

Abstract
Background
In spite of its rather specific name, glycogen synthase kinase-3 (GSK-3) is an eclectic cellular regulator that modulates an array of processes from nuclear transcription, to neurological functions and metabolism. The enzyme is also a focal point for diverse signaling pathways that act to suppress its activity.
Objectives
Here, we review recent evidence supporting the important role GSK-3 plays in glucose homeostasis and discuss the therapeutic potential of inhibiting this enzyme in the treatment of diabetes and insulin resistance.
Results/Conclusion
Despite it’s pleiotropic nature, GSK-3 has significant promise as a target for diabetes due to functional partitioning of the enzyme, tissue-selectivity and acute dosage-dependency of effects of inhibition – suggesting useful therapeutic windows.
Keywords: Diabetes, Glucose tolerance, Glycogen Synthase Kinase-3, Insulin, Metabolism
1. Introduction
Glycogen synthase kinase-3 (GSK-3) is a serine/threonine protein kinase at the epicenter of the control of numerous cellular processes including glycogen metabolism, gene transcription, mRNA translation, cytoskeletal regulation, cell cycle progression and apoptosis (reviewed in [1]). Strong evidence implicates dysregulation of GSK-3 in the pathogenesis of diabetes, Alzheimer’s disease, bipolar disorder and cancer [2]. In mammals GSK-3 is present as two ubiquitously expressed homologues, GSK-3α and GSK-3β (51 and 46 kDa, respectively, see figure 1). These two isoforms share 98 % sequence identity within a central catalytic domain [3], although within the C-terminal 76 residues, sequence identity falls to 36 %. A structural distinction occurs within the N-terminus, where GSK-3α has an elongated glycine-rich domain [4]. In addition to GSK-3α and GSK-3β, a splice variant has been identified in brain [5]. This isoform, termed GSK-3β2, contains a 13 amino acid insert within the kinase domain [5]. Mice with a homozygous deletion of the GSK-3β gene typically die late in embryogenesis, between E13.5–16.5, as a result of severe liver apoptosis due to enhanced TNFα sensitivity [6], although rescue of this effect by genetic elimination of the TNF signaling system still results in death at or near birth due to other developmental defects (Kerkela et al., in revision). In contrast, mice engineered to lack GSK-3α are viable and fertile [7], suggesting distinct physiological functions of the GSK-3 isoforms, at least during development.
Figure 1.
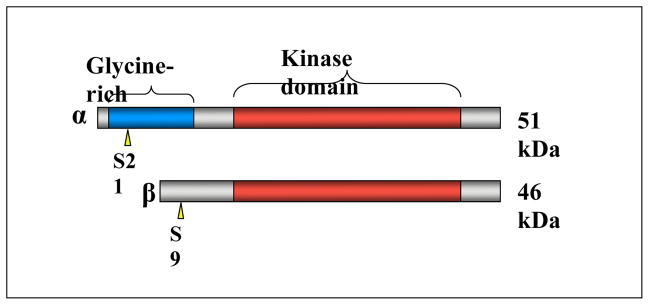
Schematic diagram of mammalian GSK-3α and GSK-3β. The N-terminal regulatory phosphorylation sites are shown by yellow arrow heads. The GSK-3α specific glycine-rich N-terminal extension is shown in blue and the highly conserved protein kinase domains are shown in red.
1.1 Regulation of GSK-3 by insulin
GSK-3 is an unusual protein kinase in that it displays high, “constitutive” activity in unstimulated cells and is rapidly inactivated upon a variety of cellular stimulations. The most thoroughly documented pathway for the inactivation of GSK-3 is in response to insulin and is mediated by protein kinase B (PKB, also termed Akt), which lies upstream of GSK-3 [8]. Insulin binding to its receptor induces a receptor conformational change resulting in receptor tyrosine kinase activation and recruitment of SH2-domain containing proteins including insulin receptor substrate-1 (IRS-1, as depicted in figure 2). One of the best documented downstream targets of IRS-1 is the lipid kinase phosphoinositide 3-kinase (PI3K) which phosphorylates the 3′-position of the inositol ring of phosphatidylinositols (PtdIns) generating PI(3,4)P2 and PI(3,4,5)P3 at the plasma membrane [9]. This PtdIns accumulation at the plasma membrane results in the recruitment of pleckstrin homology (PH) domain containing proteins including phosphoinositide-dependent kinase-1 (PDK-1) and PKB. Activation of PKB requires phosphorylation of two regulatory sites namely Thr308 and Ser473. The kinase responsible for Thr308 phosphorylation is PDK-1 whereas the Ser473 kinase is currently thought to be the mammalian target of rapamycin (mTOR) [10]. Activated PKB phosphorylates a key serine residue located within the N-terminus of GSK-3 (Ser21of GSK-3α and Ser9 of GSK-3β) [4]. Phosphorylation of this site results in transient inactivation of GSK-3 and permits the dephosphorylation of GSK-3 substrates by phosphatases. An arginine residue located at position 96 (Arg96), in addition to Arg180 and Lys205, creates a charged pocket within the GSK-3 kinase domain. This pocket binds pre-phosphorylated substrates and accounts for the unusual predilection of GSK-3 for targets that have been “primed” by other kinases (see Section 2) [4]. Phosphorylation of the N-terminal regulatory site of GSK-3 causes it to act as a high-affinity pseudo-substrate by competitively binding the Arg96 site of GSK-3, preventing interaction of the kinase with primed substrates, in effect interfering with its activity [4]. Mutation of Arg96 to alanine (R96A) not only inhibits phosphorylation of GSK-3 substrates but also prevents the phosphorylated N-terminal Ser9 from inhibiting GSK- 3’s affinity for primed substrates, suggesting that the Arg96-associated pocket plays a key role in promoting the interaction between GSK-3 and primed substrates, as well as its phosphorylated N-terminus (Frame et al., 2001). In the absence of insulin, phosphorylation of Ser21 and Ser9 is reversed by protein phosphatases such as protein phosphatase 2A and protein phosphatase 1 (PP1) [11] and hence, GSK-3 remains constitutively active until further stimulation of the tissue by insulin or other PI3K agonists.
