

Abstract
Recent studies have identified Disrupted-In-Schizophrenia-1 (DISC1) as a strong genetic risk factor associated with schizophrenia. Previously, we have reported that a mutation in the second exon of the DISC1 gene (leucine (L) to proline (P) at amino acid position 100, L100P) leads to the development of schizophrenia-related behaviors in mice. Glycogen synthase kinase-3 (GSK-3) is a serine/threonine protein kinase that interacts with the N-terminal region of DISC1 (aa 1-220) and has been implicated as an important downstream component in the etiology of schizophrenia.
Here, for the first time, we show that pharmacological and genetic inactivation of GSK-3 reverses Pre-Pulse Inhibition (PPI) and Latent Inhibition (LI) deficits as well as normalizing the hyperactivity of Disc1-L100P mutants. In parallel to these observations, interaction between DISC1 and GSK-3α and β is reduced in Disc1-L100P mutants. Our data provide genetic, biochemical and behavioral evidence for a molecular link between DISC1 and GSK-3 in relation to psychopathology and highlights the value of missense mutations in dissecting the underlying and complex molecular mechanisms of neurological disorders.
Keywords: DISC1, GSK-3, TDZD-8, genetic mouse model, Schizophrenia
INTRODUCTION
Schizophrenia has a complex psychopathology that includes positive, negative and cognitive clinical symptoms (reviewed in Tamminga and Holcomb, 2005). However, the etiology of this disease remains largely unknown. In recent years, significant effort has been made to characterize the underlying causes of schizophrenia and this work has revealed that there is indeed a strong genetic component to the illness (over 130 genes have been reported to predispose to schizophrenia), important influence of environmental factors, neurodevelopmental defects, as well as dysfunction of different neurotransmitter systems (Ross et al., 2006; Harrison and Weinberger, 2005; Lisman et al., 2008). Several independent genetic studies have confirmed DISC1 (Disrupted-in-Schizophrenia-1) as a one of the most substantiated risk factors for schizophrenia (reviewed in Harrison and Weinberger, 2005; Chubb et al., 2008; Brandon et al., 2009; Jaaro-Peled et al., 2009). Indeed, the DISC1 gene has been identified at one breakpoint of a chromosomal t(1;11) (q42.1;q14.3) translocation that co-segregates in a Scottish family with major mental illness, including schizophrenia, bipolar disorder, and major depression (Millar et al., 2000; Blackwood et al., 2001). In vivo, DISC1 protein appears to function as a molecular scaffold that interacts with a variety of protein substrates (Camargo et al., 2007) that influences processes such as cytoskeletal organization, cell cycle, signal transduction, intracellular transport/exocytosis, and development of the central nervous system (Chubb et al., 2008; Brandon et al., 2009; Jaaro-Peled et al., 2009). Only a few of the DISC1 interactors, such as cAMP-specific phosphodiesterase 4B (PDE4B) (Millar et al., 2005; Murdoch et al., 2007), NudE-like (NUDEL), lissencephaly-1 (LIS1) (Taya et al., 2007), have been investigated in detail and most of these were selected due to their neuronal function or/and involvement in the etiology of psychiatric illness (Chubb et al., 2008). So, distinct PDE4 isoforms, interacting with specific sites/signaling complexes, regulate cAMP responses in the cell (reviewed in Houslay et al., 2005, 2007; Boswell-Smith et al., 2006), and, hence, many aspects of neuronal function. PDE4 has been reported as risk factor for schizophrenia (Millar et al., 2005; Pickard et al., 2007; Tomppo et al., 2009), and it’s inhibitor, rolipram, acts as a cognitive enhancer (Barad et al., 1998; Zhang et al., 2004; Davis and Gould, 2005), antidepressant (O’Donnell and Zhang, 2004; Zhang et al., 2006) and antipsychotic (Maxwell et al., 2004; Kanes et al., 2007; Lipina and Roder, 2010).
The accumulation of all these data has facilitated the generation of DISC1 genetic mouse mutant models to study schizophrenia (Clapcote et al., 2007; Hikida et al., 2007; Li et al., 2007a; Kvajo et al., 2008; Pletnikov et al., 2008; Shen et al., 2008). We have previously shown that mutation of murine DISC1 (mutations in exon 2 of the DISC1 gene result in substitution of leucine (L) to proline (P) at amino acid position 100, denoted as Disc1-L100P line) leads to behavioral and pharmacological phenotypes modeling important aspects of human psychopathology (Clapcote et al., 2007). Furthermore, this L100P mutation in DISC1 leads to reduced binding with PDE4B and PDE4D (Clapcote et al., 2007; Murdoch et al., 2007). Rolipram treatment effectively reversed the schizophrenia-relevant behaviors of Disc1-L100P mutant mice (Clapcote et al., 2007). These studies provided the first indications that the inability of DISC1 to correctly function as a molecular scaffold could be an important factor in the etiology of schizophrenia (Millar et al., 2005; Pickard et al., 2007; Tomppo et al., 2009).
Recently, glycogen synthase kinase-3β (GSK-3β) has been identified as a novel DISC1 interactor (Mao et al., 2009). GSK-3 is highly conserved serine/threonine protein kinase that is ubiquitously expressed as two isoenzymes: GSK-3α and GSK-3β, encoded by distinct genes (Woodgett, 1990). GSK-3 is unusual as a protein kinase as it is active under basal conditions (Woodgett, 1994; Sutherland et al., 1993; Stambolic and Woodgett, 1994) and is rapidly inhibited by diverse signals such as insulin, Wnts, Sonic Hedgehog (Shh) and Notch signaling pathways (reviewed in Frame and Cohen, 2001; Grimes and Jope, 2001; Kockeritz et al., 2006). For example, insulin leads to rapid inactivation of GSK-3 by promoting phosphorylation at a N-terminal serine residue (Ser 21 of GSK-3α and Ser 9 of GSK-3β) (reviewed in Patel et al., 2004; Kockeritz et al., 2006) by protein kinase B (PKB/Akt) (Cross et al., 1995). Within the Wnt pathway, GSK-3 function is modulated independently of serine phosphorylation such that the β-catenin phosphorylation is suppressed. Thus, multiple mechanisms have evolved to regulate GSK-3 function and to provide specificity within diverse signaling pathways eliciting their biological effects (McNeill and Woodgett, 2010).
GSK-3 is widely expressed in the brain (Perez-Costas et al., 2010), and has been implicated in fundamental neuronal functions, such as neurodevelopment (Kim et al., 2009), neurotransmitter function (Li et al., 2007b; Beaulieu et al., 2004), neuroinflammation (Beurel et al., 2009) and synaptic plasticity (Peineau et al., 2007). Whilst there are no disease-associated mutations in the GSK-3 genes, there is accumulating evidence that implicates deregulation of GSK-3 in the pathology of Alzheimer’s disease and in neuropsychiatric disorders, including bipolar disorder and schizophrenia. GSK-3 inhibitors have been beneficially used to correct neurological conditions in animals such as bipolar disorder and depression (Gould et al., 2004; Kaidanovich-Beilin et al., 2004; Rosa et al., 2008, Beaulieu et al., 2008), Alzheimer’s Disease (Munoz-Montano et al., 1997; Noble et al., 2005; Engel et al., 2006; Huang and Klein, 2006) and schizophrenia (Beaulieu et al., 2004).
Indeed there is evidence of impaired Akt/GSK-3β signaling pathway in subjects with schizophrenia (reviewed in Lovestone et al., 2007; Koros and Dorner-Ciossek, 2007; Beaulieu et al., 2009; Freyberg et al., 2010). This pathway has been implicated in the mechanisms of action of antipsychotic and psychoactive drugs (Mai et al., 2002; Svenningsson et al., 2003; Emamian et al., 2004; Beaulieu et al., 2004; Li et al., 2007b). Phosphorylation of GSK-3β at the inhibitory site (Serine 9) is reduced in the peripheral lymphocytes and brains of schizophrenia patients (Emamian et al., 2004). Overexpression of GSK-3β in mice produced manic-like behavior (Prickaerts et al., 2006). Moreover, recent genetic studies found direct association between GSK-3 polymorphism and schizophrenia (Souza et al., 2008; Benedetti et al., 2010).
