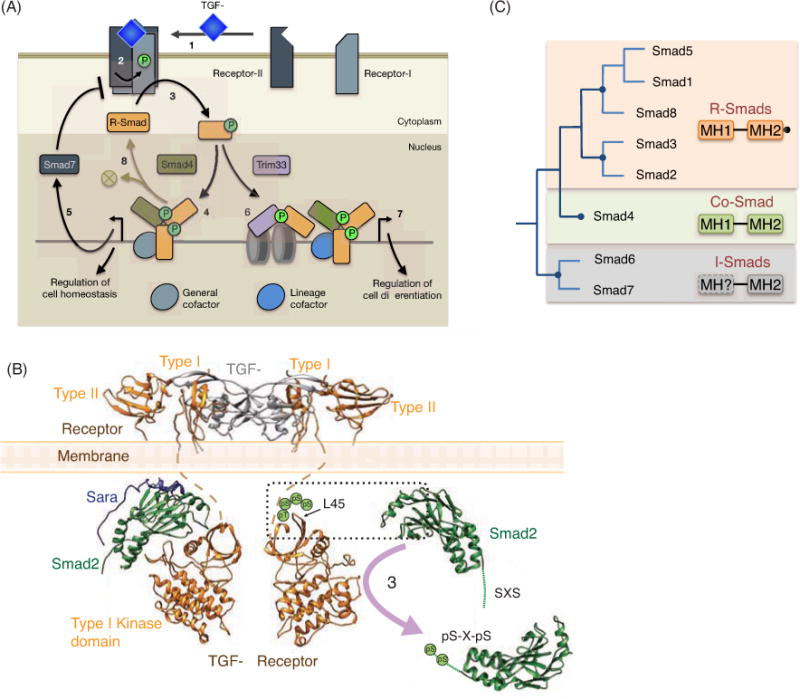
A. Schematic representation of the TGF-β/Smad pathway.1. The ligand TGF-β binds and brings together two pairs of type I and type II receptors, which are transmembrane Ser/Thr kinases. 2. In the receptor complex, the type II receptor phosphorylates and activates the type I receptor kinase. 3. The type I receptor phosphorylates and activates R-Smads (generally Smad2 and Smad3 in the case of TGF-β, activin, nodal and myostatin receptors; Smad1, Smad5 and Smad8 in the case of BMP receptors). 4. Phosphorylated R-Smads form trimeric complexes with Smad4 for transcriptional regulation. The genes to be regulated are targeted by the Smad complex in partnership with various DNA binding cofactors. Transcriptional regulation additionally involves the recruitment of histone and DNA modifying enzymes, nucelosome positioning complexes, and CDK8/9 components of transcriptional Mediator and Elongation complexes (not shown; Massagué 2012). 5. Target genes that regulate cell homeostasis include Smad7, an inhibitory Smad that provides a universal feedback loop for negative regulation of this pathway. 6. Receptor-activated Smad2/3 alternatively form a complex with Trim33, a reader of repressive H3K9me3 histone marks. 7. In collaboration with the Smad-Trim33 complex and lineage-specific DNA binding cofactors factors (e.g. FoxH1 for mesoderm differentiation), the R-Smad-Smad4 complex regulates master differentiation genes (e.g. goosecoid in the case of mesoderm differentiation). 8. Activated Smads are targeted by phosphatases for additional rounds of signaling or by ubiquitin ligases for proteasome-mediated degradation.
B. Model representing TGF-β (gray) bound to the receptor components type I and type II (gold). The MH2 domain of Smad2 (green) is bound to the adaptor protein SARA (blue). Smad2 is modeled to be in the proximity of the receptor kinase, for its activation through phosphorylation (step 3 in Figure 1A). Phosphorylated Smad2 residues are represented as two green circles.
C. The Smad protein family in Vertebrates. The proteins are grouped according to their function.
Amino acid sequence conservation across species is remarkably high for all the central components of this pathway. It is as if the TGF-β/Smad pathway is present in the last common ancestor of metazoans. Expert reviews have covered the molecular basis of TGF-β/Smad signal transduction [3–5], the contextual nature of TGF-β/Smad signaling [2], Smad-independent forms of TGF-β signaling [6, 7], and the biology of the TGF-β/Smad pathway in stem cells and embryo development [8–10], in immunity [11] and in cancer [12, 13]. Here we complement this knowledge with insights from newly available genome sequences of over one hundred metazoan species including archaic humans (GoLife, http://goo.gl/K6NSMf, Tree of Life, http://www.tolweb.org/tree/), the genomic variation of modern humans (1000 Genomes) and massive mutational data from clinical tumor samples (The Cancer Genome Atlas (TCGA); the cBioPortal, and the Catalogue of Somatic Mutations in Cancer, COSMIC).
Leveraging this unprecedented opportunity we highlight the patterns of Smad sequence conservation and variability. We present the natural variation and cancer mutations of human Smad sequences in the context of Smad sequence conservation during metazoan evolution. We place this information within the framework of available X-ray and NMR structures of Smad protein domains bound to interacting proteins and DNA (Table 1). Finally, we provide a web application (http://smad.maciasnmr.net/) that can be navigated to view sequence alignments as well as wild type and mutated complexes and models displaying the tumor mutations and variants.
Table 1.
PDB codes for the complexes of Smad proteins.
| Smad1 | Smad2 | Smad3 | Smad4 | Smad7 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yap1 | Smurf1 | DNA | Smad4 | Sara | Nedd4L | Pin1 | Smad4 | DNA | Smad2 | Smad3 | Ski | DNA | Yap1 | Nedd4L | Smurf1 | Smurf2 |
|
| ||||||||||||||||
| 2lay | 2laz | 3kmp | 1u7v | 1dev | 2lb2 | 2lb3 | 1u7f | 1ozj | 1u7v | 1u7f | 1mr1 | 3qsv | 2ltv | 2lty | 2ltx | 2ltz |
| 2lax | 2lb0 | 2laj | 1mhd | 2ltw | 2djy | |||||||||||
| 2law | 2lb1 | |||||||||||||||
The TGF-β pathway in brief
The TGF-β cytokines are dimers of two identical subunits linked, in most cases, by a disulfide bond. The basic TGF-β signaling mechanism involves binding of the dimer to membrane receptor serine kinases (Figure 1A). Of approximately five hundred serine/threonine kinases encoded in the human genome only twelve are cell-surface receptors, and all function as receptors for the TGF-β family. The receptors form two sub-families, the type I TGF-β receptor subfamily with 7 members in humans, and the type II receptor subfamily with 5. Ligand binding triggers the assembly of a complex that includes two type I receptors and two type II receptors. Within the complex, the type II receptor subunits phosphorylate the type I at multiple serine and threonine residues located N-terminally to the type I receptor kinase domain [5, 14]. Thus activated, the type I receptor kinases recruit and phosphorylate Smad proteins for signal transduction. This phosphorylation targets two serine residues in the Smad C terminus sequence, pSer-X-pSer, creating an acidic tail that drives the formation of Smad transcriptional complexes (Figure 1B, model of the receptor with bound receptor-regulated Smads [R-Smads], adapted from [5, 15].
Smad proteins consist of two globular domains connected by a linker region (Figure 1C). The main function of the Smad N-terminal domain, or “Mad homology 1” (MH1) domain, is to bind DNA (Figures 2A, 2B). The C-terminal domain, or MH2 domain, mediates protein-protein interaction with numerous regulator and effector proteins, including the TGF-β receptors, certain cytoplasmic anchor proteins, lineage-specific DNA-binding cofactors, and chromatin modifiers. The latter include co-activator histone acetylases, co-repressor deacetylates, the histone reader TRIM33, the nucleosome remodeling complex core subunit BRG1, and others (Figures 2A, 2C) [2, 16]. Amino acid sequence conservation across species is remarkably high for all Smad proteins (sequence alignments are displayed in the web-app).
Figure 2. Overview of the Smad domains.