Figure 2.

Insulin induces receptor autophosphorylation and association of IRS molecules, which then recruit PI3K to the plasma membrane. Subsequent production of PI(3,4,5)P3 allows signaling downstream of PI3K. PKB translocates to the plasma membrane where it is activated via phosphorylation by PDK1/2. GSK-3α/β is subsequently phosphorylated and inactivated by PKB thus preventing phosphorylation of down-stream substrates such as glycogen synthase. Glycogen synthase is activated by dephosphorylation by protein phosphatase 1 (PP1).
1.2 Regulation of GSK-3 by other signaling pathways
Wnts are secreted, cysteine-rich, glycoprotein ligands that are essential for embryonic development due to their involvement in cell fate, growth, differentiation and migration (reviewed in [12]). β-catenin is a member of the catenin family and functions in cell adhesion at the plasma membrane by anchoring cadherins during cell adherens junction formation [13]. In addition to a function in cell adhesion, β-catenin plays an essential role in canonical Wnt signaling. β-catenin is an effector molecule responsible for activating the T-cell factor/lymphoid enhancer-binding-factor-1 (TCF/LEF) family of architectural transcription factors by serving as a transactivator and associating with DNA bound TCF/LEF [14].
In the absence of Wnt, a small fraction of cellular GSK-3 (~5–10 %) is present in a multi-protein structure termed the destruction complex which comprises β-catenin, axin1/2 (scaffold proteins), and the tumour suppressor, adenomatous polyposis coli (APC) [15]. β-catenin, like many other GSK-3 substrates, must be pre-phosphorylated by a priming kinase before it becomes competent for phosphorylation by GSK-3. The priming kinase in this instance is casein kinase-1 (CK1) which phosphorylates β-catenin at Ser45 prior to phosphorylation of Thr41, Ser37 and Ser33 by GSK-3 [16]. Phosphorylation of β-catenin at Thr41 and Ser37 forms a phospho-degron that targets it for ubiquitination and subsequent proteosomal degradation by the β-TrCP ubiquitin ligase [17, 18]. Binding of Wnt to its seven transmembrane pass receptor, Frizzled and the LRP5/6 co-receptor, causes reorganization of the destruction complex and phosphorylation of LRP5/6 by GSK-3. β-catenin is no longer effectively phosphorylated by the destruction complex and therefore accumulates within the cytoplasm and translocates to the nucleus where it initiates transcription of Wnt target genes by binding to DNA bound TCF/LEF [14].
Gain-of-function mutations of β-catenin, including mutation of the CK1 priming residue or sites phosphorylated by GSK-3, have been found in various cancers including skin, colorectal, liver, prostate, endometrial and ovarian carcinomas (reviewed in [19]). However, recent studies utilizing embryonic stem cells containing an allelic series of GSK-3 expression (0–4 functional GSK-3 alleles), have demonstrated that loss of all 4 copies of GSK-3 is required for deregulation of β-catenin expression [1]. Of note, GSK-3α knockout mice do not develop tumors [7]. This is consistent with the fact that only a small fraction (<10 %) of GSK-3 is found in association with the destruction complex. Hence, even genetic inactivation of 3 of the 4 alleles of GSK-3 allows sufficient residual protein kinase to target and control β-catenin [1].
2. GSK-3 substrates
GSK-3 is known to have numerous substrates that are involved in a wide variety of cellular functions including glycogen metabolism, RNA transcription, protein translation and cell cycle progression [12]. An important feature required by the majority of GSK-3 substrates is the presence of a priming phosphate located four residues C-terminal to the phosphorylation site [20]. These priming sites are recognized due to adjacent repeats of consensus sequences, the shortest of which is Ser/Thr-X-X-X-Ser/Thr-(P)-, where the first serine or threonine is the target site for phosphorylation by GSK-3, X can be any amino acid (although the serine/threonine most proximal residue is very often proline) and the later serine or threonine is the priming site [21]. One of the first documented GSK-3 substrates was glycogen synthase (GS), the rate-limiting enzyme in glycogen synthesis [22]. GSK-3 phosphorylates four of nine GS regulatory serine residues (Ser641, Ser645, Ser649 and Ser653) that play a critical role in inhibiting GS activity [22]. For GS, priming phosphorylation is achieved by casein kinase II at Ser657 [4, 23] (see figure 3). Subsequent phosphorylation of Ser653 by GSK-3 then acts as the priming site for phosphorylation of Ser649, which then primes the next serine, etc. When fully phosphorylated, GS adopts an inactive state and glycogen synthesis is inhibited. However, in response to insulin, the N-terminal GSK-3 sites (Ser21 or Ser9) become phosphorylated and block access of substrate proteins to the active site of the enzyme [4]. This interferes with phosphorylation of primed substrates of GSK-3, and, in the case of GS, enables its dephosphorylation and activation by PP1 [24] hence promoting synthesis of glycogen.