More recent data indicate the potential involvement of DISC1 within the Akt/GSK-3 and Wnt/GSK-3 axis. DISC1 directly interacts with the Akt binding partner Girdin (Enomoto et al., 2009;), preventing Akt activation in vitro, affecting the proliferation of neuronal precursors. Meanwhile, Mao with colleagues (2009) recently found that when over-expressed, the N-terminus of DISC1 directly interacted with GSK-3β, suppressing its activity and perturbing its ability to regulate the Wnt/β-catenin pathway and proliferation of neuronal progenitors. Furthermore, administration of the relatively non-specific, arylindolemaleimide class of small molecule GSK-3 inhibitors (reviewed in Meijer et al., 2004), SB216763, rescued the behavioral effects of lentivirally-induced DISC1 suppression in the adult dentate gyrus (Mao et al., 2009). These and other data support a role for DISC1 as a conductor mediating protein-protein interactions and regulation of multiple cellular signaling pathways, and that perturbation in the dynamics of these functions could lead to abnormalities such as those observed in schizophrenia.
Given the biochemical and pharmacological link of DISC1 to the Akt/GSK-3, GSK-3/Wnt/β-catenin axis, alongside the fact that N-terminus of DISC1 (aa 1–220) is required for GSK-3 interaction, we sought to investigate the in vivo consequence of the Disc1-L100P mutation on GSK-3 function and its impact on schizophrenia-like behavior displayed in Disc1-L100P mouse mutants.
In this study we show that the schizophrenia-like behavior of Disc1-L100P mouse mutants is reversed by the administration of the specific, non-ATP competitive GSK-3 inhibitor, TDZD-8 (Martinez et al., 2002). Furthermore, genetic deletion of GSK-3α was equally as effective in reversing the behavioral defects in the Disc1-L100P mice. Finally, we demonstrate that whilst the enzymatic activity of both GSK-3 isoforms remained unaltered, the DISC1 x GSK-3 interactions were significantly impaired by the Disc1-L100P mutation.
Together, these results demonstrate an important in vivo role for interaction between DISC1 and GSK-3 and provide the basis of utilizing ENU missense variants for dissecting molecular mechanisms and establishing the basis of action of psychopharmacologicals.
MATERIALS AND METHODS
Animals
Disc1-L100P homozygous mutants and their wild type littermates were generated as previously described (Clapcote et al., 2007). Disc1-L100P mice were backcrossed for 12–16 generations to C57BL/6 mice. Experiments were performed with 12–16 week old male mice. Male mice were tested at 12–14 weeks old in the open field, PrePulse Inhibition (PPI) and Acoustic Startle Response and at 16 weeks of age, in latent inhibition (LI). GSK-3α heterozygous mice were backcrossed for 6 generations to C57BL/6 mice (MacAulay et al., 2007; Kaidanovich-Beilin et al., 2009) and were bred with Disc1-L100P mice and the resultant offspring intercrossed to obtain wild type, heterozygous and homozygous Disc1-L100P littermates having none or one copies of the GSK-3α gene. Groups of three to five same-gender littermates were housed in filtered polycarbonate cages under controlled temperature (21 ± 1° C), lighting (lights on: 7 am – 7 pm) and humidity (50–60%). The animals were given ad libitum sterile food (Purina mouse chow) and water, except in the latent inhibition experiments (see below). All animal procedures were approved by the Animal Management Committee of Mount Sinai Hospital and followed the requirements of the Province of Ontario Animals for Research Act 1971 and the Canadian Council on Animal Care.
Protein preparation
Mouse striatal tissues were homogenized in RIPA buffer containing 50 mM Tris-Cl, pH 7.6, 150 mM NaCl, 2 mM EDTA, 1 mM PMSF plus 1% Igepal CA-630, 0.5~1% sodium deoxycholate, 1% Triton X-100, and protease inhibitor mixture (5 μl/100 mg of tissue; Sigma). After centrifugation at 10.000 x g at 4°C for 20 min, the supernatant was extracted and protein concentrations were measured.
Co-immunoprecipitation and Western Blot analysis
Twenty μl of protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA) were washed once with RIPA buffer. Solubilized mouse striatal tissues (500~700 μg of protein) were pre-incubated with 20 μl of protein A/G agarose for 1 hour at 4°C to reduce non-specific binding. The beads were incubated in the presence of primary antibodies, anti-DISC1 (Invitrogen, cat# 40-6800, Carlsbad, CA) or rabbit IgG (Cat# A0545, Sigma Aldrich, St. Louis, MO, 1~2 μg), in RIPA buffer for 4 hours at 4°C followed by the addition of solubilized striatal extracts. The mixture was then incubated for 12 hours at 4°C with gentle rotation. Pellets were washed four times in the RIPA buffer each time for 5 minutes. Samples were boiled for 5 minutes at 100°C in 20 μl 2XSDS sample buffer or incubated at 37°C for 45 minutes to move the IgG band up to ~100 kDa, and then subjected to SDS-PAGE. Approximately 50 μg of tissue-extracted protein was used as a positive control. Proteins were then transferred electrophoretically to a 0.2-μm nitrocellulose membrane. Nonspecific binding sites were blocked by incubating the blots in 5% non-fat dry milk for 1 hour at room temperature and incubated with an appropriate primary antibody, anti-GSK-3α (Cat# 9338) or anti-GSK-3β (Cat #9315, Cell Signaling Technology, Danvers, MA), at 4°C overnight with a gentle rotation. Total GSK-3α or GSK-3β bound to DISC1 as well as direct DISC1 (anti-DISC1, cat# LS-B2805, Lifespan Biosciences, Seattle, WA) immunoprecipitation levels were determined using horseradish peroxidase-linked secondary antibodies and enhanced chemiluminescence detection. The intensity of each protein band was quantified by densitometry using ImageJ software. Data were normalized to direct IP proteins and presented as means ± S.E.M.
Phosphorylation of GSK-3 at Ser21/Ser9, Y276/216 and protein levels of GSK-3α/β and β-catenin (Western Blot analysis)
Tissue extracts from striatum and hippocampus were prepared by homogenization in 1% SDS solution supplemented with 10nM okadaic acid, 1mM sodium-orthovanadate and Protease Inhibitors Cocktail tablets (Roche Applied Science) and boiled for 10 min, followed by centrifugation at 16,000 g for 5 min at 15°C to remove particulate matter. For the analysis of cytosolic β-catenin, frozen striatum and hippocampus tissue were homogenized using hypotonic buffer containing 50 mM Tris pH7.4, 1 mM EDTA, 25 mM NaF, 1 mM Na-orthovanadate, 5 mM Na beta-glycerophosphate, phosphatase inhibitor cocktail (Sigma), and a protease inhibitor cocktail tablet (Roche). Tissue homogenates were cleared by ultracentrifugation at 80,000 rpm for 30 min at 4°C, and protein concentration was determined by a Bio-Rad DC protein assay. The supernatants were stored at -80°C until analysis. Equal amounts of protein extracts (20–40 μg), determined by BCA protein assay (Thermo Scientific), were boiled with Laemmli sample buffer and were separated on 10% SDS PAGE and transferred to PVDF membrane (Millipore). Blots were immunostained overnight at 4°C with the following antibodies: anti-phospho-GSK-3α/β Ser21/9 (1:1000, Cell Signaling Technology); anti-phospho-GSK-3α/β Y276/Y216 (1:2000, Millipore); anti-GSK-3 (1:1000, Invitrogen), anti-GSK-3α (1:1000, Cell Signaling Technology, clone (anti-β-catenin (1:2000, BD Biosciences) and anti-GAPDH (1:10000, Abcam). Immune complexes were detected using appropriate peroxidase-conjugated secondary antibodies along with a chemiluminescent reagent (ThermoScientific).
