

Abstract
Asthma is characterized by airway inflammation and airflow obstruction from human airway smooth muscle (HASM) constriction due to increased local bronchoconstrictive substances. We have recently found bitter taste receptors (TAS2Rs) on HASM, which increase [Ca2+]i and relax the muscle. We report here that some, but not all, TAS2R agonists decrease [Ca2+]i and relax HASM contracted by G-protein coupled receptors (GPCRs) that stimulate [Ca2+]i. This suggests both a second pathway by which TAS2Rs relax, and, a heterogeneity of the response phenotype. We utilized eight TAS2R agonists and five procontractile GPCR agonists in cultured HASM cells. We find that heterogeneity in the inhibitory response hinges on which procontractile GPCR is activated. For example, chloroquine inhibits [Ca2+]i increases from histamine, but failed to inhibit [Ca2+]i increases from endothelin-1. Conversely, aristolochic acid inhibited [Ca2+]i increases from endothelin-1 but not histamine. Other dichotomous responses were found when [Ca2+]i was stimulated by bradykinin, angiotensin, and acetylcholine. There was no association between [Ca2+]i inhibition and TAS2R subtype, nor whether [Ca2+]i was increased by Gq- or Gi-coupled GPCRs. Selected studies revealed a correlation between [Ca2+]i inhibition and HASM cell-membrane hyperpolarization. To demonstrate physiologic correlates, ferromagnetic beads were attached to HASM cells and cell stiffness measured by magnetic twisting cytometry. Consistent with the [Ca2+]i inhibition results, chloroquine abolished the cell stiffening response (contraction) evoked by histamine but not by endothelin-1, while aristolochic acid inhibited cell stiffening from endothelin-1, but not from histamine. In studies using intact human bronchi, these same differential responses were found. Those TAS2R agonists that decreased [Ca2+]i, promoted hyperpolarization, and decreased HASM stiffness, caused relaxation of human airways. Thus TAS2Rs relax HASM in two ways: a low-efficiency de novo [Ca2+]i stimulation, and, a high-efficiency inhibition of GPCR-stimulated [Ca2+]i. Furthermore, there is an interaction between TAS2Rs and some GPCRs that facilitates this [Ca2+]i inhibition limb.
Introduction
Asthma is a disease characterized by airway inflammation and airflow limitation caused by contraction of airway smooth muscle (ASM). Contraction of ASM is due to local accumulation of agonists such as acetylcholine (Ach) and histamine, which activate G-protein coupled receptors (GPCRs) on ASM [1,2]. Indeed, the bronchoconstrictive GPCRs all increase [Ca2+]i via coupling to Gαq, or less commonly, Gαi [1]. Thus a number of GPCR antagonists acting at these receptors are used for treating asthma, and are considered “indirect” bronchodilators. The only class of direct bronchodilators is composed of agonists for ASM β2-adrenergic receptors (β2ARs), which couple to Gαs, increase cAMP, and relax ASM through a series of events mediated by protein kinase A. The use of β-agonists, however, is associated with tachyphylaxis (tolerance) [3], increased bronchial hyperresponsiveness [4,5], interindividual variability [6], and worsening asthma and mortality [7–9].