Figure 3.
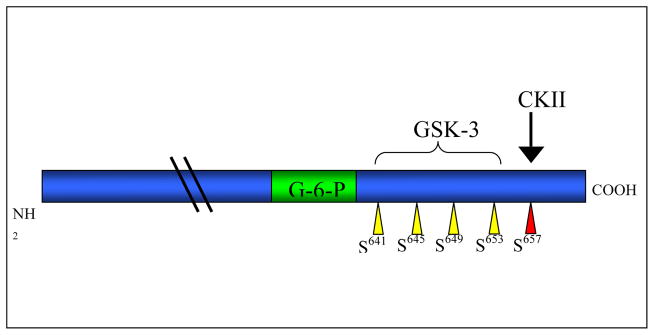
Schematic diagram of human skeletal muscle glycogen synthase. The casein kinase II (CKII) priming phosphorylation is shown in red. The GSK-3 target sites are highlighted in yellow and the glucose 6-phosphate binding site in green.
3. GSK-3 and insulin resistance
Insulin resistance is a key factor in the pathogenesis of obesity and Type 2 diabetes mellitus (T2DM) [25] and is characterized by the failure of muscle, liver and adipose tissue to efficiently respond to physiological concentrations of insulin. Numerous complications are attributed to T2DM including heart disease and stroke, vision loss, thyroid disease and limb amputation and with over 300 million people estimated to have T2DM by 2025, this will prove a huge financial burden on global health care systems.
Numerous studies implicate GSK-3 in the pathogenesis of insulin resistance and T2DM. GSK-3α/β expression and activity have been found to be significantly elevated in muscle biopsies of T2DM patients [26]. Examination of C57B1/6J mice fed a high-fat diet to induce obesity and T2DM, revealed elevated GSK-3 activity in epididymal fat tissue compared to control mice fed a regular chow diet [27]. Since GSK-3 is constitutively active in the basal state of cells, its inactivation by insulin is considered important for a normal insulin response and failure to achieve this inactivation may contribute to the pathogenesis of T2DM. Some studies have suggested that activation of glycogen synthase may not be impaired in T2DM, rather that the decreased glycogen storage is due to reduced transport [28]. Other studies have suggested inactivation if GSK-3 is not necessarily affected in insulin resistance and that glycogen synthase activity is affected by other kinases [29, 30].
3.1 Glucose transport
A major contributor to T2DM is impaired glucose transport from the blood stream into peripheral tissues. Several studies have implicated GSK-3 in the hormonal activation of glucose transport. This hypothesis is elegant as it would co-ordinate glucose transport into the cell with conversion of glucose into glycogen. Lithium, a non-selective GSK-3 inhibitor [31] has been shown to stimulate glucose uptake in isolated rat muscle [32, 33], adipose tissue [34], 3T3-L1 adipocytes [35, 36] and L6 muscle cells [35]. Furthermore, lithium has been shown to activate GS activity in isolated rat adipocytes [34], isolated rat muscle [32, 37], murine 3T3-L1 adipocytes [35, 36] and L6 muscle cells [35]. However, lithium is a rather non-selective GSK-3 inhibitor and has been shown to have effects on numerous signaling pathways. Since, lithium stimulation of GS activity is PI3K-dependent and more specific, potent, small molecule inhibitors of GSK-3 have also been shown to activate GS [35], it is likely that inhibition of GSK-3 is responsible for the activation of GS activity observed in response to lithium. However, it has recently been suggested that the effect of lithium on stimulation of glucose transport is not a result of GSK-3 inhibition. Rather, it has been proposed that lithium promotes glucose uptake via p38 MAPK activation [38, 35]. Therefore, the role of GSK-3 in the hormonal activation of glucose transport remains unclear.
Further investigation into the role of GSK-3 in glucose uptake has been investigated using more specific, ATP-competitive GSK-3 inhibitors. Cohen et al. recently assessed a panel of GSK-3 inhibitors and found the most potent and specific in vitro GSK-3 inhibitor to be CT 99021 [39]. Chronic inhibition of GSK-3 in human muscle cells using CT98014 and CHIR 98023 stimulated glucose transport to a level comparable to that observed with either lithium or insulin [40]. The effects of GSK-3 inhibition on glucose uptake in diabetic models has also been assessed. Incubation of soleus muscle isolated from Zucker diabetic fatty (ZDF) rats with CHIR 98014 was found to enhance insulin stimulated glucose transport [41]. Furthermore, administration of CT98014 [37, 38] and both acute and long-term treatment with CT118637 [42, 43] (respectively), also resulted in enhanced insulin-stimulated glucose transport in muscle of ZDF rats.