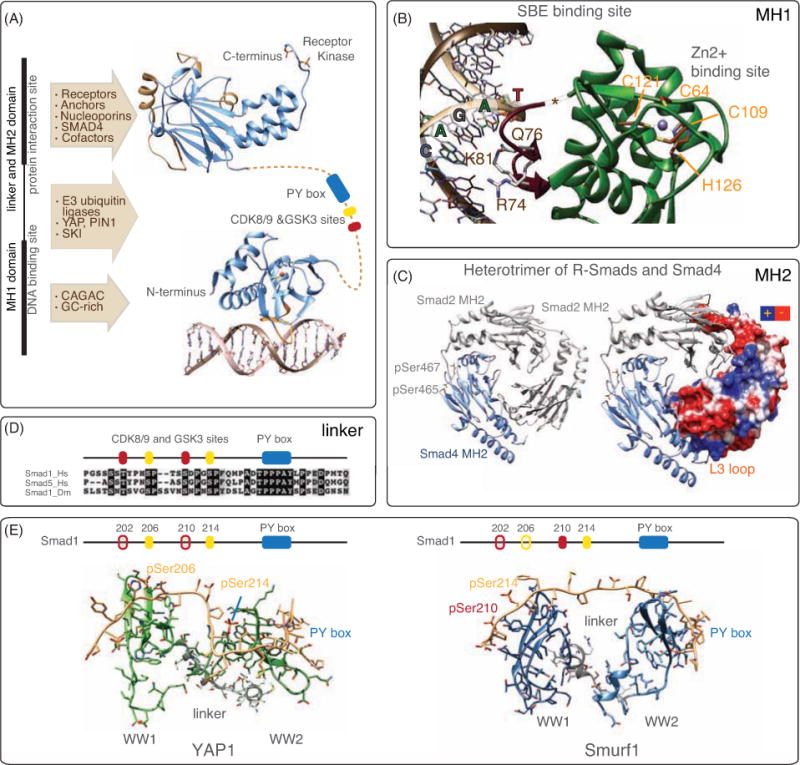
A. Structural domains present in R-Smads. The elements of secondary structure present in the MH1 and MH2 domains are depicted using a ribbon representation and are colored in blue. The hairpin that interacts with the DNA is shown in orange and the different binding sites characteristic of the MH2 domain are shown in light brown. The pair of phosphorylated serines characteristic of activated R-Smads are shown with the full atom representation. The linker connecting the MH1 and MH2 domains is represented as a dotted line and includes the PY motif in blue and the phosphorylated (CDK8/9) and (GSK3) sites in yellow and in red respectively.
B. Details of the MH1 domain. Residues that recognize the dsDNA SBE motif of Smad3 are displayed and labeled in dark brown while the residues involved in the recognition of the Zn2+ (gray sphere) are labeled in orange. The position of the insert encoded by exon3 in the Smad2 MH1 domain is marked with an asterisk.
C. Details of the MH2 domain: The heterotrimer of two Smad2 MH2 domains (gray) and one Smad4 MH2 domain (blue) is shown as cartoons. Phosphorylated serines 465 and 467 are displayed and labeled (left panel). One of the Smad2 monomers is shown with the surface electrostatic charge distribution highlighted. The location of the positive patch characteristic of the L3 loop is indicated (right panel).
D. Sequence alignment of the Smad1/5 human linker and its comparison to the drosophila Smad1 counterpart. Identical and conservative differences are shaded in black and in gray respectively. The CDK8/9, and GSK3 phosphorylation sites and the PY motif are represented as boxes on top of the alignment (yellow, red and blue respectively, as in panel A).
E. Structures of the pair of WW domains present in YAP and in Smurf1 bound to the Smad1 linker. While YAP recognizes the CDK8/9 and the PY motif for Smad activation, the interaction with the E3 ubiquitin ligase Smurf1 requires the presence of both CDK8/9 and GSK3 phosphorylation sites. The phosphorylated positions recognized by YAP and by Smurf1 are represented as full boxes and labeled, while the other phosphorylated positions, which are not bound by these proteins, are represented as light boxes.
The Smad family in humans includes eight members, five of which (Smads 1, 2, 3, 5 and 8), called R-Smads, contain the C-terminal Ser-X-Ser motif for phosphorylation by the type I receptors [2]. Signaling specificity is determined by the choice of type I and type II receptors that each particular TGF-β family member can bind and drive into a complex. With some notable exceptions [17], the receptors for TGF-β, nodal, activin, and myostatin (the TGF-β branch of the family) signal through Smad2 and Smad3, whereas the BMPs and anti-Müllerian hormone (the BMP branch) signal through Smads 1, 5 and 8.
Receptor-phosphorylated R-Smads in vitro can form homotrimers and also heterotrimers with Smad4. Trimers of one Smad4 molecule and two receptor-phosphorylated R-Smad molecules are thought to be the predominant effectors of TGF-β transcriptional regulation (Figure 1A, 2B). Smad4 functions as a partner with all the R-Smads and is required for most gene responses to the TGF-β superfamily. Smad4 is therefore called a Co-Smad, and its role may be to recruit specific transcriptional co-regulators to the Smad transcriptional complex. Whether R-Smad2 homotrimers also occur in vivo and mediate certain gene responses that require R-Smads but not Smad4 are questions under investigation. The remaining two members of the family, Smad6 and Smad7, are inhibitory Smads (I-Smads) that interact with activated receptors and R-Smads to suppress their activity. Smad7 expression is induced by TGF-β and BMP as a negative feedback loop that is conserved across cell types and metazoan species. Additionally, I-Smads respond to pathways that oppose TGF-β signaling.
The interdomain linker region of Smads is an entry point for positive and negative regulatory inputs. On reaching the nucleus, receptor-activated R-Smads bound to Smad4 undergo two rounds of phosphorylation events in the linker (Figure 2D). The first round is catalyzed by the cyclin-dependent kinases CDK8 and CDK9, which are part of the transcriptional Mediator and Elongation complexes, respectively. CDK8/9-mediated phosphorylation maximizes the transcriptional activity of R-Smads by favoring interactions with co-activators [18]. CDK8/9 additionally prime Smads for phosphorylation by glycogen synthase kinase-3 (GSK3) [19]. GSK3 switches the linker region from a binding site for co-activators to a binding site for HECT family E3 ubiquitin ligases that mark R-Smads for proteasome-mediated degradation (Figure 2E) [20, 21]. Thus, these phosphorylation events lead R-Smads to peak transcriptional activity followed by degradation, constituting an activation-turnover switch in the Smad signaling cycle.
The receptor-mediated phosphorylation of R-Smads can be reversed by PPM1 [22] or other protein phosphatases that limit TGF-β and BMP signaling, and the CDK8/9 phosphorylations by SCP1/2/3 phosphatases that prolong Smad transcriptional action [23, 24]. The CDK8/9 sites in the Smad linker region also function as phosphorylation sites for MAP kinases (as subsequently for GSK3) in response to regulatory crosstalk inputs from receptor tyrosine kinase and Wnt pathways [19, 23, 25, 26].
Smads in the metazoan common ancestry
The TGF-β pathway, with essentially all of its key components–ligands, type I and type II receptors, R-Smads, Co-Smad and I-Smads–is represented in the genome of all metazoans sequenced to date, but it is not evident in other organisms. The level of sequence conservation of individual Smad proteins across species is extremely high. This conservation of components and sequences suggests that the general functions and operating logic of the TGF-β pathway emerged with metazoans and have remained generally intact ever since.
Of course, the families of TGF-β ligands, receptors, and Smad proteins have undergone expansion and some sequence variation to provide regulatory capacity of the growing complexity and diversity of animal life [27–29] (see web-app). Vertebrates have eight Smads (five R-Smads, Smad4, and two I-Smads). Invertebrates have four (two R-Smads, Smad4, and one I-Smad), with the exception of sponges, which lack I-Smads and contain multiple duplications of other Smads [30–32]. Modern and archaic humans have identical Smad protein sequences [33].