GSK-3 activity assay
Mice striatum and hippocampus tissues were homogenized in 50mM Tris buffer, containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM glycerophosphate, 50 mM NaF, 5 % glycerol, 1 % Triton x-100, and with phosphatase inhibitor cocktail (Sigma) and protease inhibitor cocktail tablet (Roche) using a 2 ml Kontes Tissue grinder. Tissue homogenates were cleared by centrifugation at 16,000 g for 15 min at 4 °C and protein concentration determined by BioRad DC protein assay. 250 mg of striatum or hippocampus lysates was incubated with either rabbit IgG (1 mg), 2.5 μl of GSK-3α (Cell Signaling Technology #4818 clone D80D1) or 2.5 μl of GSK-3β (Cell Signaling Technology #9315) overnight at 4°C with gentle rotation followed by the addition of 10 ml packed volume of protein G Sepharose for a further incubation at 4°C for 4h. The pellets were washed 3 times with lysate buffer and once with kinase assay wash buffer, containing 8mM MOPS, 0.2mM EDTA, 0.1mM EGTA. 10mM MgAc, 2.5mM 2- glycerophosphate before being assayed at 30° C for 15 min in a 50 μl reaction volume containing 8 mM MOPS (pH 7.4), 0.2 mM EDTA, 15 mM MgCl2, 0.1 mM EGTA, 2.5 mM protein kinase A inhibitor (PKI, Upstate Biotech), 10 mM magnesium acetate, 5 mM Na β- glycerophosphate, 25 mM glycogen synthase 2 peptide (Upstate Biotech) and initiated with 2 mCi/ml [γ-32P] ATP (6000 Ci/mmol). The reactions was stopped by placing samples on ice, briefly spun and 40 μl of the reaction mixture was spotted onto 2 cm2 squares of Whatman phosphocellulose P81 paper that was washed five times with 0.75% phosphoric acid and once with acetone. Incorporation of radioactivity in the peptide was determined by using a Beckman LS 6500 liquid scintillation counter. GSK-3 kinase activity is expressed relative to that for WT control (which is set at 100% for each GSK-3 isoform assayed) and corrected for potential differences in immunoprecipitated GSK-3 amount. Values are the means ± SEM of results for four different tissue samples.
Behavioral Studies
Behavioral tests were done between 9:00am and 4pm. All pharmacological experiments were performed on drug-naïve Disc1-L100P male mice and their wild-type (WT) littermates. Experimental groups of genetic crosses between Disc1-L100P and GSK-3α deficient mice were sex balanced. Animals were allowed a minimum 5 days interval between tests. Physical assessments of Disc1-L100P mutants and GSK-3 deficient mice have been previously described (Clapcote et al., 2007; Kaidanovich-Beilin et al., 2009) and did not detect any physical, sensory functions and reflex deficits in the studied animals. We also found that fur condition, whisker presence, eye blink, ear twitch and righting reflex were normal in the studied mouse genetic crosses, assessed as described (Miyakawa et al., 2001). Priori to all experiments mice were acclimatized to the experimental room for 30 min. The behavioral equipment was cleaned with 70% ethanol between mice.
Locomotor activity
Each mouse was placed in a 41cm x 41cm x 33 cm3 activity monitoring chamber (AccuScan Instruments Inc, Ohio, USA), and activity was measured for 30 min by an automated recording system connected to infrared beams (VersaMax Animal Activity Monitoring System, Ohio, USA). Data were analyzed by two-way ANOVAs followed by Fisher least significance difference (LSD) post-hoc test.
Prepulse Inhibition and Latent Inhibition
Deficits in attention and information processing are considered a central feature of schizophrenia, leading to stimulus overload, cognitive fragmentation, and thought disorders (Perry and Braff, 1994). The prepulse inhibition and latent inhibition are the most common methods to quantify information-processing deficits in schizophrenia with a reasonable amount of face, predictive, and construct validity (Geyer and Ellenbroek, 2003).
Prepulse Inhibition (PPI) of Acoustic Startle Response (ASR)
PPI procedure was conducted as previously described (Lipina et al., 2005). Five types of trials were used. Pulse alone trials consisted of a single white noise burst (120 dB, 40ms). Prepulse + pulse trials (PP69P, PP73P, PP81P) consisting of a prepulse of noise (20 ms at 69, 73, or 81 dB respectively) followed 100 ms after prepulse onset by a startling pulse (120 dB, 40 ms). No-stimulus trials consisted of background noise only (65 dB). Sessions were structured as follows: 1) 15-min acclimation at background noise level; 2) five Pulse trials; 3) ten blocks each containing all five trials (Pulse, PP69P, PP73P, PP81P, No-stimulus) in pseudorandom order; 4) five Pulse trials. The force intensity for each trial was recorded as the startle level. The percentage PPI induced by each prepulse intensity was calculated as [1-(startle amplitude on prepulse trial)/(startle amplitude on pulse alone)]*100%. PPI data were analyzed by two-way ANOVA with repeated measures followed by Fisher least significance difference (LSD) post-hoc test.
Latent Inhibition (LI) of fear conditioning
LI was measured as previously described (Lipina et al., 2005). Prior to the beginning of each LI experiment, water was removed from the cages for 24 h and mice were then trained to drink in the experimental chamber for 5 days, 15 minutes per day (Pre-Training session). On each daily pre-training session, mice were acclimated to the chamber without access to the sipper tube for 5 minutes then the guillotine door was opened. Latency to the first lick and number of licks were recorded for 15 min. The conditioning chambers had clear Plexiglas walls (ENV-307W) and removable floors consisting of either metal rods, used on pre-exposure and conditioning days or a flat piece of aluminum used on Pre-training session, baseline drinking, and test days, and were equipped with a bottle with a metal tip (sipper tube). On the Pre-exposure and Conditioning accesses to the bottle were prevented by a guillotine door. The LI procedure was conducted on days 6–9 and consisted of Pre-exposure, Conditioning, Lick Retraining and Test sessions.
Pre-exposure
The pre-exposed (PE) mice received 40, 85 dB-white noise presentations with an inter-stimulus interval of 60 s. The non-pre-exposed (NPE) mice were confined to the chamber for an identical period of time without receiving the stimuli.
Conditioning
All mice received fear conditioning to the noise stimulus on that day. Two noise-shock pairings were used to produce LI in control animals (Lipina et al., 2005) and 4 noise-shock pairings were used in the experiment with TDZD-8 on C57BL/6 mice in order detect facilitation of LI. Five minutes after the start of the session, a 10-s white noise was followed by a 1-s 0.37 mA foot shock. The noise-shock pairings were given 5 min apart and after the last pairing, mice were left in the experimental chamber for an additional 5 min. Mice received 15 min access to water in their home cages after pre-exposure and conditioning sessions.
During the Test session each mouse was placed in the chamber and when the mouse completed 75 licks, the noise was presented and lasted until the mouse reached lick 101. Time to first lick, time to complete licks 50–75 (before noise onset; A period) and time to complete licks 76–101 (after noise onset; B period) were recorded. The degree of lick suppression was calculated as a suppression ratio A/(A+B). A lower suppression score indicates a stronger suppression of drinking. LI consists of lower suppression of drinking (higher suppression ratio) in the pre-exposed compared to the non-pre-exposed mice.
Drugs
4-benzyl-2-methyl-1,2,4,-thiadiazolidine-3,5-dione (TDZD-8) (2.5 mg/kg, 7.5 mg/kg and 15 mg/kg, i.p, Calbiochem) was dissolved in saline containing 0.3% Tween-20 (BioRad). Doses of TDZD-8 were chosen based on the literature (Beaulieu et al., 2004) and preliminary experiments. TDZD-8 was given with 5 min as injection-test intervals. The drug was administered in a volume 10 ml/kg.
Data analysis
Statistical analyses were completed using Statistica (Statsoft, Tulsa, OK, USA). Biochemical data from immunoblotting and enzymatic activity were analyzed by two-tailed t test for independent samples. Locomotor activity data were analyzed by two-way ANOVA with the main factors genotype and drug treatment or genotype and gender as a between-subject factors and testing intervals as a repeated measurement factor. The percentage of inhibition of startle and basic startle response was analyzed with two-way ANOVA with genotype and drug treatment effects or genotype and gender as a between-subjects factor, and prepulse intensity as a repeated measurement factor. Suppression ratios data obtained in LI experiments were analyzed by three-way ANOVAs with main factors of pre-exposure (0, 40), genotype (Disc1-L100P; WT) and drug treatment (vehicle; TDZD-8) or pre-exposure (0, 40), genotype (Disc1-L100P+/+-GSK3α+/+; Disc1-L100P+/−/GSK-3α+/+; Disc1-L100P−/−/GSK-3α+/+; Disc1+/+/GSK-3α+/−; Disc1-L100P+/−/GSK-3α+/−; Disc1-L100P−/−/GSK-3α+/−) and gender (male; female). No statistical analyses detected significant gender effects and, hence, data were combined between sexes. Fisher least significance difference test (LSD) was used for post-hoc comparisons when ANOVAs yielded statistically significant main effects or interactions with significance set at p < 0.05. Values in figures are expressed as mean ± S.E.M.