Conversely, over-expression of wild type GSK-3β in 3T3-L1 [44] or expression of constitutively-active GSK-3β in 3T3-L1 adipocytes [44] or L6 muscle cells [35] or GSK-3α/β in mouse models [45] had no affect on basal or insulin-stimulated glucose uptake. More recent studies using genetic ablation of GSK-3α showed no effect on basal or insulin stimulated glucose transport in isolated soleus or EDL muscle [7] or isolated adipose tissue (MacAulay & Woodgett, unpublished data). Genetic loss of GSK-3β in muscle was found to have no effect on basal or insulin-stimulated glucose uptake in isolated soleus or EDL [46]. Taken together these genetic models demonstrate that GSK-3 likely plays no direct role in the hormonal stimulation of glucose transport. Given that the inhibitor studies demonstrate increased insulin-stimulated glucose transport in response to GSK-3 inhibition, with no reported effect on basal uptake, the observations are likely due to enhanced insulin sensitivity in response to GSK-3 inhibition.
3.2 Glycogen metabolism
The other major contributor to the pathophysiology of T2DM is impaired glycogen storage. The two rate limiting steps in glucose metabolism are glucose transport into the tissue and conversion of the glucose to glycogen by GS. Although loss of GSK-3 does not result in an increase in glucose transporter copy number or rate of glucose transported into skeletal muscle, is it sufficient to regulate whole body glucose metabolism, in particular, glycogen synthesis?
Several ATP-competitive inhibitors of GSK-3 have been utilized to investigate the contribution of GSK-3 to glucose metabolism and insulin resistance. Administration on CHIR 98023 in Zucker diabetic fatty rats improved glucose tolerance and elevated GS activity in liver and muscle accompanied by increased hepatic glycogen accumulation [47]. The GSK-3 inhibitor CHIR 98014 led to enhanced glucose sensitivity [41] and activated GS in CHO-IR cells, primary rat hepatocytes and EDL and soleus from both lean and fatty Zucker rats [37, 41]. GSK-3 inhibition using CT118637 in Zucker diabetic fatty rats improved whole body glucose tolerance and insulin sensitivity [43]. Furthermore, basal and insulin stimulated GS activity was significantly elevated in EDL and soleus muscle of lean and obese Zucker rats following CT118637 treatment [43].
A novel peptide inhibitor of GSK-3, L803-mts, has recently been developed. Long-term administration of L803-mts to ob/ob mice improved glucose tolerance and insulin sensitivity [25]. However, L803-mts did not sufficiently inhibit GSK-3 to induce GS activation [25]. Interestingly, administration of L803-mts to high-fat fed C57B1/6J mice did result in enhanced hepatic GS activity [48]. In line with these studies, over-expression of human GSK-3β in skeletal muscle of mice results in impaired glucose tolerance and suppressed GS activity and glycogen synthesis, further supporting a role for GSK-3 in T2DM [49]. Genetic ablation of GSK-3α results in improved glucose (figure 4A) and insulin sensitivity (figure 4B) and this is accompanied by elevated hepatic GS activity and glycogen accumulation under fasted conditions and following a glucose load (figure 4C) [7]. Furthermore, although hepatic loss of GSK-3β does not alter glucose tolerance or insulin sensitivity, skeletal muscle-specific inactivation of GSK-3β does result in improved glucose and insulin sensitivity. Insulin-stimulated GS activity was found to be increased in skeletal muscle of muscle tissue-specific GSK-3β knockout animals and this was accompanied by elevated fasted and fed muscle glycogen content (Patel & Woodgett, in press). Therefore, these studies suggest that although inhibition of GSK-3 does not affect skeletal muscle glucose uptake, it is sufficient to activate GS, improve glucose tolerance and insulin sensitivity and increase glycogen storage [7, 46].
Figure 4.

Glucose tolerance, insulin sensitivity and hepatic glycogen deposition in GSK-3α knockout animals. Mice were fasted overnight and injected intraperitoneally (i.p.) with (A and C) 2 mg/g glucose and (B) 1 mU/g insulin. Blood glucose concentration was determined at indictated times. (C) 2 hours post glucose administration liver tissue was extracted, acid hydrolyzed and glycosyl units assayed. Figures A and B demonstrate enhanced glucose and insulin sensitivity in response to GSK-3α knockout. Figure C shows elevated fasted and glucose-stimulated hepatic glycogen deposition in GSK-3α knockout animals. (A) n ≥ 12 animals, *p < 0.001. (B) n=8 animals, *p < 0.05. (C) n=8 animals, *p < 0.05 versus WT for fasted; *p < 0.001 versus WT for 2 hr glucose. For more detailed experimental method description please refer to [7].
In addition to phosphorylation by GSK-3, GS is known to be regulated by several other mechanisms. This is nicely illustrated by the fact that in animals in which the PKB inactivating phosphorylation sites (serine 21 and 9 of GSK-3α and β respectively) are mutated to alanine, insulin is still able to promote glycogen synthesis [50]. This finding suggests that in the absence of a means to inactivate GSK-3 via phosphorylation, tissues may adapt to retain responsiveness to insulin, perhaps through insulin-stimulated changes in glucose-6-phosphate (G-6-P) acting to activate GS via allosteric mechanisms. GS is activated by dephosphorylation by PP1 [24]. This dephosphorylation can be facilitated by G-6-P. Binding of G-6-P to GS results in a conformation change which allows better binding of PP1 and hence G-6-P allosterically activates GS by facilitating PP1 binding [24,51]. In addition to G-6-P both glucose and glycogen are also thought to regulate the activity status of GS [51, 52]. GS activity is also regulated indirectly by glycogen phosphorylase given that glycogen phosphorylase allosterically regulates PP1 activity [53]. The relative importance of each of these mechanisms affecting GS is not known although the dominance of the effects of GSK-3 inhibitors suggest, for example, that compensatory inactivation of PP1 does not occur. It is possible that selective allosteric activators of GS could be developed as an alternative or complementary therapeutic strategy to GSK-3 inhibition.