There is a high level of sequence identity in Smad4 and the R-Smads, not only within mammals but also across all vertebrates. The few identified differences cluster in structural loops in the MH1 and MH2 domains, and in the inter-domain linker region. For example, the human Smad4 sequence differs from that of the elephant shark (representing cartilaginous fishes) by only 5 residues, and from that of ray-finned fishes by approximately 20. The sequence variability is higher in I-Smads. Among mammals, the Smad7 sequence presents more than 10 variable amino acid residues, most of them in the N-terminal region, which is very divergent from the MH1 domains of R-Smads and Smad4. The number of Smads encoded in different metazoan genomes, and the extent of protein sequence identity of Smad4 are shown in Figure 3. We selected Smad4 for this comparison because it is represented by a single gene in most metazoans.
Figure 3. Distribution of Smad protein in metazoans.
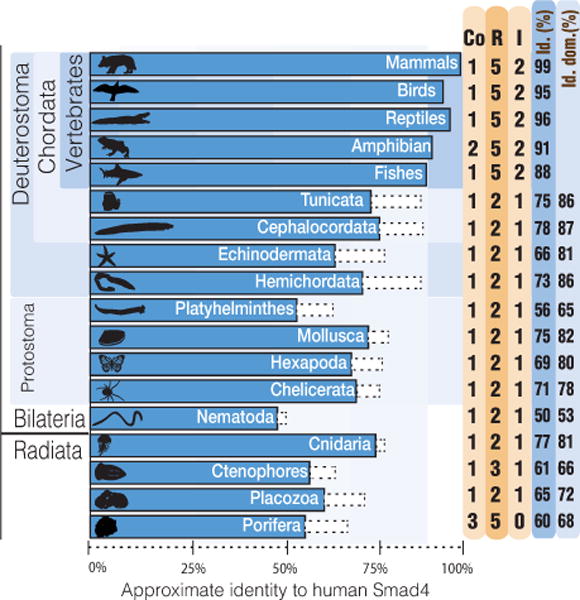
The number of Smads (R represents R-Smads, Co Smad4, I I-Smads) is annotated next to each phylum. The length of the boxes is approximately proportional to the level of Smad4 sequence identity with the human reference sequence. Dotted line boxes represent the value excluding the inter-domain linker region. The percentages of identity are given in the last two columns. The list of species used in the comparisons is shown in the Supplementary Table 1 and in the web-app.
Smad polymorphisms and somatic mutations
For every Smad protein and interacting partner with available structures (Table 1), we collected the non-synonymous single nucleotide polymorphism (SNP) variations of modern humans deposited in the ENSEMBL database and aligned them to the corresponding human reference genome sequence GRCh38 (Genome Reference Consortium build 38) in the web application. To facilitate the localization of sequence differences, we demarcated the domain boundaries and the elements of secondary structure on the top of the sequence in the schemes presented in the Figures.
Human-to-vertebrate differences are marked with green bars on the linear domain schemas (Figures 5–9). Human polymorphisms are marked with blue arrows. In general, R-Smads and I-Smads present fewer polymorphisms (about 10) than Smad4 (about 50). Among the R-Smads, the Smad1 and Smad5 contain fewer differences than Smad2 and Smad3. The amino acid differences are distributed along the sequence, affecting loops as well as elements of secondary structure.
Figure 5. Structure of the Smad4 MH1 domain bound to the SBE dsDNA.
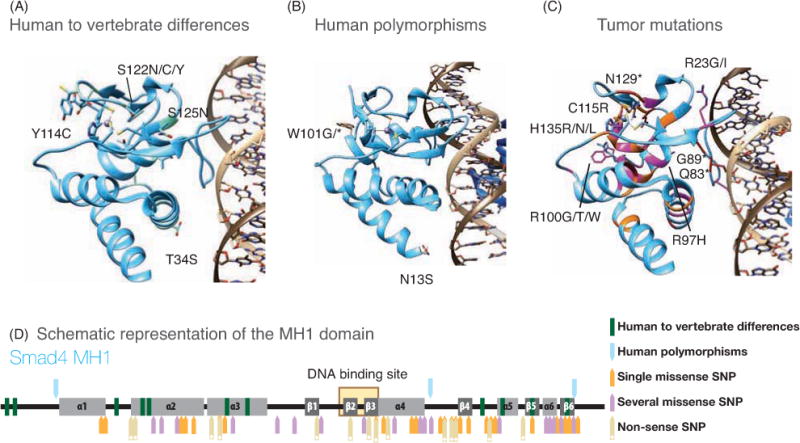
Smad4 MH1 domain is displayed in cyan. The Zn2+ is represented as a gray sphere. The complexes of Smad1 and Smad3 with the SBE dsDNA are shown in Supplementary Figures 1 and 2.
A. Differences observed in vertebrates. The residues corresponding to the human sequence are superimposed to the variants and labeled.
B. Non-synonymous SNPs in human population as identified in the 1000 Genomes project. The residues corresponding to the reference sequence are superimposed to the variants and labeled. The W100 to stop codon allele corresponds to a case of potential juvenile polyposis syndrome.
C. Tumor mutations identified in the Catalogue of Somatic Mutations in Cancer project, (COSMIC). The residues corresponding to the reference sequence are superimposed to the mutations identified in tumors. For the representation we have generated a Smad4 sequence combining all described mutations independently of tumor site. Due to the high number of mutants described for Smad4 only some selected mutants are represented with side-chains. The remaining positions with mutations are colored in the backbone: blue (reference sequence), orange (a position is mutated to a single amino acid), purple (when a given position is mutated to different amino acids) and red, when the mutation introduces a stop codon.
D. Map of the MH1 domain. The elements of secondary structure of the domain are represented schematically. Non-synonymous SNPs and tumor-associated mutations are represented as arrows and Vertebrate differences with the human sequence as bars (color code is shown in the figure).
Figure 9. Tumor mutations in the Smad7 PY site.
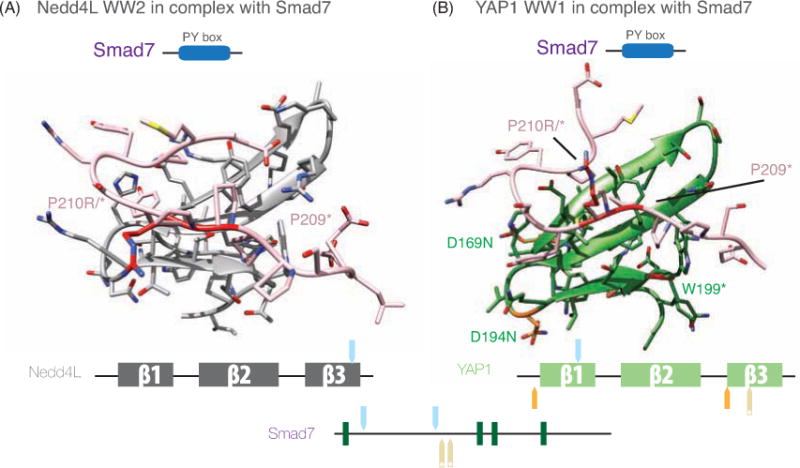
A. Nedd4L and Smad7: Cartoon representation of Nedd4L WW2 domain in complex with the Smad7 PY peptide (misty rose), displaying the elements of secondary structure (graphite) and the residues of the Nedd4L WW2 domain that participate in the interaction with the peptide. Human to Vertebrate differences and SNPs and tumor mutations are shown as Supplementary Figure 7.
B. YAP and Smad7 YAP WW1 domain (residues 163–206, blue) with the Smad7 [PY] peptide (misty rose), tumor mutations are displayed and labeled.
Below the complexes each protein sequence is represented schematically, with SNPs and tumor mutations highlighted as in Figure 5.
The existence of inactivating Smad mutations in cancer has long been known. Smad4 was independently identified as a gene of interest called Deleted in Pancreatic Carcinoma locus 4 (DPC4) based on the high frequency of somatic deletions and mutations that affect the gene in pancreatic adenocarcinomas [34]. Smad4 and Smad2 inactivating mutations were identified in colorectal carcinomas also early on [35, 36] (see web-app), providing genetic evidence for the tumor-suppressor nature of these genes. Most Smad point mutations associated with cancer are loss of function mutations that either target functional elements or affect the overall stability of the protein.