These issues have led to our search for other drug targets that promote human ASM (HASM) relaxation [10]. We found that bitter taste receptors (TAS2Rs) are expressed on HASM cells, and when activated cause marked relaxation [11,12]. These findings have been corroborated by several other groups [13–16] although there remains some debate over the mechanism of action. TAS2Rs are broadly tuned receptors that display relatively low apparent affinities (μM to mM range) for the vast majority of currently recognized agonists [17]. In pharmacological studies in HASM using agonists for the most highly expressed TAS2R subtypes, we demonstrated that TAS2R stimulated [Ca2+]i mobilization [11]. Intracellular cAMP levels remained unchanged in HASM exposed to TAS2R agonists [11]. This signaling is consistent with the pathway described for TAS2R in taste cells, where TAS2R couple to gustducin, and its βγ subunit activates phospholipase C, generating inositol 1,4,5-trisphosphate (IP3). IP3 acting on its receptor releases Ca2+ from the endoplasmic reticulum, and in taste cells this leads to release of neurotransmitter, activation of a transient receptor potential (TRP) channel, and depolarization of the cell membrane [18]. Such depolarization in the ASM cell would be expected to cause ASM contraction. However, TAS2R agonists relax ASM, and in fact cause hyperpolarization of the membrane [11], and thus the signaling of TAS2R in ASM diverges from that observed in taste cells [19,20]. Of note, TAS2R agonists cause membrane hyperpolarization and ASM relaxation of isolated cells as well as intact airways at baseline, i.e., in the absence of any procontractile stimulus [11]. However, the majority of physiological studies that we [11,12,21–24] and others [14–16] have performed with human, nonhuman primate, mouse, or guinea pig have been under circumstances where the muscle is contracted with receptor agonists such as Ach and histamine. These conditions more closely resemble the pathogenic settings of airflow obstruction in asthma. These spasmogens act at their cognate receptors to also increase [Ca2+]i which ultimately activates myosin light chain kinase which phosphorylates regulatory light chains of myosin and evokes cross-bridge cycling and tension generation [25,26]. We have shown that the increase in [Ca2+]i caused by TAS2Rs, though, appears to be compartmentalized, which may be the basis for its relaxation effect, as compared to Gq/Gi-coupled receptors which evoke a more global increase in [Ca2+]i and contract ASM [11]. In studies of mouse ASM cells, it has recently been shown that TAS2Rs can also inhibit [Ca2+]i that has been elevated by procontractile GPCR agonists [15]. We hypothesized that TAS2Rs act to inhibit [Ca2+]i that has been stimulated by GPCRs, and that this response requires lower concentrations of TAS2R agonist compared to the low-efficiency [Ca2+]i stimulatory pathway. We proposed that upon activation with contractile GPCR agonists, the TAS2R signaling from this pathway will antagonize cell membrane depolarization, promote a decrease in single cell stiffness, and cause relaxation of human airways. Based on the heterogeneity of the [Ca2+]i-inhibition response observed in initial studies, we further hypothesized that the capacity for TAS2Rs to inhibit [Ca2+]i and relax ASM (by this mechanism), is dependent on which GPCR is acting to stimulate [Ca2+]i and contract the airway. Thus in the current work we utilized multiple TAS2R agonists for the three highest expressing TAS2R subtypes in HASM, and agonists for multiple Gq-and/or Gi-coupled GPCRs which increase [Ca2+]i. We indeed demonstrate a dual mechanism for TAS2R signaling, where the inhibitory pathway hinges upon how [Ca2+]i is increased in the ASM cell.
Materials and Methods
HASM Cells and Intact Bronchi
Primary HASM cultured cells were established from airways from deceased nonasthmatic individuals obtained from the National Disease Research Interchange (NDRI) (Philadelphia, PA, http://ndriresource.org) as described [23] and isolated as previously reported [27]. Cells were maintained in HAM’s F12 medium with 10% fetal bovine serum, 1% penicillin and streptomycin, 1% L-glutamine, 1.7 mM CaCl2, 12 mM NaOH, and 25 mM HEPES at 37°C in 95% air, 5% CO2. Cells were studied at passages 5–8. As previously noted, these cultures represent virtually 100% smooth muscle without epithelial or other cell types [28], and at these passage numbers the cells maintain pharmacologic and physiologic properties [22,29,30]. HASM cell viability after TAS2R agonist exposure was determined using the Vybrant and LIVE fluorescence assays (Life Technologies). Intact human bronchi were also obtained from NDRI and prepared as described [11]. The use of these tissues was in accordance with the guidelines of the Institutional Review Boards of the University of South Florida, Johns Hopkins University, and Thomas Jefferson University.