3.3 GSK-3 and gluconeogenesis
In addition to stimulating glucose uptake and conversion of glucose into glycogen, insulin also regulates blood glucose concentration by inhibiting hepatic glucose output. Inhibition of hepatic glucose output by insulin occurs largely through PEPCK (phosphoenolpyruvate carboxykinase) [54, 55] and G-6-Pase (glucose-6-phosphatase) [56]. Dysregulation of PEPCK and G-6-Pase has been shown to contribute to the development of T2DM [57]. In models of T2DM, insulin fails to inhibit PEPCK and G-6-Pase. Inhibition of GSK-3, using Li, SB-216763 and SB-415286, results in repression of PEPCK and G-6-Pase [58]. IGFBP-1 (insulin-like growth factor-binding protein-1) expression is also inhibited in H4IIE-C3 cells using Li, SB-216763 and SB-415286 [59]. Furthermore, GSK-3 over-expression in hepatoma cells decreases the sensitivity of IGFBP-1 to insulin [59]. These studies further suggest GSK-3 as a potential therapeutic target for the treatment of T2DM. Interestingly, mice expressing constitutively active GSK-3, in which GSK-3 is not inactivated by insulin, PEPCK, G-6-Pase and IGFBP-1 were inactivated normally in response to feeding [60]. This later study suggests that inactivation of GSK-3 by PKB is not required for the regulation of PEPCK, G-6-Pase and IGFBP-1 by insulin [60].
3.4 GSK-3 and Insulin Receptor Substrates
The increased insulin sensitivity observed in response to GSK-3 inhibition has been attributed to signaling though IRS-1. Following priming at Ser336, GSK-3 has been shown to phosphorylate IRS-1 at Ser332 [61]. Serine phosphorylation of IRS-1 is thought to impair tyrosine phosphorylation by the insulin receptor [62], thus attenuating insulin signaling. This hypothesis is further substantiated by the observation that mutation of Ser332 and Ser336 to alanine enhanced insulin stimulated IRS-1 tyrosine phosphorylation and PKB activation [61]. IRS-1 phosphorylation status has also been examined in models of diabetes and insulin resistance. Inhibition of GSK-3 in muscle of Zucker diabetic fatty rats using CT118637 increased insulin-stimulated IRS-1 tyrosine phosphorylation, with no effect on insulin receptor β tyrosine phosphorylation [43] This enhanced IRS-1 tyrosine phosphorylation was accompanied by increased IRS-1-associated p85 activity [42]. Short-term inhibition of GSK-3 with CT98014, increased insulin-stimulated insulin receptor β tyrosine phosphorylation and IRS-1 tyrosine phosphorylation in soleus of Zucker diabetic fatty rats and decreased insulin-stimulated Ser307 phosphorylation [37]. Furthermore, incubation of cultured human muscle cells with CHIR98023 resulted in increased IRS-1 protein expression in a time- and concentration-dependent manner [63]. The elevated insulin sensitivity and insulin-stimulated PKB and GSK-3β phosphorylation observed in liver in response to genetic loss of GSK-3α was attributed to enhanced insulin signaling through IRS-1. This was based on the observation that IRS-1 protein expression was significantly higher in liver tissue of GSK-3α knockout animals [7].
Heterozygous loss of the insulin receptor or homozygous loss of IRS-2 promotes insulin resistance and diabetes through effects on pancreatic β-islet cells [64, 65]. Crossing of these animals with mice heterozygous for GSK-3β substantially restored glucose sensitivity, protected the β-islet cells and prevented the onset of diabetes. Moreover, tissue-specific inactivation of GSK-3β only in the β-islet cells was sufficient to protect against IRS-2-induced diabetes [66]. These findings indicate a cell-specific function of GSK-3β in islets and suggest this enzyme interacts with IRS-2 signaling in this tissue.
4. Expert opinion
Numerous lines of evidence have accumulated linking GSK-3 to the development of insulin resistance and T2DM which can each be used to build a case for GSK-3 inhibition for the treatment of T2DM. However, GSK-3 inhibitors available to date do not discriminate between GSK-3α and GSK-3β, and therefore, it is unclear as to which isoform (or both) may be responsible for the improved insulin sensitivity and glucose metabolism observed in response to GSK-3 inhibition.