The Cancer Genome Atlas (TCGA) and other tumor sequence data annotated in the Catalogue of Somatic Mutations in Cancer (COSMIC project, cancer.sanger.ac.uk) [37] reveal missense and non-sense mutations in Smad coding regions, as well as mutations in non-coding regions and gene copy number alterations (CNA) including large deletions. Large deletions may additionally target neighboring tumor suppressor loci in the same chromosomal region [38, 39]. A cBioPortal-based plot summarizing all tumor-associated Smad genetic alterations (Figure 4A) shows a concentration of genetic alterations in Smad2, Smad3 and particularly in Smad4, in gastric (18%), colorectal (24%) and pancreatic adenocarcinomas (33%) (Figure 4B).
Figure 4. Frequency of alterations in Smad2, 3 and 4 by cancer type.
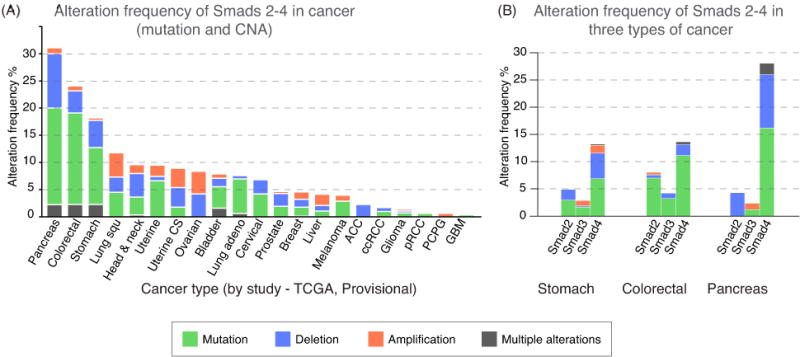
A. Plot displaying the frequency of genetic alteration (point mutations, deletions, amplifications, or multiple alterations) in Smad2, Smad3 and Smad4 combined, in different types of cancer. Data were derived from TCGA datasets (The Cancer Genome Atlas, cancergenome.nih.gov) at the time of this writing. Analysis was done using cBioPortal (www.cbioportal.org). Cancer type abbreviations are listed in Supplementary Table 2.
B. Plot showing the alteration frequency of Smad 2, 3 and 4 in pancreatic, gastric and colorectal cancers.
Point mutations in Smads are present in tumors of different origins (3% of all sequenced tumors, COSMIC database). Up to 15 non-synonymous substitutions (missense and nonsense mutations) have been reported in I-Smads, 30–35 in R-Smads, and up to 150 in Smad4 (see web-app and Figures 4 and 5). A full quarter of the amino acids in Smad4 (130 of 528 residues) are mutated in cancer, including nonsense mutations in 50 different codons. Smad binding proteins for which structural information exists (discussed later) are less frequently mutated in cancer, although some of the mutations affect the Smad binding interface (Figures 7 and 8). For each Smad and their binding proteins we have generated a linear domain structure schematic indicating all the somatic non-synonymous substitutions that introduce single amino acid mutations or stop codons, independent of tumor type (highlighted in red in the web app).
Figure 7. Model of the MH2 domain of Smad7.
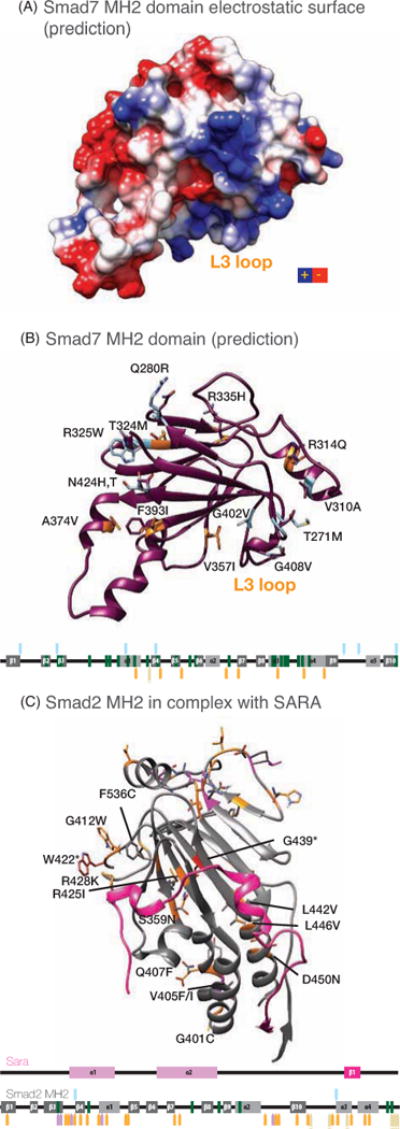
The model of Smad7 has been generated using the Swiss Model homology-modeling server and the structure of Smad4 MH2 domain as template.
A. Electrostatic charge distribution of the Smad7 MH2 domain predicted using UCSF-Chimera. The positively charged patch required for the interaction with Smads and with the receptor (known as L3 loop) is labeled.
B. Predicted secondary structure elements of the Smad7 MH2 domain, using the same orientation as in A. Non-synonymous SNPs and tumor mutations are displayed (blue and orange respectively) and labeled. Map of the MH2 domains of Smad7 with the elements of secondary structure is represented schematically and the positions with SNPs and tumor mutations are colored as in Figure 5.
C. Structure of Smad2 MH2 in complex with Sara. Smad2 is shown in grey, while Sara is depicted in fuchsia. Tumor mutations are labeled and colored in orange on the structure. Side-chains for the wild type and mutated are shown as sticks. A schematic map of Sara is shown underneath the structure, with SNPs, tumor mutations and human-to-vertebrate variations represented as in Figure 5.
Figure 8. Complexes with the Smad1 and Smad3 linkers.
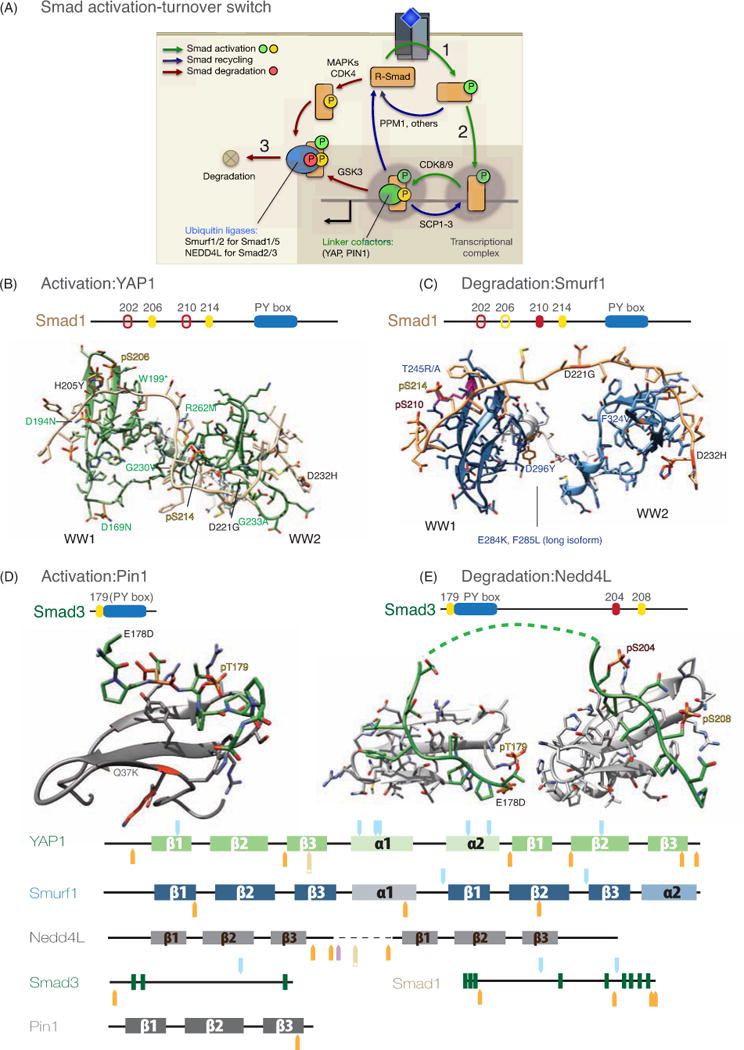
A. Smad activation-turnover switch. Upon receptor-mediated phosphorylation at the C-terminus (1), R-Smads form complexes with Smad4 that are retained in the nucleus (2). In the complex, the linker region of R-Smads is phosphorylated by CDK8 and CDK9, which create binding sites for WW domain containing transcriptional cofactors including YAP1, Pin1, and probably others. CDK8/9 phosphorylation leads the Smad complex to peak transcriptional activity, but it primes the linker region for phosphorylation by GSK3. GSK3-mediated phosphorylation creates binding sites for WW domain containing HECT ubiquitin ligases, which target R-Smads for proteasome-mediated degradation (3). SCP1-3 phosphatases reverse R-Smad linker phosphorylation likely for repeated cycles of transcriptional action, whereas PPM1 and/or other phosphatases reverse C-terminal phosphorylation. The CDK8/9 phosphorylation sites also serve as sites for R-Smad phosphorylation by mitogen activated protein kinases (MAPKs) and cell cycle kinases (CDK4).