Results
Effects of Pharmacological inhibition of GSK-3 on behavior of Disc1-L100P mutants
Given there is accumulative evidence pointing towards using GSK-3 inhibitors as therapy for neurological diseases such as bipolar disorder and depression (Gould et al., 2004; Kaidanovich-Beilin et al., 2004; Rosa et al., 2008, Beaulieu et al., 2008), Alzheimer’s Disease (Munoz-Montano et al., 1997; Noble et al., 2005; Engel et al., 2006; Huang and Klein, 2006) and schizophrenia (Beaulieu et al., 2004), we sought to investigate the effect of GSK-3 inhibition on the schizophrenic phenotypes exhibited by Disc1-L100P mutant mice. TDZD-8 is a potent and selective inhibitor of GSK-3. Unlike most other synthetic inhibitors, TDZD-8 does not interfere with ATP binding and acts as a noncompetitive inhibitor of the substrate binding site (Martinez et al., 2002). We evaluated the effect of in vivo GSK-3 inhibition on hyperactivity, PPI and LI deficits in Disc1-L100P mutant mice.
First, we performed the open field test and measured the distance traveled in 30 min by WT and Disc1-L100P mice under vehicle-control and TDZD-8 treated conditions (2.5 mg/kg, 7.5 mg/kg, 15.0 mg/kg). Compared to WT mice, Disc1-L100P mutant mice showed abnormally elevated motor activity, as detected by two-way ANOVA: a significant main effect of genotype [F1, 50 = 17.6, p < 0.001], drug treatment [F3, 50 = 21.9, p < 0.001], testing interval [F5, 250 = 35.7, p < 0.001], and gene x drug [F3, 50 = 11.5, p < 0.01], gene x testing interval [F5, 250 = 2.7, p < 0.05], drug x testing interval [F15, 250 = 5.9; p < 0.001] and gene x drug x testing interval interactions [F5, 250 = 2.21, p < 0.01]. Vehicle-treated Disc1-L100P mutant mice showed the expected increase in motor activity compared to WT (p’s < 0.001 at 0–10 min; p’s < 0.05 at 10–25 min), confirming previous findings (Clapcote et al., 2007). The amount of time spent and distance traveled near margins, and in the center of the open field during the first five minutes was not different between the genotypes (data not shown). TDZD-8 induced a dose-dependent effect on both Disc1-L100P and WT mice. The drug had no effect at the lowest dose (2.5 mg/kg) on mice of both genotypes, but effectively normalized hyperactivity in Disc1-L100P mutants at 7.5 mg/kg (all p’s < 0.001) without effects on WT animals (p > 0.05) and decreased locomotion in both WT and Disc1-L100P mice at 15 mg/kg (all p’s < 0.001) (Fig. 1A–C).
Fig. 1.
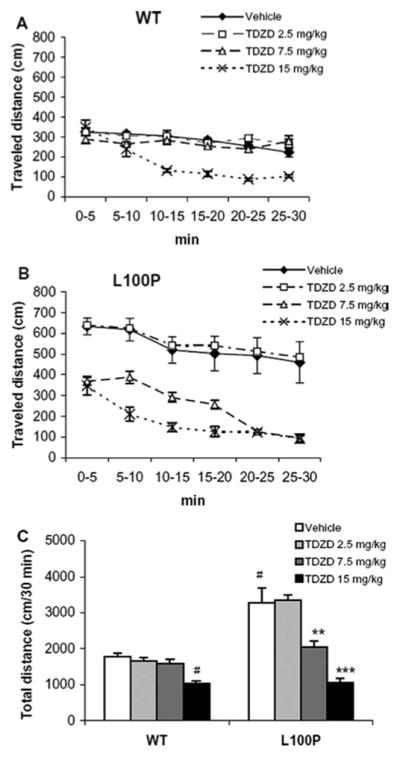
A–C. Effects of TDZD-8 (0mg/kg, 2.5 mg/kg, 7.5 mg/kg and 15 mg/kg) on locomotor activity in WT and Disc1-L100P mutants. Locomotor activity curve in (A) - WT (n = 6–8 per group) and (B) - Disc1-L100P mice (n = 6–9 per group) depicted with 5 min intervals over 30 min, expressed as traveled distance in centimeters (cm); (C) total locomotor activity. # - p < 0.001 in comparison with vehicle-treated WT mice; ** - p < 0.01; *** - p < 0.001 – in comparison with vehicle-treated mice within Disc1-L100P group.
Next, we performed prepulse inhibition analysis (PPI) in WT and Disc1-L100P mice, treated with TDZD-8 (Fig. 2A). Administration of TDZD-8 at 7.5 mg/kg effectively reversed the PPI defects in the Disc1-L100P mice, restoring them to within WT parameters (Fig. 5A; p’s < 0.001), without an effect on ASR [F1, 45 = 0.004, p > 0.05] (Fig. 2B), There was a significant effect on prepulse intensities [F2, 90 = 33.2, p < 0.01], genotype [F1, 45 = 64.2, p < 0.001], drug treatment [F2, 45 = 41.5, p < 0.001], and gene x drug interactions [F1, 45 = 31.8, p < 0.001]. Notably, the GSK-3 inhibitor reversed the PPI deficits in Disc1-L00P mutants, indicating that TDZD-8 appeared to behave similarly to antipsychotic drugs.
Fig. 2.

A–D. Effects of TDZD-8 on (A) sensorimotor gating, (B) Acoustic Startle Response (n = 7–12 per group), (C) Latent Inhibition (n = 8–9 per group) in WT and Disc1-L100P mice and on LI in C57BL/6 mice with 4 conditioning trials (n = 8–10 per group) (D). (A) Figure depicts percent PPI at three prepulses (69, 73 and 81 dB) in WT and Disc1-L100P mice. Background noise was set of 65 dB. (B) Figure depicts the magnitude of the acoustic startle response in WT and Disc1-L100P mice. Startle intensity was 120 dB. # - p < 0.001 in comparison with vehicle-treated WT mice; * - p < 0.001 in comparison with vehicle-treated Disc1-L100P mutants. (C). Mean suppression ratios of PE and NPE WT and Disc1-L100P mice. TDZD-8 at 7.5 mg/kg was administrated before preexposure and conditioning sessions and reversed the LI deficit in Disc1-L100P mice due to the improved capacity to ignore irrelevant tone of PE group (p < 0.001). * - p < 0.001 in comparison with PE group; # - p < 0.001 in comparison with PE vehicle-treated Disc1-L100P mice. (D). Mean suppression ratios of C57BL/6 mice in LI procedure with 40 PE and 4 conditioning trials. TDZD-8 at 7.5 mg/kg facilitated the disrupted LI (p < 0.001). * - p < 0.001 – in comparison with PE group, # - p < 0.001 in comparison with PE vehicle-treated C57BL/6 mice.
Fig. 5.