To date, the majority of studies published have focused on GSK-3β. This is most likely due to the observation that the rat GSK-3β isoform rescued the shaggy (Drosophila GSK-3 homologue) mutation in Drosophila, whereas GSK-3α did not [67], perhaps for trivial reasons such as expression efficiency. GSK-3β embryos die late in embryogenesis which indicates that GSK-3α is not sufficient to compensate for GSK-3β and also suggesting functional diversity between the two isoforms. Genetic ablation of GSK-3α results in enhanced glucose tolerance and insulin sensitivity [7]. This insulin sensitization is thought to be as a result of increased IRS-1 expression (as discussed above), and, consequently, insulin-stimulated PKB and GSK-3β phosphorylation was found to be significantly higher in liver samples of GSK-3α knockout animals compared to wild type animals [7]. Loss of GSK-3α did not affect insulin-signaling in the muscle. The enhanced glucose tolerance is thought to be attributed to the increased GS activity and glycogen deposition observed in the liver. No alteration was observed in GS activity or glycogen accumulation in the muscle tissue of GSK-3α knockout animals. Furthermore, specific inactivation of GSK-3β in the liver has no effect on insulin signaling or glycogen metabolism, whereas inactivation of GSK-3β in skeletal muscle results in significantly elevated GS activity and muscle deposition of glycogen [46]. Interestingly, whereas insulin activates GS in muscle of animals containing constitutively active GSK-3α (GSK-3α21A/21A), constitutively active GSK-3β (GSK-3β9A/9A) prevents insulin-stimulated activation of GS [46]. Specific inactivation of GSK-3β in pancreatic β-cells is sufficient to rescue insulin resistance and diabetes caused by inactivation of both alleles of IRS-2 [66]. Taken together these findings strongly suggest that GSK-3α and GSK-3β have unique tissue-specific functions with respect to glycogen metabolism.
From these data it is plausible that GSK-3α and GSK-3β do not function equally in the insulin signaling cascade in insulin-responsive tissues. The studies by MacAulay et al., Patel et al. and McManus et al., suggest that GSK-3α functions as the hepatic GS kinase regulating glycogen synthesis and deposition primarily in the liver. Conversely, GSK-3β has a minimal role in glycogen metabolism in the liver tissue. In contrast, GSK-3β plays an important role in skeletal muscle tissue and β-islet cells where its knockout leads to enhanced GS activity and glycogen accumulation or insulin responsiveness respectively. GSK-3α does not appear to be an important regulator of glycogen metabolism in the muscle.
The major contributing factor in the pathogenesis of T2DM is insulin resistance which is subsequently accompanied with impaired glucose transport and conversion of glucose into glycogen. Given that selective, global inhibition of GSK-3α significantly improves insulin sensitivity and hepatic glycogen deposition, GSK-3α maybe a good therapeutic target for the treatment of T2DM. However, this begs the question of GSK-3α/β isoform specific inhibitors which, to date, have not been developed. Given the highly conserved ATP binding pocket of GSK-3α and GSK-3β, generation of isoform specific inhibitors may prove to be extremely challenging. However, GSK-3β inhibition in skeletal muscle and b-islet cells also modulates glucose metabolism. Hence pan-GSK-3 inhibitors are likely to result in combinatorial effects in multiple tissues. It is also likely that a wide therapeutic window exists, at least with respect to induction of Wnt signaling since effects on glucose metabolism are manifested by <50 % inhibition of GSK-3 whereas effects on β-catenin are not observed until ~80 % of the kinase inhibition is achieved.
In summary, a role for GSK-3 in the development of insulin resistance has been established. GSK-3 inhibition has been shown to enhance insulin sensitivity and promote glycogen synthesis, suggesting GSK-3 as a viable therapeutic target for the treatment of T2DM. Recent data shows that GSK-3α and GSK-3β have distinct physiological functions in insulin-sensitive tissues, particularly in glycogen metabolism. These data reveal a complexity of function previously unappreciated but do not necessarily complicate the therapeutic potential of GSK-3 inhibitors in treating diabetes.
Acknowledgments
KM is supported by a Canadian Diabetes Association postdoctoral fellowship. JRW is a CIHR Senior Investigator and is supported by grants from the Canadian Diabetes Association and Canadian Institutes of Health Research (MOP-74711).
Abbreviations
- APC
adenomatous polyposis coli
- CK1
casein kinase-1
- EDL
extensor digitorum longus
- GS
glycogen synthase
- GSK-3
glycogen synthase kinase-3
- IRS-1
insulin receptor substrate-1
- mTOR
mammalian target of rapamycin
- PDK-1
phosphoinositide-dependent kinase-1
- PH
pleckstrin homology
- PI3K
phosphoinositide 3-kinase
- PKB
protein kinase B
- PP1
protein phosphatase 1
- PtdIns
phosphatidylinositol
- TCF/LEF
T-cell factor/lymphoid enhancer-binding-factor-1
- T2DM
type-2 diabetes mellitus
- ZDF
Zucker diabetic fatty
References
- *1.Doble BW, Patel S, Wood GA, et al. Functional redundancy of GSK-3α and GSK-3β in Wnt/β-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–71. doi: 10.1016/j.devcel.2007.04.001. This paper demonstrates the redundancy of the two isoforms of GSK-3 with respect to Wnt signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobio. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 3.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/Factor A. EMBO J. 1990;9:2431–38. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–27. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 5.Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3β. J Neurochem. 2002;81:1073–83. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- 6.Hoeflich KP, Luo J, Rubie EA, et al. Requirement for glycogen synthase kinase-3β in cell survival and NF-kB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- **7.Macaulay K, Doble BW, Patel S, et al. Glycogen synthase kinase 3α-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–37. doi: 10.1016/j.cmet.2007.08.013. This paper demonstrates insulin sensitization caused by whole body inactivation of GSK-3α. [DOI] [PubMed] [Google Scholar]