B. NMR model of the complex between the human YAP WW1-WW2 pair and the of the Smad1 linker (199–233 di-phosphorylated at S206 and S214) showing the tumor mutations of YAP and Smad1. The schematic representation of Smad1 is shown on top of the complex and the PY box and the phosphorylated residues are labeled. The pair of WW domains and the linker of YAP are shown in green and Smad1 as beige sticks.
C. NMR model of the complex between the WW domain pair of the ubiquitin ligase Smurf1 (shown in slate) and the fragment of Smad1 di-phosphorylated at positions S210 and S214 (shown in beige). The tumor mutations observed for Smurf1 and Smad1 are labeled in blue and black respectively. The phosphorylated residues of Smad1 are also labeled.
D. Solution structure of the Pin1 WW domain bound to the Smad3 pT179[PY] motif. The WW domain is shown as a ribbon representation shown in dark gray and Smad3 is shown as sticks (green). This complex is displayed using the same orientation as that of the Nedd4L WW2 complex (E) to highlight that these WW domains bind to the pT179[PY] site in opposite orientations. The schematic representation of the binding region of Smad3 is shown on top of the complexes of Pin1 and Nedd4L.
E. Nedd4L: NMR-Model of the Nedd4L WW2-WW3 pair in complex with the Smad3 linker tri-phosphorylated at T179, S204 and S208. Nedd4L is shown in gray and Smad3 as green sticks. Smad3 and Nedd4L residues mutated in tumors are indicated.
Tumor mutations of YAP are annotated in green and the corresponding mutations of Smad1 in black. Polymorphisms and the human to vertebrates differences are shown in Supplementary Figures 4 (YAP), 5 (Smurf1) and 6 (Nedd4L).
At the bottom of the figure we represent each protein sequence schematically, with SNPs and tumor mutations highlighted as in Figure 5.
Currently there are no structures of full-length Smad proteins. Structural information exists for the MH1 domain [40–43] and MH2 domain of several R-Smads and Smad4 [44–48] and for the linker region of Smad2 and Smad3 [20, 21]. In the sections that follow we discuss the structural information available and the location and structural impact of variant amino acid residues and mutations based on structural models of the wild-type proteins. To facilitate comparisons, the figures display the side chains of the human reference sequence (GRCh38) on the structure deposited in the PDB, superimposed to side chain differences in vertebrates, human polymorphisms, and tumor mutations, using the SCwrl4 software [49].
In summary, amino acid differences in R-Smad and Co-Smad during evolution are infrequent, suggesting that a high level of functional and structural optimization was achieved in the ancestors of this gene family. However, the frequency of Smad mutations in certain cancers suggests a strong selective pressure to inactivate the pathway, and provides a wealth of information of the functional importance of certain amino acid residues and structural domains of Smad proteins.
The Smad MH1 domain and its DNA binding function
In R-Smads and Smad4, the MH1 domain recognizes a double-stranded DNA sequence motif 5′-CAGAC, called the Smad binding element (SBE). The MH1 domain can also interact with motifs that are more G/C-rich than CAGAC, both in vitro and in vivo, [8, 41, 50] but these interactions are not yet characterized with an atomic detail.
X-ray crystal structures are currently available for the MH1 domains of Smad3 [43], Smad1 [41] and Smad4 [40] in complex with the SBE DNA ((Figure 2B, 5A and Supplementary Figures 1, 2); see also the web-app). In all three cases the MH1 domain consists of four-helices and three sets of antiparallel beta hairpins, one of which is used to interact with DNA. Among vertebrates (excluding ray-finned fishes), the MH1 domain presents only 7 variable residues out of 135 in Smad3 and only one out of 140 in Smad4. These differences are all located in loops. MH1 domains of the other R-Smads are similarly conserved. The corresponding N-terminal region of the I-Smads, Smad6 and Smad7, which is divergent from canonical Smad MH1 domains, is less conserved across species with 10 variant residues among mammals alone.
The fold of the Smad MH1 domain is not found in other proteins. Two of the beta hairpins (β1–β4 and β5–β6) are stabilized through the coordination of a Zn2+ ion with three cysteines and a histidine residue (Figure 2B). These four residues, and their role in the coordination of Zn2+, are hallmarks of the MH1 domain and are invariable in all Smad sequences. A similar Zn2+ coordination arrangement has been detected in homing endonucleases [51]. Several non-synonymous human polymorphisms are located in the proximity to the Zn2+ binding site (Figure 5B and Supplementary Figures 1 and 2). The calculated models for these polymorphisms do not predict any alterations in the main fold of the MH1 domain. Non-conservative mutations have been identified in the MH1 domains of Smad1, Smad3 and Smad4 in cancer (Figure 5 and Supplementary Figures 1 and 2). Two residues (Cys115 and His132) in the Smad4 MH1 domain that directly participate in the Zn2+coordination are mutated in colorectal tumors. These mutations likely compromise Zn2+ binding and alter the domain fold. MH1 domain nonsense mutations in Smad4 occur in tumors, and in the germline of an individual with juvenile polyposis syndrome.
The third hairpin, formed by the β2 and β3 strands, mediates the interaction with DNA. The hairpin is accommodated into the major groove of the DNA and provides specific contacts with three nucleotides (3′-CAGAC/T-5′, contacted nucleotides in bold) using three conserved residues, Arg74, Gln76 and Lys81 (numbers refer to human Smad3). The sequence of this hairpin is identical in R-Smads and Smad4 of all vertebrates and many invertebrates, but it presents differences in the Smad2 orthologue of Hexapoda (insects and wingless arthropods) that may denote a different mode of DNA binding by this protein. Mutations in and around the DNA binding hairpin occur in gastrointestinal and pancreatic tumors, including stop codon mutations (Figure 5C). Other tumor-associated Smad mutations cluster in the second and fourth helices of the MH1 domain helical bundle. These mutations can affect the packing of the helices, and the overall stability of the protein. Functional characterization of several of these mutations (L43S, G65V and R100T) in Smad4 showed poor DNA binding activity in vitro [52] and increased protein instability [53]. R100T was also found to force intra- or intermolecular interactions that trap the protein in inactive conformations [54]. Recent data has shown that R100 is also mutated to Gly and to Trp in colorectal tumors. The variable residues among vertebrates, the non-synonymous polymorphisms and the most salient tumor-association mutations affecting the MH1 domain are highlighted in Figure 5A–D.
An unsolved mystery in this field is the DNA binding function in Smad2. Vertebrates have two Smad2 isoforms that are generated by alternative splicing of a 30-residue segment encoded by exon 3. This segment is highly conserved, and is located right next to the DNA binding hairpin (Figure 2B, the position of the insert is shown with an asterisk). The Smad2 variant that incorporates the exon 3 insert is the most abundantly expressed form in mammalian embryos and adult tissues. This protein was reported to have very limited DNA binding activity [43, 55]. However, Smad2-null mice have a more severe early embryonic lethal phenotype than do Smad3-null or Smad4-null mice [56]. It seems strange that such an important Smad family member would lack DNA binding activity. A deeper understanding of Smad2 interaction with target genes is necessary in order to define the role of Smad2 in transcriptional complexes and how it may interact with DNA in this context.