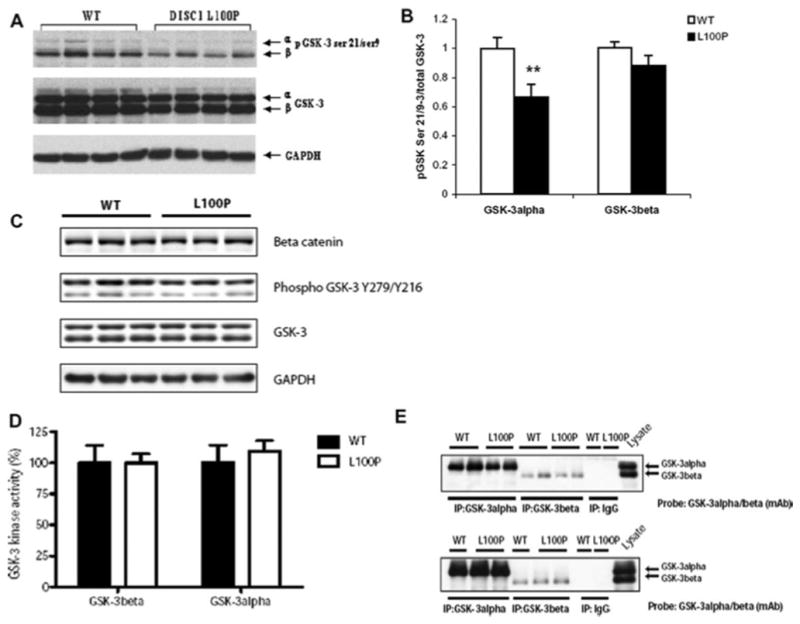
A–D. Biochemical characterization of GSK-3 functional state in the Striatum of Disc1-L100P mice. (A) Representative immunoblots are shown, probed with an antibody against total GSK-3α and GSK-3β and with antibodies to p-GSK-3α at Ser21, p-GSK-3β at Ser9 as well as GAPDH as a loading control. (B) Optical densities are shown, reflecting normalized phosphorylation of GSK-3α and GSK-3β over the total GSK-3α and GSK-3β, respectively (n = 7–15 per group). ** - p < 0.01 – in comparison with WT (C). Representative immunoblots are shown, probed with an antibody against β-catenin, total GSK-3α and GSK-3β and with antibodies to p-GSK-3α at Tyr 276, p-GSK-3β at Tyr 216, as well as GAPDH as a loading control. Equal loading of the lanes was assessed using an anti-GAPDH antibody and the blots are representative of 2 independent experiments. (D). GSK-3 kinase activities in WT and L100P Disc1 mutant mice. Striatum tissues were extracted from WT and L100P Disc 1 mice, lysed with Buffer H and immunoprecipitated for GSK-3α or GSK-3β with their activities assayed as described in materials and methods. GSK-3 kinase activity is expressed relative to that for WT control (which is set at 100% for each GSK-3 isoform assayed) and corrected for potential differences in immunoprecipitated GSK-3 amount (E). Values are the means +/− SEM of results for four different tissue samples.
Figure 2C depicts the mean suppression ratios of the pre-exposed (PE) and non-pre-exposed (NPE) groups of Disc1-L100P and WT mice in 2 drug conditions (vehicle; TDZD-8 at 7.5 mg/kg). The eight experimental groups did not differ in their times to consume water between 50–75 licks (all p’s > 0.05; overall mean A period = 8.4 sec). Three-way ANOVA found a main effect of pre-exposure [F1, 60 = 260.2, p < 0.001], genotype [F1, 60 = 57.4, p < 0.001], gene x drug interactions [F1, 60 = 48.2, p < 0.001] and drug x pre-exposure interactions [F1, 60 = 11.9, p ≤ 0.01]. As can be seen, there was no LI in vehicle-treated Disc1-L100P animals (p > 0.05), whereas Disc1 mutant mice that received TDZD-8 exhibited LI (p < 0.001) similar to vehicle-treated WT and TDZD-8-treated WT (both p’s < 0.001).
The capacity of TDZD-8 to reverse the PPI deficit and disrupted LI associated with the Disc1-L100P mutation suggest an antipsychotic action. To investigate this further, we asked whether TDZD-8 (at 7.5 mg/kg as effective dose) could also facilitate LI in C57BL/6 mice with 40 pre-exposure and 4 conditioning trials, a process that disrupts LI in mice (Lipina et al., 2005) mimicking antipsychotic-like action shown for rats (e.g. Weiner et al., 1997) and mice (Lipina et al., 2005; Lipina and Roder, 2010). This effect is specific and selective for drugs with known antipsychotic activity and is not produced by a wide range of non-antipsychotic drugs (Dunn et al., 1993). The four experimental groups did not differ in their times to consume water between 50–75 licks (all p’s > 0.05; overall mean A period = 9.2 sec). There was significant effect of pre-exposure [F1, 52 = 94.7, p < 0.001], drug treatment [F1, 52 = 28.7, p < 0.001] and their interaction [F1, 52 = 9.8, p < 0.01]. LI was disrupted by the extended number of conditioning trials (4) in vehicle-treated mice (Fig. 2D p > 0.05), however LI was exhibited by TDZD-8-treated C57BL/6 mice under the same conditions (Fig. 2D p < 0.001), indicating that mice receiving TDZD-8 were less suppressed than the control group and ignored the conditioning stimulus as irrelevant.
Effects of Genetic inactivation of GSK-3α on behavior of Disc1-L100P mutants
The studies above are consistent with pharmacological inhibition of GSK-3 reversing the schizophrenia-related behavioral phenotypes observed in Disc1-L100P mutant mice. We sought to further investigate the specific role of GSK-3 within this process, by selectively crossing mice that lack a single allele of GSK-3α to WT and Disc1-L100P mutants.
As displayed earlier in the open field test, Disc1-L100P homozygous mutants showed hyperactivity. However, genetic inhibition of one allele of GSK-3α significantly decreased the ambulation of the Disc1-L100P mutants to the level of WT control mice. Figures 3A–B depicts the distance traveled in 30 minutes in the studied mice in the open field. ANOVA detected a significant effect of genotype [F5, 64 = 8.4, p < 0.001], testing interval [F 5, 320 = 43.8, p < 0.001] and gene x testing interval interactions [F 25, 320 = 10.3, p < 0.001]. Disc1-L100P homozygous mutants showed hyperactivity (all p’s < 0.001 at all testing intervals; (p’s < 0.001, Fig. 3A,B) whereas genetic inhibition of one allele of GSK-3α significantly decreased their ambulation to the level of the control mice (all p’s > 0.05).
Fig. 3.

A–D. Genetic inactivation of GSK-3α rescued schizophrenia-related phenotypes of Disc1-L100P mutant mice. (A–B) Ambulation in the open field of Disc1-L100P mice (WT, HET or MUT) carrying one or both GSK-3α allele (n = 8–17 per group). * - p < 0.01 – in comparison with WT mice; # - p < 0.001 - in comparison with Disc1-L100P MUT. (C) percent of PPI at three prepulses of ASR in genetic crosses between Disc1-L100P and GSK-3α-HET mice (n = 9–17 per group). ** - p 0.01, *** - p < 0.001 - in comparison with WT mice; # - p < 0.01, ## - p < 0.001 - in comparison with Disc1-L100P HET and Disc1-L100P MUT, respectively. (D) Mean suppression ratios of PE and NPE mice (n = 7–14 per group). PE groups of Disc1-L100P HET and MUT animals without one GSK-3α allele expressed LI. * - p < 0.001 – in comparison with PE group within each genotype, # - p < 0.001 – in comparison with PE group of Disc1-L100P HET or Disc1-L100P MUT. Balanced groups of male and female mice were used and gender effect was not detected in any of the behavioral tests, hence data from different genders were analyzed together. Numbers of animals (n) for each condition are indicated.
Furthermore, the PPI deficit of Disc1-L100P heterozygous and homozygous animals was significantly reversed at all three prepulses (p’s < 0.01 and p’s < 0.001, respectively) by genetic reduction of GSK-3α (Fig. 3C). ANOVA revealed significant effects of prepulse intensities [F2, 134 = 24.2, p < 0.001] and genotypes [F5, 67 = 27.5, p < 0.001]. There was a genotype effect on the ASR [F5, 67 = 19.4, p < 0.001], but, in agreement with the TDZD-8 studies, the lack of a GSK-3α allele did not alter the ASR in Disc1-L100P heterozygous and homozygous mice (Table 1).
Table 1.
Effects of genetic inactivation of GSK-3α on ASR of Disc1-L100P mutant mice
| Disc1 | GSK-3α | ASR |
|---|---|---|
| WT | WT | 1108.3 ± 121.3 (N = 10) |
| L100P+/− | WT | 389.6 ± 85.2 # (n = 10) |
| L100P−/− | WT | 401.3 ± 99.5 # (n = 10) |
| WT | GSK-3α+/− | 1210.4 ± 112.5 (n = 17) |
| L100P+/− | GSK-3α+/− | 405.3 ± 87.3 # (n = 9) |
| L100P−/− | GSK-3α+/− | 393.7 ± 65.2 # (n = 14) |
p < 0.001 – in comparison with control mice (post-hoc LSD test, ANOVA).