- *8.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. Describes a molecular mechanism for insulin-dependent inactivation of GSK-3. [DOI] [PubMed] [Google Scholar]
- 9.Foster FM, Traer CJ, Abraham SM, Fry MJ. The phosphoinositide (PI) 3-kinase family. J Cell Sci. 2003;116:3037–40. doi: 10.1242/jcs.00609. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 11.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagafuchi A, Takeichi M. Transmembrane control of cadherin-mediated cell adhesion: a 94 kDa protein functionally associated with a specific region of the cytoplasmic domain of E-cadherin. Cell Reg. 1989;1:37–44. doi: 10.1091/mbc.1.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Noort M, Clevers H. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev Biol. 2002;244:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- 15.Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol. 2005;58:225–36. doi: 10.1136/jcp.2003.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amit S, Hatzubai A, Birman Y, et al. Axin-mediated CKI phosphorylation of β-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–76. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3β. Curr Biol. 1998;8:573–81. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- 19.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]
- 20.Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem. 1987;262:14042–8. [PubMed] [Google Scholar]
- 21.Roach PJ. Multisite and hierarchal protein phosphorylation. J Biol Chem. 1991;266:14139–42. [PubMed] [Google Scholar]
- 22.Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–20. doi: 10.2174/1566524024605761. [DOI] [PubMed] [Google Scholar]
- 23.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–27. [PubMed] [Google Scholar]
- 24.Brady MJ, Saltiel AR. The role of protein phosphatase-1 in insulin action. Recent Prog Horm Res. 2001;56:157–73. doi: 10.1210/rp.56.1.157. [DOI] [PubMed] [Google Scholar]
- 25.Kaidanovich-Beilin O, Eldar-Finkelman H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J Pharmacol Exp Ther. 2006;316:17–24. doi: 10.1124/jpet.105.090266. [DOI] [PubMed] [Google Scholar]
- 26.Nikoulina SE, Ciaraldi TP, Carter L, et al. Impaired muscle glycogen synthase in type 2 diabetes is associated with diminished phosphatidylinositol 3-kinase activation. J Clin Endocrinol Metab. 2001;86:4307–14. doi: 10.1210/jcem.86.9.7872. [DOI] [PubMed] [Google Scholar]
- 27.Eldar-Finkelman H, Argast GM, Foord O, et al. Expression and characterization of glycogen synthase kinase-3 mutants and their effect on glycogen synthase activity in intact cells. Proc Natl Acad Sci USA. 1997;93:10228–33. doi: 10.1073/pnas.93.19.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelley DE, Mandarino LJ. Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J Clin Invest. 1990;86:1999–2007. doi: 10.1172/JCI114935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hojlund K, et al. Increased phosphorylation of skeletal muscle glycogen synthase at NH2-terminal sites during physiological hyperinsulinemia in type 2 diabetes. Diabetes. 2003;52:1393–402. doi: 10.2337/diabetes.52.6.1393. [DOI] [PubMed] [Google Scholar]
- 30.Gaster M, et al. The primary defect in glycogen synthase activity is not based on increased glycogen synthase kinase-3α activity in diabetic myotubes. Biochem Biophys Res Commun. 2004;319:1235–40. doi: 10.1016/j.bbrc.2004.05.109. [DOI] [PubMed] [Google Scholar]
- 31.Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–5. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- 32.Furnsinn C, Noe C, Herdlicka R, Roden M, et al. More marked stimulation by lithium than insulin of the glycogenic pathway in rat skeletal muscle. Am J Physiol. 1997;273:E514–20. doi: 10.1152/ajpendo.1997.273.3.E514. [DOI] [PubMed] [Google Scholar]
- 33.Tabata I, Schluter J, Gulve EA, Holloszy JO. Lithium increases susceptibility of muscle glucose transport to stimulation by various agents. Diabetes. 1994;43:903–7. doi: 10.2337/diab.43.7.903. [DOI] [PubMed] [Google Scholar]
- 34.Cheng K, Creacy S, Larner J. ‘Insulin-like’ effects of lithium ion on isolated rat adipocytes. II. Specific activation of glycogen synthase. Mol Cell Biochem. 1983;56:183–9. doi: 10.1007/BF00227219. [DOI] [PubMed] [Google Scholar]
- 35.MacAulay K, Hajduch E, Blair AS, et al. Use of lithium and SB-415286 to explore the role of glycogen synthase kinase-3 in the regulation of glucose transport and glycogen synthase. Eur J Biochem. 2003;270:3829–38. doi: 10.1046/j.1432-1033.2003.03777.x. [DOI] [PubMed] [Google Scholar]
- 36.Orena SJ, Torchia AJ, Garofalo RS. Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem. 2000;275:15765–72. doi: 10.1074/jbc.M910002199. [DOI] [PubMed] [Google Scholar]
- 37.Henriksen EJ, Kinnick TR, Teachey MK, et al. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2003;284:E892–900. doi: 10.1152/ajpendo.00346.2002. [DOI] [PubMed] [Google Scholar]