The Smad MH2 domain and its protein-protein interactions
The MH2 domain mediates the interaction of R-Smads with activated TGF-β receptors, and with partner Smads after receptor-mediated phosphorylation of the Ser-X-Ser motif (Figures 2A, C). The MH2 domain is also a binding platform for cytoplasmic anchors, DNA-binding cofactors, histone modifiers, chromatin readers, and nucleosome positioning factors. MH2 domain structures have been determined for Smad4 [46], Smad2 [48] and Smad3 [44]. The MH2 fold is defined by two sets of antiparallel beta-strands (six and five strands respectively) arranged as a beta-sandwich flanked by a three helical bundle on one side and by a set of large loops and a helix on the other side (Figures 2A, 2C and Figure 6).
Figure 6. Heterotrimer of Smad3 and Smad4 MH2 domains.
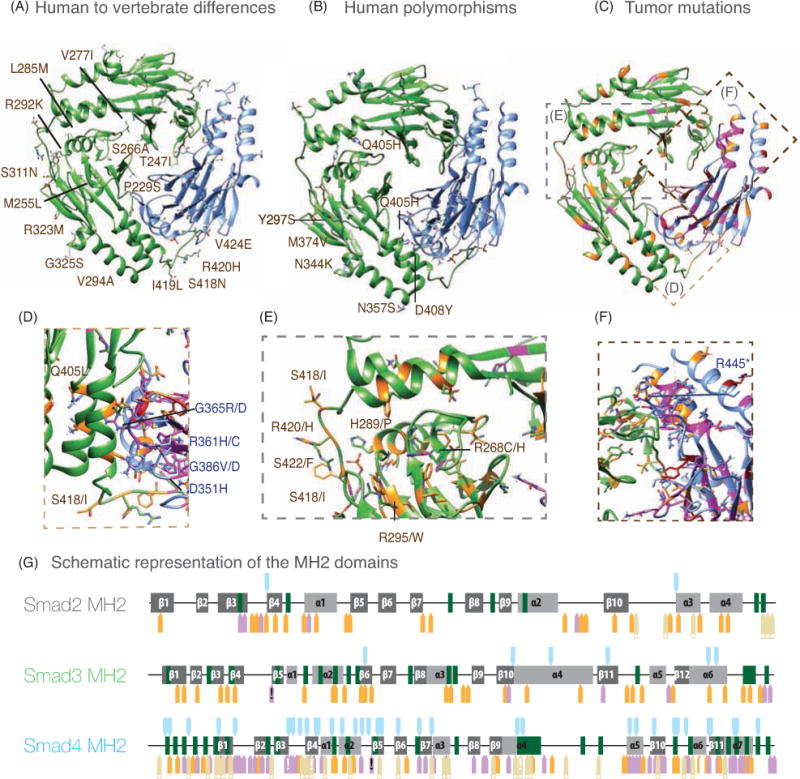
The Smad3 pair of MH2 domains is shown in green and the Smad4 MH2 is displayed in blue. The complex of Smad2 and Smad4 is shown in Supplementary Figure 3.
A. Human to vertebrate differences annotated only in the Smad3 MH2 domain for simplicity.
B. Non-synonymous SNPs of Smad3 and Smad4. As in A, only the Smad3 SNPs are annotated.
C–F. Positions with Tumor mutations are highlighted in the backbone of Smad3 and Smad4 MH2 domains. Close-up views (D–F) of the three interfaces displaying the side-chains of wild type and mutated residues are represented and labeled. For Smad4 only the most frequently found mutations are labeled. Dashed lines are used to demarcate the regions expanded in the close-up views.
G. Maps of the MH2 domains of Smad2, Smad3 and Smad4. The elements of secondary structure of the domain are represented schematically. Positions with Human to Vertebrate differences, Non-synonymous SNPs and Tumor mutations are represented as arrows (colored as in Figure 5). Arrows with a white (!) correspond to the residues labeled in panels D and F while arrows with a black (!) correspond to the equivalent residue in Smad3 and Smad4. These residues are frequently mutated in tumors.
In Deuterostomes, the MH2 sequence is found only in Smad proteins. Protostomes, however, also contain MH2 domains in Expansion and Rebuf proteins, functionally unrelated to Smads [57–59]. The sequence identity of the MH2 domain of Expansion and Rebuf to Smad MH2 domains is low (16%), but the overall domain fold is conserved, except for the presence of an extra alpha-helical region. The MH2 domain exhibits structural and surface electrostatic potential similarity (but no sequence similarity) to the forkhead-associated (FHA) and the interferon-regulatory factor-3 (IRF-3) domains [45, 60].
The MH2 domain of R-Smads contains a positively charged patch next to the L3 loop (Figure 2C) that is believed to interact with the L45 loop of type I receptor subunits [61]. The positively charged patch present in R-Smads is also predicted in the Smad7 MH2 domain (Figures 7A, 7B), potentially explaining the basis for the competition of I-Smads and R-Smads for binding to the receptor. Recognition of R-Smads by type I receptors is achieved with the assistance of Smad-binding proteins such as the Smad anchor for receptor activation (SARA) [62] and the related protein endofin [63]. SARA binds to the MH2 domain of Smad2 and 3 to facilitate phosphorylation by the TGF-β or activin/nodal receptors. Endofin is an anchor for Smad1 to facilitate its phosphorylation by BMP receptors. SARA and endofin contain a phospholipid-binding FYVE finger domain that tethers the proteins on the cytoplasmic surface of early endosomes.
Phosphorylation of the Ser-X-Ser C-terminal motif enhances the formation of Smad MH2 trimers. In the trimers, each MH2 domain interacts with the other two by using distinct interfaces. A set of large loops and the first α-helix on one side of a MH2 domain interact with a three-helical bundle of a neighboring MH2 domain, following a head-to-tail arrangement with three-fold symmetry (Figures 2C, 6 and Supplementary Figure 3). A loop-helix region that is thought to mediate R-Smad binding to the pSer-X-pSer motif of activated type I receptors mediates binding to the pSer-X-pSer motif on a vicinal R-Smad in the trimer. This phosphoserine-binding interface is conserved in all Smad proteins.
The differences in the MH2 domain of vertebrates and the non-synonymous human SNPs are few and conservative (Figure 6A, B). However, many MH2 residues are affected by mutations in cancer. A large number of such mutations have been described, particularly in Smad4, and this number is rapidly increasing as a result of routine tumor genome sequencing in the clinic. Many of the mutations target trimerinterface residues or residues that are critical for secondary structure elements (Figure 6C–G). We have annotated in the figures the mutant residues in Smad2 and Smad3 reported to date, and only the most frequently mutated residues in Smad4. All positions are highlighted in the schematic representations of the secondary structural elements (Figure 6C–G; Supplementary Figure 3).
In Smad4, the G365, R361, G386 and D351 residues all fall in the trimer interface and together account for 1 in every 9 Smad4 mutations in cancer. Several mutations in Smad2 and Smad3 also map to the MH2 trimer interface regions of these proteins and a few in equivalent residues to these found in Smad4 (represented with a ! symbol in the Figures). In breast, kidney, lung and pancreatic carcinomas certain mutations cause truncation of the Smad2 C-terminal tail, preventing receptor-mediated activation. Other mutations affecting Smad2 affect the SARA binding site. The structure of the Smad-binding domain of SARA in complex with the Smad2 MH2 domain is shown Figure 7C [47], with the tumor mutations affecting the SARA-binding surface indicated.