HASM [Ca2+]i Measurements
For measurement of [Ca2+]i mobilization, we used the no-wash Fluo-4 Direct Calcium Assay kit (Life Technologies) according to the manufacturer’s instructions. Briefly, cells seeded in 96-well plates (40,000 cells/well) were loaded with the Ca2+ sensitive fluorescence indicator Fluo-4 and probenecid (2.5 mM) in Hank’s balanced salt solution containing (in mM), CaCl2 (1.3), MgCl2.6 H2O (0.5), MgSO4.6 H2O (0.4), KCl (5.3), KH2PO4 (0.4), NaHCO3 (4.2), NaCl (137.9), Na2HPO4 (0.3), D-Glucose (5.5), and HEPES (20). After 30 min incubation in the dark at 37°C under 5% CO2 / 95% air atmosphere followed by 30 min at 25°C in air and darkness, drugs were added, and the increase in [Ca2+]i recorded over 120 sec (unless indicated otherwise) using the FlexStation3 plate reader (Molecular Devices). Fluorescence (excitation 485 nm, emission 525 nm, cut-off 515 nm) was measured every 1.52 sec. Baseline fluorescence background (Fo) was captured for 16–19 sec before the addition of procontractile agonist plus or minus TAS2R agonists (50 μL, 5x each). When present, the final concentration of DMSO or DMF was below 0.25%, a concentration that did not affect the baseline signal. Calcium response (ΔF, arbitrary units) was calculated by subtracting basal fluorescence signal (Fo, average of the first 10 readings) from the agonist peak value of fluorescence signal (F). The effect of co-stimulation with TAS2R agonists on [Ca2+]i response evoked by procontractile agonists was expressed as percentage of ΔF in the presence (ΔFa) of the TAS2R agonist relative to the ΔF in the absence (ΔF0) of the TAS2R agonist [100 x (1- ΔFa / ΔF0)].
Cell Membrane Potential Assay
HASM cells plated in 96-well plates (40,000 cells/well) were studied using the FLIPR membrane potential dye BLUE (Molecular Devices). The sensitivity of this assay and comparison to patch-clamp recordings have been previously reported [31–33]. Cells were incubated with dye in Hanks’ balanced salt solution supplemented with 20 mM HEPES for 10 min at 25°C in the dark. Baseline fluorescence (excitation 530 nm, emission 565 nm, cut-off 550 nm) was measured for 16 sec before addition of agonists using the FlexStation3 instrument. Signals were acquired every 2 sec for 120 sec. Increase or decrease in fluorescence after cell stimulation with various agonists indicates cell membrane depolarization or hyperpolarization, respectively. Change in fluorescence is expressed as F—Fo (ΔF) as above.
Magnetic Twisting Cytometry (MTC)
Dynamic changes in cell stiffness were measured in isolated HASM using forced motions of functionalized beads anchored to the cytoskeleton through cell surface integrin receptors, as described in detail previously [11,34]. The increase or decrease in cell stiffness is considered an index of smooth muscle contraction and relaxation, respectively, as has been previously described [34]. For each individual HASM cell, baseline stiffness was measured for the first 60 sec and after drug(s) addition, stiffness was measured continuously for the next 60 sec. For each cell, drug-induced changes in cell stiffness were normalized to its baseline stiffness prior to drug administration.
Intact Airway Physiology
Third or fourth order bronchi from human lungs were dissected and cut into rings of 5 mm in length. They were studied in an isometric myograph (AD Instruments, Colorado Springs, CO) as previously described [11]. Briefly, rings were fitted between a fixed wire and a transducer-coupled wire in Krebs solution at 37°C bubbled with 95% O2 and 5% CO2. A passive tension of 5 mN was applied to the rings and tension recorded over the next 15 min to assure a stable baseline. Procontractile agonists were added to the bath at the indicted concentrations and measurements of force obtained until the maximal response was observed (typically 10 min). Then TAS2R agonists were added at the indicted concentrations, and tension measured over the next 10 min or until the maximal decrease in tension was observed.