- 38.Henriksen EJ, Teachey MK. Short-term in vitro inhibition of glycogen synthase kinase 3 potentiates insulin signaling in type I skeletal muscle of Zucker Diabetic Fatty rats. Metabolism. 2007;56:931–8. doi: 10.1016/j.metabol.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bain J, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikoulina SE, Ciaraldi TP, Mudaliar S, et al. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51:2190–8. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- 41.Ring DB, Johnson KW, Henriksen EJ, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–95. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- 42.Dokken BB, Henriksen EJ. Chronic selective glycogen synthase kinase-3 inhibition enhances glucose disposal and muscle insulin action in prediabetic obese Zucker rats. Am J Physiol Endocrinol Metab. 2006;291:E207–213. doi: 10.1152/ajpendo.00628.2005. [DOI] [PubMed] [Google Scholar]
- 43.Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in pre-diabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–94. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- 44.Summers SA, Kao AW, Kohn AD, et al. The role of glycogen synthase kinase 3β in insulin-stimulated glucose metabolism. J Biol Chem. 1999;274:17934–40. doi: 10.1074/jbc.274.25.17934. [DOI] [PubMed] [Google Scholar]
- **45.McManus EJ, Sakamoto K, Armit LJ, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–83. doi: 10.1038/sj.emboj.7600633. Elegant test of the effects of phosphorylation site mutants of GSK-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *46.Patel S, Doble B, et al. Tissue-specific role of glycogen synthase kinase-3β in insulin action. Mol Cell Biol. 2008 doi: 10.1128/MCB.00763-08. (in press). Tissue-specific effects of GSK-3β in muscle versus liver knockout. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cline GW, Johnson K, Regittnig W, et al. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes. 2002;51:2903–10. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- *48.Rao R, Hao CM, Redha R, et al. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia. 2007;50:452–460. doi: 10.1007/s00125-006-0552-5. Selective effect of inhibition of GSK-3 in reversing insulin resistance but not hypertensive effects of high fat diet. [DOI] [PubMed] [Google Scholar]
- 49.Pearce NJ, Arch JR, Clapham JC, et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3β on a muscle-specific promoter. Metabolism. 2004;53:1322–30. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 50.Bouskila M, Hirshman MF, et al. Insulin promotes glycogen synthesis in the absence of GSK3 phosphorylation in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E28–35. doi: 10.1152/ajpendo.00481.2007. [DOI] [PubMed] [Google Scholar]
- 51.Leloir LF, et al. Biosynthesis of glycogen from uridine diphosphate glucose. Arch Biochem Biophys. 1959;81:508–20. doi: 10.1016/0003-9861(59)90232-2. [DOI] [PubMed] [Google Scholar]
- 52.Halse R, et al. Control of glycogen synthesis by glucose, glycogen, and insulin in cultured human muscle cells. Diabetes. 2001;50:720–6. doi: 10.2337/diabetes.50.4.720. [DOI] [PubMed] [Google Scholar]
- 53.Kelsall IR, et al. The hepatic PP1 glycogen-targeting subunit interaction with phosphorylase a can be blocked by C-terminal tyrosine deletion or an indole drug. FEBS Lett. 2007;581:4749–53. doi: 10.1016/j.febslet.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 54.O’Brien RM, Granner DK. PEPCK gene as model of inhibitory effects of insulin on gene transcription. Diabetes Care. 1990;13:327–39. doi: 10.2337/diacare.13.3.327. [DOI] [PubMed] [Google Scholar]
- 55.Sutherland C, O’Brien RM, Granner DK. New connections in the regulation of PEPCK gene expression by insulin. Philos Trans R Soc Lond B Biol Sci. 1996;351:191–9. doi: 10.1098/rstb.1996.0016. [DOI] [PubMed] [Google Scholar]
- 56.Foster JD, Pederson BA, Nordlie RC. Glucose-6-phosphatase structure, regulation, and function: an update. Proc Soc Exp Biol Med. 1997;215:314–32. doi: 10.3181/00379727-215-44142. [DOI] [PubMed] [Google Scholar]
- 57.Patel S, Lipina C, Sutherland C. Different mechanisms are used by insulin to repress three genes that contain a homologous thymine-rich insulin response element. FEBS Lett. 2003;549:72–6. doi: 10.1016/s0014-5793(03)00774-9. [DOI] [PubMed] [Google Scholar]
- 58.Lochhead PA, et al. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001;50:937–46. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- 59.Finlay D, et al. Glycogen synthase kinase-3 regulates IGFBP-1 gene transcription through the thymine-rich insulin response element. BMC Mol Biol. 2004;5:15. doi: 10.1186/1471-2199-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lipina C, et al. Analysis of hepatic gene transcription in mice expressing insulin-insensitive GSK3. Biochem J. 2005;392:633–9. doi: 10.1042/BJ20051046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem. 2005;280:4422–8. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- 62.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci U S A. 1997;94:9660–64. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nikoulina SE, Ciaraldi TP, Abrams-Carter L, et al. Regulation of glycogen synthase activity in cultured skeletal muscle cells from subjects with type II diabetes: role of chronic hyperinsulinemia and hyperglycemia. Diabetes. 1997;46:1017–1024. doi: 10.2337/diab.46.6.1017. [DOI] [PubMed] [Google Scholar]
- 64.Bruning JC, Winnay J, Bonner-Weir S, et al. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–72. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- 65.White MF. Regulating insulin signaling and β-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84:725–37. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- **66.Tanabe K, Liu Z, Patel S, et al. Genetic deficiency of glycogen synthase kinase-3β corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6:e37. doi: 10.1371/journal.pbio.0060037. Rescue of insulin resitance caused by suppression of IR and IRS2 function by β-islet cell inactivation of of GSK-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruel L, Bourouis M, Heitzler P, et al. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature. 1993;362:557–60. doi: 10.1038/362557a0. [DOI] [PubMed] [Google Scholar]