The Smad linker action-turnover switch
The recently elucidated mechanism of Smad action-coupled turnover has revealed the role of the inter-domain linker region in the Smad transcriptional cycle (Figure 8A). This region contains phosphorylation sites and a Pro-Tyr (PY) motif that are involved in regulating peak Smad transcriptional activity as well as the subsequent elimination of Smad molecules that participate in transcription [18, 20, 21]. The linker region has the highest concentration of amino acid differences among vertebrate Smads. However, the phosphorylation sites and the PY motif present in the linker sequences of R-Smads are highly conserved in metazoans. These sites are binding targets for various proteins containing WW domains. WW domains are 38–40 amino acid residue units folded as an antiparallel triple stranded β-sheet [64].
After receptor activation, R-Smad-Smad4 complexes in the nucleus are further phosphorylated in the linker region by CDK8 and CDK9, and subsequently by GSK3 [18, 19, 21]. In the BMP pathway, the CDK8/9 phosphorylation of Smad1 at positions S206 and S214 creates binding sites that are recognized by the WW domains of YAP [18], a transcriptional effector that is negatively regulated by the Hippo pathway [65–67]. These phosphorylations also prime T202 and S210 for subsequent phosphorylation by GSK3, which switches the Smad1 binding preference from YAP to the E3 HECT-domain ubiquitin ligase Smurf1.
YAP and Smurf1 each contain a pair of WW domains. Although WW domains can bind singly to either PY or pSer-Pro motifs, YAP and Smurf1 bind to Smad1 by the ability of their WW pair to collaboratively bind the canonical PY motif plus a phosphorylated motif in the Smad1 linker (Figures 8B, 8C) [18, 20, 21]. Thus, YAP binds the CDK8/9-phosphorylated pS206 in the Smad1 with its WW1 and the PY motif with its WW2 domain (Figure 8B). After the additional phosphorylation of Smad1 by GSK3, Smurf1 becomes the preferred binding partner because it recognizes the pair of phosphorylated residues pS210 and pS214 plus the PY motif (Figure 8C). Similarly, CDK8/9 phosphorylation of Smad2 and Smad3 in the TGF-β pathway creates binding sites for the WW domain of Pin1, and subsequent phosphorylations by GSK3 switches the binding preference for the two WW domains of the HECT ubiquitin ligase Nedd4L (Figures 8D, 8E) [18, 20].
A human Smad1 polymorph has isoleucine in position 214, which may affect the efficiency of the interactions with Smurf1 and YAP proteins (Supplementary Figures 4 and 5). Smad1 alterations in cancer include missense mutations in D221 and D232, residues that participate in key electrostatic interactions with the WW2 domain both in YAP and in Smurf1 complexes (Figures 8B, 8C). Tumor-associated mutations occur in the WW2 domain of Smurf1 [68] that may destabilize the domain folding and compromise the ability to bind Smad1 for degradation (Figure 8C). Similarly, tumor mutations in the Smad3 binding site and in the WW domain of Pin1 have been identified (Figures 8D, 8E and Supplementary Figure 6). In the case of Nedd4L WW2–WW3 region, the mutations are localized in the linker connecting both WW domains.
Structural basis for negative feedback by Smad7
TGF-β signaling activates the transcription of inhibitory Smads (Smad6 and Smad7) for negative feedback within the pathway. Smad7 competes with R-Smads for receptor binding [69]. Additionally, Smad7 helps recruit Nedd4L, Smurf1 and Smurf2 ubiquitin ligases to the TGF-β and BMP receptors [70]. Whereas the interaction of YAP and the ubiquitin ligases with R-Smads is dependent on phosphorylation and requires a WW pair, binding of these proteins to Smad7 involves a single WW domain [71].
The WW domains in Smurf1, Smurf2 and Nedd4L E3 ubiquitin ligases interact with the Smad7 PY motif in a similar manner (Figure 9A and Supplementary Figure 7). For instance, when in complex with the Nedd4L WW2 domain, the PY fragment forms an ordered hairpin (from E205 to D217), with a turn centered at positions Y211-S212-R213 (Figure 9A). Smurf1 and Smurf2 interact with the same Smad7 peptide using the WW2 and WW3 domains respectively (web-app) [71, 72]. The Smad7 segment containing the PY motif interacts with the YAP WW1 domain in a similar manner to that observed for the ubiquitin ligases. In the interaction with Smad1, YAP uses its WW1 domain to bind a phosphoserine motif, while the WW2 domain binds the Smad1 PY motif [20]. Remarkably, the YAP WW1-Smad7 structure does not support a role (or room) for a phosphate group on the Smad7 S206, which is tightly bound by W199. This residue, which is key for ligand recognition in both Smad1 and Smad7 complexes, suffers nonsense mutations in kidney cancers. Furthermore, three other tumor mutations in Smad7 cluster in the PY site, and several truncating mutations map to the WW domains in YAP (Figures 8A, 9A, 9B).
Perspectives
The core components of TGF-β pathway–the receptors and the Smad transcription factors–provide remarkable examples of structural economy for the many functions that these proteins perform. The remarkable sequence conservation across species and time suggest that these proteins are packed with functional capabilities whose presence underwent little evolutionary tinkering once the TGF-β/Smad pathway emerged as a regulatory device in ancestral metazoans. The current availability of a large number of Smad sequences reveals that amino acid differences in R-Smad and Co-Smad evolution are relatively few and largely conservative. In tumors, however, cancer cells must inactivate the tumor suppressive effects of the TGF-β pathway, and do so by accumulating mutations on its essential components. Mutation of one of the many key structural elements of a Smad protein can suffice to confer oncogenic advantage during clonal evolution of cancer cell populations.
The incidence of Smad mutations in different tumors is not merely a reflection of the overall mutational content in these tumors. For example, human melanoma genomes contain, on average, five times more mutations than do pancreatic ductal carcinoma genomes, yet Smad mutations are much more prevalent in these pancreatic cancers than in metastatic melanomas. Differences in the incidence of Smad mutations in different tumor types can be explained by the fact that certain tumors extract an advantage from retaining an intact TGF-β/Smad pathway. TGF-β/Smad signaling is tumor suppressive in most cell types, but if a cancer cell can disable the tumor suppressive output of this pathway downstream of Smads (for example, by eliminating a downstream mediator of Smad-driven apoptosis), this cell will be free to use TGF-β/Smad signaling for metastatic dissemination, organ colonization, or drug resistance.
The type of Smad mutations differs considerably in different types of cancer. For example, the incidence of Smad4 deletions versus missense mutations is higher in pancreatic cancer than in colorectal cancer. This could reflect the presence of additional, pancreas-specific tumor suppressor genes in the Smad4 chromosomal region, or differences in the mutagens that lead to tumor initiation in the different tissues. However, it could also be that different Smad missense mutations selectively disable different tumor-specific suppressive functions or spare different tumor-specific pro-metastatic functions in different cancers.
The analysis of Smad mutations in cancer may shed light on questions that have long remained unanswered. Why do Smads function as trimers? How do Smads suppress cancer? And, how do cancer cells get to use Smad signaling for metastasis? The different ways in which cancer cell neutralize TGF-β/Smad tumor suppression functions, and the analysis of the resulting Smad mutational landscape may provide valuable clues. Tumor mutations cluster both at the DNA-binding and protein interaction surfaces of Smads to perturb–likely to different extents–the structural and functional properties of the proteins. A further analysis of specific Smad mutations could provide unprecedented information on the normal as well as pathophysiological functions of this remarkably conserved and pivotal pathway.
Supplementary Material
Highlights.