Using the latent inhibition test, we examined the mean suppression ratios of PE and NPE groups. The LI deficits normally observed in the Disc1-L100P heterozygous and homozygous mice was found to be dramatically reversed by the genetic inactivation of one GSK-3α allele with the mice improving their capacity to ignore the irrelevant stimulus. Figure 3D depicts mean suppression ratios of PE and NPE groups of all studied genotypes of Disc1 x GSK-3α mice. Twelve experimental groups did not differ in their times to consume water between 50–75 licks (all p’s > 0.05; overall mean A period = 8.4 sec). Two-way ANOVA revealed a main effect of pre-exposure [F1, 109 = 99.3, p < 0.001], genotype [F5, 109 = 23.7, p < 0.001], and their interaction [F5, 109 = 12.9, p < 0.001].
Co-immunoprecipitation of DISC1 with GSK-3α and GSK-3β
Previous studies have shown that the N-terminus of DISC1 (aa 1–220) is required to interact with GSK-3 (Mao et al., 2009). We examined whether the L100P point mutation within DISC1 affected its ability to interact with GSK-3. Using co-immunoprecipitation experiments, we immunoprecipitated DISC1 from WT and Disc1-L100P striatum lysates and found that the interaction with either GSK-3α or GSK-3β was significantly reduced by the Disc1-L100P mutation (both p’s < 0.05) (Fig. 4A–C). Furthermore, in reciprocal immunoprecipitations using GSK-3α or GSK-3β antibodies, DISC1 binding was significantly reduced in Disc1-L00P lysates (both p’s < 0.05) (Fig. 4D–G).
Fig. 4.

(A–G). Characterization of the DISC1-GSK-3a/b complex formation in Disc1-L100P mutant mice. A. Mice striatal brain samples were incubated with DISC1 antibody for coimmunoprecipitation. Precipitated proteins were subject to SDS-PAGE and then immunoblotted with GSK-3α or GSK-3β antibody. Each coimmunoprecipitation was in parallel with the directly immunoprecipitated DISC1 proteins. B–C. The densitometry evaluation of coimmunoprecipitation of GSK-3α (left) and GSK-3β (right). Data are means ±SEM, and were normalized to direct IP of DISC1 (D). Mice striatal brain samples were incubated with GSK-3β antibody for coimmunoprecipitation. Precipitated proteins were subject to SDS-PAGE and then immunoblotted with DISC1 antibody. Each coimmunoprecipitation was in parallel with the directly immunoprecipitated GSK-3β proteins. (E). The densitometry evaluation of coimmunoprecipitation of DISC1. Data are means ±SEM., and were normalized to direct IP of GSK3β. (F). Mice striatal brain samples were incubated with GSK-3α antibody for coimmunoprecipitation. Precipitated proteins were subject to SDS-PAGE and then immunoblotted with DISC1 antibody. Each coimmunoprecipitation was in parallel with the directly immunoprecipitated GSK-3α proteins. (G). The densitometry evaluation of coimmunoprecipitation of DISC1. Data are means ±SEM, and were normalized to direct IP of GSK3α. All data were analyzed with t-test (n = 5 per group; * - p < 0.05 – in comparison with WT). WT: Wild type; L100P – Disc1-L100P mutants.
Effect of Disc1-L100P mutation on GSK-3 regulation
Given that the Disc1-L100P mutation reduced binding to GSK-3 and that pharmacological/genetic inactivation reversed the behavorial phenotypes in Disc1-L100P mice, we investigated whether GSK-3 function is affected.
We examined the phosphorylation status of GSK-3 in striatum and hippocampus lysates. There was a slight, but non-significant (p > 0.05) decrease of GSK-3α and GSK-3β protein levels in the striatum of Disc1-L100P compared to WT (Fig. 5A). We used the ratio of Ser9 and Ser21 density to total GSK-3β and GSK-3α protein density respectively, as a measure for phosphorylation of GSK-3. There was significantly lower phosphorylation of GSK-3α (p < 0.01) but not GSK-3β in the striatum of Disc1-L100P mutants (Fig. 5A–B), with no changes in the phosphorylation of both GSK-3α and GSK-3β in the hippocampus (Fig. 6A–B). There were no changes in the phosphorylation status of Tyr 279 and Tyr 216 of GSK-3α and GSK-3β, respectively in both striatum (Fig. 5C) and hippocampus (Fig. 6C), nor did we observe any alterations in GSK-3α or GSK-3β kinase activities, when assayed in vitro against a GSK-3 peptide substrate (Fig. 5D–E; Fig. 6D–E). To examine GSK-3 function in vivo, we examined the expression levels of the GSK-3 substrate, β-catenin. We observed no significant changes in the levels of β-catenin in striatum (Fig. 5C) or hippocampus (Fig. 6C) tissues from WT and Disc1-L100P mice, suggesting that whilst the DISC1 x GSK-3 interaction is reduced by L100P, there is no alteration in the activity of the GSK-3 isoforms, at least towards in vitro substrates or to β-catenin in vivo.
Fig. 6.

A–D. Biochemical characterization of GSK-3 functional state in the Hippocampus of Disc1-L100P mice. (A) Representative immunoblots are shown, probed with an antibody against total GSK-3α and GSK-3β and with antibodies to p-GSK-3α at Ser21, p-GSK-3β at Ser9 as well as GAPDH as a loading control. (B) Optical densities are shown, reflecting normalized phosphorylation of GSK-3α and GSK-3β over the total GSK-3α and GSK-3β, respectively (n = 7 per group). (C). Representative immunoblots are shown, probed with an antibody against β-catenin, total GSK-3α and GSK-3β and with antibodies to p-GSK-3α at Tyr 276, p-GSK-3β at Tyr 216, as well as GAPDH as a loading control. Equal loading of the lanes was assessed using an anti-GAPDH antibody and the blots are representative of 2 independent experiments. (D). GSK-3 kinase activities in WT and L100P Disc1 mutant mice. Hippocampal tissues were extracted from WT and L100P Disc 1 mice, lysed with Buffer H and immunoprecipitated for GSK-3α or GSK-3β with their activities assayed as described in materials and methods. GSK-3 kinase activity is expressed relative to that for WT control (which is set at 100% for each GSK-3 isoform assayed) and corrected for potential differences in immunoprecipitated GSK-3 amount (E). Values are the means +/− SEM of results for four different tissue samples.
DISCUSSION
Here, we describe for the first time, an important in vivo role for GSK-3 in the schizophrenia-related phenotypes of Disc1-L100P mutant mice. Previous studies have set a precedent for the utility of GSK-3 inhibitors in scenarios of certain neurological diseases and conditions. The therapeutic notion of GSK-3 inhibition has arisen from an accumulation of evidence demonstrating perturbations of GSK-3 regulated pathways and downstream substrates. For example, the GSK-3 substrate Tau is abnormally hyperphosphorylated in Alzheimer’s (Kosik, 1992), whereas over-expression of GSK-3® results in tau hyperphosphorylation and disrupted microtubules in transgenic mice (Lucas et al., 2001). Furthermore, GSK-3 phosphorylation and activity are altered in conditions of Alzheimer’s disease, bipolar disorder, depression and schizophrenia (reviewed in Gould et al., 2004; Takashima, 2009; Lovestone et al., 2007; Koros and Dorner-Ciossek, 2007; Freyberg et al., 2009; Beaulieu et al., 2009). Meanwhile, inhibitors of GSK-3, such as lithium, has been used for many years to treat mood disorders as well as demonstrating beneficial effects of reducing Abeta peptides levels and tau phosphorylation in vitro and in vivo (Noble et al., 2005; Caccamo et al., 2007; Su et al, 2004, Sereno et al., 2009).
Here, we focused our attention on characterizing the molecular link between GSK-3 and DISC1 in schizophrenia. During the course of this study, Mao with colleagues (2009) demonstrated that the GSK-3 inhibitor, SB216763, significantly reduced the abnormal behavioral phenotype caused by DISC1 down regulation in the dentate gyrus of adult mice (as tested by open-field, forced swim and elevated maze tests). We now provide strong evidence that pharmacological and genetic inactivation of GSK-3 effectively reverses the schizophrenia-relevant behavioral phenotypes observed in an ENU-induced mutation in exon 2 of mouse Disc1 (L100P).