Smad proteins are highly conserved in metazoans
Smad structures illuminate the impact of polymorphisms and cancer mutations
Many tumor mutations cluster in the interface of Smad protein complexes
Acknowledgments
We thank R. Domenech and J. Amorós for their help with the web-app, B. Degnan, and B. Venkatesh for their help with the analysis of Smad proteins in A. queenslandica and C.milii respectively, and Y. Zou for his help with cBioPortal data analysis. This work was supported by SAF2011-25119 grant (M.J.M.) and by NIH grant CA34610 (J.M.) and by the BBVA Foundation. M.J.M. is an ICREA Programme Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Web-app
References
- 1.Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–91. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Massagué J. TGF-beta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21(2):166–76. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 7.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118(Pt 16):3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 8.Beyer TA, et al. Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013;5(6):1611–24. doi: 10.1016/j.celrep.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 9.Oshimori N, Fuchs E. The harmonies played by TGF-beta in stem cell biology. Cell Stem Cell. 2012;11(6):751–64. doi: 10.1016/j.stem.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16(3):329–43. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 11.Flavell RA, et al. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikushima H, Miyazono K. TGF-beta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–24. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 13.Massagué J. TGF-beta in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrana JL, et al. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370(6488):341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 15.Qin BY, et al. Smad3 allostery links TGF-beta receptor kinase activation to transcriptional control. Genes Dev. 2002;16(15):1950–63. doi: 10.1101/gad.1002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massagué J. TGF-beta signaling in development and disease. FEBS Lett. 2012;586(14):1833. doi: 10.1016/j.febslet.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 17.Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20(9):556–67. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Alarcón C, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139(4):757–69. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentealba LC, et al. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell. 2007;131(5):980–93. doi: 10.1016/j.cell.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aragón E, et al. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011;25(12):1275–88. doi: 10.1101/gad.2060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao S, et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell. 2009;36(3):457–68. doi: 10.1016/j.molcel.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Feng XH. Regulation of TGF-beta signalling by protein phosphatases. Biochem J. 2010;430(2):191–8. doi: 10.1042/BJ20100427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sapkota G, et al. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281(52):40412–9. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 24.Wrighton KH, et al. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J Biol Chem. 2006;281(50):38365–75. doi: 10.1074/jbc.M607246200. [DOI] [PubMed] [Google Scholar]
- 25.Demagny H, Araki T, De Robertis EM. The tumor suppressor Smad4/DPC4 is regulated by phosphorylations that integrate FGF, Wnt, and TGF-beta signaling. Cell Rep. 2014;9(2):688–700. doi: 10.1016/j.celrep.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Kretzschmar M, et al. A mechanism of repression of TGF-beta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13(7):804–16. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huminiecki L, et al. Emergence, development and diversification of the TGF-beta signalling pathway within the animal kingdom. BMC Evol Biol. 2009;9:28. doi: 10.1186/1471-2148-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards GS, Degnan BM. The dawn of developmental signaling in the metazoa. Cold Spring Harb Symp Quant Biol. 2009;74:81–90. doi: 10.1101/sqb.2009.74.028. [DOI] [PubMed] [Google Scholar]
- 29.Riesgo A, et al. The analysis of eight transcriptomes from all poriferan classes reveals surprising genetic complexity in sponges. Mol Biol Evol. 2014;31(5):1102–20. doi: 10.1093/molbev/msu057. [DOI] [PubMed] [Google Scholar]
- 30.Pang K, et al. Evolution of the TGF-beta signaling pathway and its potential role in the ctenophore, Mnemiopsis leidyi. PLoS One. 2011;6(9):e24152. doi: 10.1371/journal.pone.0024152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava M, et al. The Trichoplax genome and the nature of placozoans. Nature. 2008;454(7207):955–60. doi: 10.1038/nature07191. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava M, et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466(7307):720–6. doi: 10.1038/nature09201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paabo S. The human condition-a molecular approach. Cell. 2014;157(1):216–26. doi: 10.1016/j.cell.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 34.Hahn SA, et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996;56(3):490–4. [PubMed] [Google Scholar]
- 35.Eppert K, et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86(4):543–52. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 36.Hata A, Shi Y, Massagué J. TGF-beta signaling and cancer: structural and functional consequences of mutations in Smads. Mol Med Today. 1998;4(6):257–62. doi: 10.1016/s1357-4310(98)01247-7. [DOI] [PubMed] [Google Scholar]
- 37.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skipper M. Cancer genomics: A panoramic view of cancer. Nat Rev Genet. 2013;14(11):750. doi: 10.1038/nrg3602. [DOI] [PubMed] [Google Scholar]
- 39.Watson IR, et al. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14(10):703–18. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baburajendran N, et al. Structural basis for the cooperative DNA recognition by Smad4 MH1 dimers. Nucleic Acids Res. 2011;39(18):8213–22. doi: 10.1093/nar/gkr500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baburajendran N, et al. Structure of Smad1 MH1/DNA complex reveals distinctive rearrangements of BMP and TGF-beta effectors. Nucleic Acids Res. 2010;38(10):3477–88. doi: 10.1093/nar/gkq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chai J, et al. Features of a Smad3 MH1-DNA complex. Roles of water and zinc in DNA binding. J Biol Chem. 2003;278(22):20327–31. doi: 10.1074/jbc.C300134200. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y, et al. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. 1998;94(5):585–94. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 44.Chacko BM, et al. Structural basis of heteromeric smad protein assembly in TGF-beta signaling. Mol Cell. 2004;15(5):813–23. doi: 10.1016/j.molcel.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 45.Qin BY, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10(11):913–21. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 46.Shi Y, et al. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388(6637):87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- 47.Wu G, et al. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science. 2000;287(5450):92–7. doi: 10.1126/science.287.5450.92. [DOI] [PubMed] [Google Scholar]
- 48.Wu JW, et al. Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-beta signaling. Mol Cell. 2001;8(6):1277–89. doi: 10.1016/s1097-2765(01)00421-x. [DOI] [PubMed] [Google Scholar]
- 49.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77(4):778–95. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown S, et al. Activin/Nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells. 2011;29(8):1176–85. doi: 10.1002/stem.666. [DOI] [PubMed] [Google Scholar]
- 51.Grishin NV. MH1 domain of Smad is a degraded homing endonuclease. J Mol Biol. 2001;307(1):31–7. doi: 10.1006/jmbi.2000.4486. [DOI] [PubMed] [Google Scholar]
- 52.Jones JB, Kern SE. Functional mapping of the MH1 DNA-binding domain of DPC4/SMAD4. Nucleic Acids Res. 2000;28(12):2363–8. doi: 10.1093/nar/28.12.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 2000;97(9):4820–5. doi: 10.1073/pnas.97.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hata A, et al. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388(6637):82–7. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- 55.Zawel L, et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1(4):611–7. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 56.Dunn NR, et al. Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development. 2004;131(8):1717–28. doi: 10.1242/dev.01072. [DOI] [PubMed] [Google Scholar]
- 57.Iordanou E, et al. The novel Smad protein Expansion regulates the receptor tyrosine kinase pathway to control Drosophila tracheal tube size. Dev Biol. 2014;393(1):93–108. doi: 10.1016/j.ydbio.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moussian B, et al. Deciphering the genetic programme triggering timely and spatially-regulated chitin deposition. PLoS Genet. 2015;11(1):e1004939. doi: 10.1371/journal.pgen.1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beich-Frandsen M, et al. Structure of the N-terminal domain of the protein Expansion: an ‘Expansion’ to the Smad MH2 fold. Acta Crystallographica, D. 2015;71 doi: 10.1107/S1399004715001443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takahasi K, et al. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. 2003;10(11):922–7. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 61.Lo RS, et al. The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-beta receptors. EMBO J. 1998;17(4):996–1005. doi: 10.1093/emboj/17.4.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsukazaki T, et al. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95(6):779–91. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 63.Shi W, et al. Endofin acts as a Smad anchor for receptor activation in BMP signaling. J Cell Sci. 2007;120(Pt 7):1216–24. doi: 10.1242/jcs.03400. [DOI] [PubMed] [Google Scholar]
- 64.Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002;513(1):30–7. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 65.Gaspar P, Tapon N. Sensing the local environment: actin architecture and Hippo signalling. Curr Opin Cell Biol. 2014;31:74–83. doi: 10.1016/j.ceb.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 66.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94(4):1287–312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 68.Cheng PL, et al. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69(2):231–43. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 69.Nakao A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–5. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 70.Inoue Y, Imamura T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99(11):2107–12. doi: 10.1111/j.1349-7006.2008.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aragón E, et al. Structural basis for the versatile interactions of Smad7 with regulator WW domains in TGF-beta Pathways. Structure. 2012;20(10):1726–36. doi: 10.1016/j.str.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chong PA, et al. An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J Biol Chem. 2006;281(25):17069–75. doi: 10.1074/jbc.M601493200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.