Administration of the selective GSK-3 inhibitor, TDZD-8, reversed PPI and LI deficits and normalized hyperactivity of Disc1-L100P mutant mice. Interestingly, these effects are similar to those prescribed to antipsychotics. Indeed, reversal of PPI and LI deficits is a reliable index of antipsychotic activity (Moser et al., 2000; Geyer and Ellenbroek, 2003). It is interesting to note that the efficacy of clozapine, a prescribed anti-psychotic used in schizophrenia, was similar to TDZD-8 in correcting hyperactivity and reversing PPI and LI deficits in a previous study using the Disc1-L100P mice (Clapcote et al., 2007). Furthermore, clozapine treatment has previously been shown to inhibit GSK-3β activity in SH-SY5Y cells (Aubry et al., 2009), as well as in the rat frontal cortex (Roh et al., 2007) and the mouse cortex, hippocampus, striatum, and cerebellum (Li et al., 2007b). Therefore, one could postulate that part of the action of this anti-psychotic is mediated through its ability to regulate GSK-3. Consistent with this body of literature, the present study shows that the same effect is also produced by TDZD-8. The efficacy of TDZD-8 to correct the schizophrenia-relevant endophenotypes of Disc1-L100P mutants is comparable with its effect in dopamine transporter (DAT) knockout mice to reverse hyperactivity (Beaulieu et al., 2004), validating that the doses used in this study are within range with previous studies using this compound to implicate GSK-3. More importantly, we were fully able to recapitulate the effects of TDZD-8 in Disc1-L100P mice by genetic deletion of GSK-3. Indeed, the inactivation of only one GSK-3α allele was sufficient to overcome the schizophrenia-related phenotypes of Disc1-L100P mutants. This not only supports the efficacy of TDZD-8 that has been used in previous studies (Beaulieu et al., 2004), but also demonstrates the relatively mild inhibition of GSK-3 can produce beneficial effects in neurobehavior. Indeed, mice lacking one allele of GSK-3β phenocopy behavioral effects of lithium treatment (O’Brien et al., 2004). Indeed, we observed that another GSK-3 inhibitor, lithium (Klein and Melton, 1996; Stambolic et al., 1996), mimicked the action of TDZD-8 in Disc1-L100P mutants reversing PPI deficit (data not shown). We also predict that mice with genetic loss of one allele of GSK-3β would rescue the behavioral phenotypes of the Disc1-L100P mutation, similar to the GSK-3α heterozygous mice described in this study.
Since pharmacological and genetic inhibition of GSK-3 relieves the DISC1 associated phenotypes, we next explored the molecular interactions and the consequence of the L100P mutation on GSK-3 function. Mao and colleagues (2009) recently described that DISC1 was a negative regulator of GSK-3. More specifically, using over-expression and knockdown studies, they demonstrated that DISC1 negatively affected tyrosine phosphorylation of GSK-3 (Tyr216 GSK-3β and Tyr279 GSK-3α), phosphorylation of which is important for kinase activity. Interestingly, however, abrogation of GSK-3 phosphorylation/kinase activity by DISC1 only perturbed GSK-3’s ability to phosphorylate β-catenin (canonical Wnt signaling) but not other known GSK-3 substrates. This observation suggested that DISC1 orchestrates multiple protein-protein interactions in a cell-context dependent manner and that GSK-3 and β-catenin may be intricately involved in the DISC1 scaffold.
In this study, we found that the Disc1-L100P mutation did not affect tyrosine phosphorylation of both GSK-3 isoforms and nor did we see any alterations in Wnt signaling as measured by β-catenin protein levels. Furthermore, whilst we did observe a slight reduction in serine phosphorylation of GSK-3 within the striatum, this did not reflect in any alterations of total GSK-3 activity. However, we discovered that the interaction between DISC1 and GSK-3 was significantly reduced within the striatum of the Disc1-L100P mice. It is important to note that the Disc1-L100P mutation does not alter the relative abundance of DISC1 when compared to WT expression. Therefore, the reduced interaction of GSK-3 is most likely a direct result of the L100P mutation. Indeed, to support this notion, we previously demonstrated that the L100P mutation severely impaired the ability of DISC1 to bind PDE4B, which is an important modulator of cAMP signaling (Clapcote et al., 2007). Moreover, rolipram (PDE4 inhibitor), similar to TDZD-8 in the current study, also was able to reverse schizophrenia-like phenotypes of Disc1-L100P mice (Clapcote et al., 2007). The fact that both DISC1 interactors - PDE4B and GSK-3, have common binding sites based on peptide mapping (Murdoch et al., 2007; Mao et al., 2009) suggests that altered DISC1-PDE4B-GSK-3 function is fundamentally related to the phenotypes of Disc1-L100P mutant mice. Thus, it appears that there are significant pertubations of the DISC1 scaffold caused by this single missense variation and that it is of extreme interest to examine the dynamics of these interactions further, alongside gaining structural insights.
Collectively, our data provide strong genetic and pharmacological evidence in vivo for impaired Disc1-GSK-3 interplay in schizophrenia-relevant behaviors of Disc1-L100P mutant mice, signifying the importance of the DISC1-GSK-3 complex in the psychopathology. Our findings also highlight the value of missense variants in the mouse for revealing selective effects on specific interactors and behaviors that can contribute to understanding molecular mechanisms in psychopathology.
Acknowledgments
We thank Dr. David Porteous for the helpful discussions of the manuscript. Contributions of each co-author are as follows: TVL, OKB and JR designed experiments, OKB generated crossbred mice, TVL performed pharmaco-behavioral experiments, OKB performed immunoblotting analysis of striatum and hippocampus, SP performed GSK-3 kinase assays and MW performed immunoprecipitation experiments; TVL, OKB, SP and JW wrote and edited the manuscript. This work was supported by CIHR grant FRN 74711 to JW, and CIHR GMH 79044 to JR. Portions of the work were presented as poster (249.6/N22) at the annual meeting of Society for Neuroscience (SfN-2009).
References
- Aubry JM, Schwald M, Ballmann E, Karege F. Early effects of mood stabilizers on the Akt/GSK-3beta signaling pathway and on cell survival and proliferation. Psychopharmacology (Berl) 2009;205:419–429. doi: 10.1007/s00213-009-1551-2. [DOI] [PubMed] [Google Scholar]
- Barad M. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-term potentiation and improves memory. PNAS. 1998;95:15020–15025. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR, Caron MG. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105(4):1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK-3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Poletti S, Radaelli D, Bernasconi A, Cavallaro R, Falini A, Lorenzi C, Pirovano A, Dallaspezia S, Locatelli C, Scotti G, Smeraldi E. Temporal lobe grey matter volume in schizophrenia is associated with a genetic polymorphism influencing glycogen synthase kinase 3-beta activity. Genes Brain Behav. 2010 Jan 25; doi: 10.1111/j.1601-183X.2010.00566.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation. 2009;6:9. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders--cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Br J Pharmacol. 2006;147:S252–257. doi: 10.1038/sj.bjp.0706495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK, Sawa A. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci. 2009;29(41):12768–12775. doi: 10.1523/JNEUROSCI.3355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669–1675. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ. Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- Chubb JE, Brashaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC1 locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar KJ, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ, Roder JC. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Davis JA, Gould TJ. Rolipram attenuates MK-801-induced deficits in latent inhibition. Behav Neurosci. 2005;119:595–602. doi: 10.1037/0735-7044.119.2.595. [DOI] [PubMed] [Google Scholar]
- Dunn LA, Atwater GE, Kilts CD. Effects of antipsychotic drugs on latent inhibition – sensitivity and specificity of an animal behavioral model of clinical drugs action. Psychopharmacology. 1993;112:315–323. doi: 10.1007/BF02244927. [DOI] [PubMed] [Google Scholar]
- Engel T, Hernández F, Avila J, Lucas JJ. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Asai N, Namba T, Wang Y, Kato T, Tanaka M, Tatsumi H, Taya S, Tsuboi D, Kuroda K, Kaneko N, Sawamoto K, Miyamoto R, Jijiwa M, Murakumo Y, Sokabe M, Seki T, Kaibuchi K, Takahashi M. Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron. 2009;63(6):774–787. doi: 10.1016/j.neuron.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359(Pt 1):1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry. 2010;167:388–396. doi: 10.1176/appi.ajp.2009.08121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer MA, Ellenbroek B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1071–1079. doi: 10.1016/j.pnpbp.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Gould TD, Quiroz JA, Singh J, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry. 2004;9:734–755. doi: 10.1038/sj.mp.4001518. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andradé M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104: 14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Baillie GS, Maurice DH. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res. 2007;100:950–966. doi: 10.1161/01.RES.0000261934.56938.38. [DOI] [PubMed] [Google Scholar]
- Huang HC, Klein P. Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Current Drug Targets. 2006;7:1389–1397. doi: 10.2174/1389450110607011389. [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32:485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Roh M-S. Glycogen synthase kinase-3 (GSK-3) in psychiatric diseases and therapeutic interventions. Current Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry. 2004;55(8):781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina T, Keizo T, Eed M, Satoko H, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, MacAulay K, Doble B, Henkelman M, Miyakawa T, Roder JC, Woodgett JR. Abnormalities in brain structure and behavior in GSK-3 mutant mice. Molecular Brain. 2009;2:35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanes SJ, Tokarczyk J, Siegel SJ, Bilker W, Abel T, Kelly MP. Rolipram: a specific phosphodiestirase 4 inhibitor with potential antipsychotic activity. Neuroscience. 2007;144(1):239–246. doi: 10.1016/j.neuroscience.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Duan X, Liu C, Jang M-H, Gui JU, Powanpongkul N, Kang E, Song H, Ming G-I. Disc1 regulates new neuron development in the adult brain via modulation of Akt-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P, Melton D. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockeritz L, Doble B, Patel S, Woodgett JR. Glycogen synthase kinase-3 – an overview of an over-achieving protein kinase. Current Drug Target. 2006;7:1377–1388. doi: 10.2174/1389450110607011377. [DOI] [PubMed] [Google Scholar]
- Koros E, Dorner-Ciossek C. The role of glycogen synthase kinase-3β in schizophrenia. Drug News Perspect. 2007;20:437–445. doi: 10.1358/dnp.2007.20.7.1149632. [DOI] [PubMed] [Google Scholar]
- Kosik KS. Alzheimer’s disease: a cell biological perspective. Science. 1992;256:780–783. doi: 10.1126/science.1589757. [DOI] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M, Gogos JA. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, Hennah W, Peltonen L, Lönnqvist J, Huttunen MO, Kaprio J, Trachtenberg JT, Silva AJ, Cannon TD. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A. 2007a;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007b;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- Lipina T, Labrie V, Weiner I, Roder J. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology. 2005;179:54–67. doi: 10.1007/s00213-005-2210-x. [DOI] [PubMed] [Google Scholar]
- Lipina T, Roder J. A new model of the disrupted latent inhibition on C57BL/6J mice. Psychopharmacology. 2010;208:487–498. doi: 10.1007/s00213-009-1749-3. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007;30:142–149. doi: 10.1016/j.tins.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20: 27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in Schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3b/b-Catenin Signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3β (GSK-3β) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s Disease. J Med Chem. 2002;45:1292–1299. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- Maxwell CR, Kanes SJ, Abel T, Siegel SJ. Phosphodiesterase inhibitors: a novel mechanisms for receptor-independent antipsychotic medications. Neuroscience. 2004;129(1):101–107. doi: 10.1016/j.neuroscience.2004.07.038. [DOI] [PubMed] [Google Scholar]
- McNeill H, Woodgett JR. When pathways collide: collaboration and connivance among signaling proteins in development. Nature Reviews Molecular Cell Biology. 2010 doi: 10.1038/nrm2902. doi:10.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11:763–775. doi: 10.1002/hipo.1092. [DOI] [PubMed] [Google Scholar]
- Moser PC, Hitchcock JM, Lister S, Moran PM. The pharmacology of latent inhibition as an animal model of schizophrenia. Brain Res Rev. 2000;33:275–307. doi: 10.1016/s0165-0173(00)00026-6. [DOI] [PubMed] [Google Scholar]
- Muñoz-Montaño JR, Moreno FJ, Avila J, Diaz-Nido J. Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett. 1997;411:183–188. doi: 10.1016/s0014-5793(97)00688-1. [DOI] [PubMed] [Google Scholar]
- Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, Porteous DJ, Millar JK, Houslay MD. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociate by elevated intracellular cAMP levels. J Neurosci. 2007;27:9513–9524. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JM, Zhang HT. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4) Trends Pharmacol Sci. 2004;25:158–163. doi: 10.1016/j.tips.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochem Soc Trans. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Doble BW, Macaulay K, Sinclair E, Drucker DJ, Woodgett J. Tissue-specific role of glycogen synthase kinase beta in glucose homoeostasis and insulin action. Molecular Cellular Biology. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Costas E, Gandy JC, Melendez-Ferro M, Roberts RC, Bijur GN. Light and electron microscopy study of glycogen synthase kinase-3beta in the mouse brain. PLoS One. 2010;27:e8911. doi: 10.1371/journal.pone.0008911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Perry W, Braff DL. Information-processing deficits and thought disorder in schizophrenia. Am J Psychiatry. 1994;151:363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- Pickard BS, Thomson PA, Christoforou A, Evans KL, Morris SW, Porteous DJ, Blackwood DH, Muir WJ. The PDE4B gene confers sex-specific protection against schizophrenia. Psychiatr Genet. 2007;17:129–133. doi: 10.1097/YPG.0b013e328014492b. [DOI] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice overexpressing glycogen synthase kinase 3β: a putative model of hyperactivity and mania. Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh MS, Seo MS, Kim Y, Kim SH, Jeon WJ, Ahn YM, Kang UG, Juhnn YS, Kim YS. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2007;39:353–360. doi: 10.1038/emm.2007.39. [DOI] [PubMed] [Google Scholar]
- Rosa AO, Kaster MP, Binfaré RW, Morales S, Martín-Aparicio E, Navarro-Rico ML, Martinez A, Medina M, García AG, López MG, Rodrigues AL. Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1549–1556. doi: 10.1016/j.pnpbp.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SAJ, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla TA. novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G, St Clair D. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated DISC1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza RP, Romano-Silva MA, Lieberman JA, Meltzer HY, Wong AH, Kennedy JL. Association study of GSK3 gene polymorphisms with schizophrenia and clozapine response. Psychopharmacology (Berl) 2008;200:177–186. doi: 10.1007/s00213-008-1193-9. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signaling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–68908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A. Drug development targeting the glycogen synthase kinase-3beta (GSK-3beta)-mediated signal transduction pathway: role of GSK-3beta in adult brain. J Pharmacol Sci. 2009;109:174–178. doi: 10.1254/jphs.08r29fm. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Holcomb HH. Phenotype of schizophrenia: a review and formulation. Mol Psychiatry. 2005;10(1): 27–39. doi: 10.1038/sj.mp.4001563. [DOI] [PubMed] [Google Scholar]
- Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, Kuroda S, Kuroda K, Shimizu M, Hirotsune S, Iwamatsu A, Kaibuchi K. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J Neurosci. 2007;27(1):15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomppo L, Hennah W, Lahermo P, Loukola A, Tuulio-Henriksson A, Suvisaari J, Partonen T, Ekelund J, Lönnqvist J, Peltonen L. Association between genes of Disrupted in schizophrenia 1 (DISC1) interactors and schizophrenia supports the role of the DISC1 pathway in the etiology of major mental illnesses. Biol Psychiatry. 2009;65(12):1055–1062. doi: 10.1016/j.biopsych.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner I, Shadach E, Barkai R, Feldon J. Haloperidol- and clozapine-induced enhancement of latent inhibition with extended conditioning: implications for the mechanism of action of neuroleptic drugs. Neuropsychopharmacology. 1997;16:42–50. doi: 10.1016/S0893-133X(96)00145-5. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/Factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol. 1994;5:269–275. [PubMed] [Google Scholar]
- Zhang HT, Zhao Y, Huang Y, Dorairaj NR, Chandler LJ, O’Donnell JM. Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into CA1 subregion of the rat hippocampus. Neuropsychopharmacology. 2004;29:1432–1439. doi: 10.1038/sj.npp.1300440. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Zhao Y, Huang Y, Deng C, Hopper AT, De Vivo M, Rose GM, O’Donnell JM. Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl) 2006;186(2):209–217. doi: 10.1007/s00213-006-0369-4. [DOI] [PubMed] [Google Scholar]