Drugs and Chemicals
Unless otherwise indicated, reagents were purchased from Sigma-Aldrich. Procontractile GPCR agonists used were histamine (3 μM), endothelin-1 (ET-1, 1 μM), bradykinin (BK, 5 nM), angiotensin II (Ang II, 100 μM), and acetylcholine (ACh, 1 mM). TAS2R agonists (1 nM to 2 mM) included aristolochic acid (AA), chloroquine (CQ), diphenhydramine (DPD), flufenamic acid (FFA), quinine (QUI), saccharin (SAC), strychnine (STRY), and yohimbine (YOH). Stock solutions were prepared in water or vehicle (DMSO or DMF) and diluted (5x final concentration) in calcium buffer or membrane potential buffer. Cell culture reagents including media, antibiotics, and fetal bovine serum were from Lonza and Life Technologies.
Data Analysis
Results are expressed as the mean, standard error (SE) and the number of experiments (N). IC50 and EC50 values were obtained from concentration-effect non-linear regression sigmoid curves fitted using Prism (Graph Pad Software, La Jolla, CA). When biphasic curves were observed, values within the ascending portion were excluded, with the IC50 determined with data points within the descending portion of the curve. MTC results were analyzed using a nested effect method as described [35] using SAS V9.2 (SAS Institute Inc., Cary, NC). Statistical analyses for other studies consisted of student’s t-tests, performed with Prism and Excel. Two-tailed P values less than 0.05 were considered statistically significant. Figures were generated with Prism and Excel. In figures where gaps in the fitted line or axis appear, the lower concentration is baseline or “no drug”.
Results
TAS2R Agonists Stimulate [Ca2+]i with Low Potency
The three highest expressing TAS2Rs in human ASM cells are TAS2R10, 14, and 31 [11]. We thus utilized CQ, YOH and STRY (TAS2R10), FFA, DPD (TAS2R14), AA and SAC (TAS2R 31), and QUI (TAS2R 10, 14, 31) for activating one or more of these three TAS2Rs. We used the Ca2+-sensitive fluorescent dye Fluo-4 to monitor changes in [Ca2+]i in HASM cells. We first built upon our previous findings by determining the EC50 for each TAS2R agonist for stimulating [Ca2+]i in HASM cells (S1 Table). These experiments were performed in the absence of co-incubation with procontractile agonists. As indicated, the EC50 values for these agonists for stimulating [Ca2+]i in HASM are relatively high. However, these are consistent with results from [Ca2+]i studies obtained using TAS2R cDNA-transfected cells [17] as well as taste bud cells [36].
Chloroquine Inhibits the [Ca2+]i Increase from Activation of Histamine Receptors in HASM Cells
Stimulation of confluent HASM cells with histamine induced a rapid, dose-dependent, increase in [Ca2+]i, with an EC50 value of 0.6 ± 0.3 μM (not shown). Co-treatment of cells with 3 μM histamine and increasing concentrations of the TAS2R10 agonist CQ showed a dose-dependent decrease in the peak [Ca2+]i beginning with 1 μM CQ and up to concentrations of ~1 mM CQ (Fig 1A). At higher concentrations of CQ, an increase in [Ca2+]i was observed (Fig 1A). Thus a biphasic [Ca2+]i response to CQ was evident in the presence of histamine stimulation (Fig 1B). Non-linear regression analyses of the inhibitory limb of the curve revealed an IC50 = 14.8 ± 5.8 μM (N = 4) (Fig 1C). Of note, CQ stimulates [Ca2+]i, in the absence of histamine with a EC50 of ~450 μM (Fig 1D and S1 Table). Together, these data indicate that CQ is more potent in inhibiting histamine-stimulated [Ca2+]i than in de novo stimulation of [Ca2+]i in HASM.
Fig 1. Biphasic effect of the TAS2R10 agonist CQ on HASM cell [Ca2+]i mobilization.

(A) Representative dose-response and time course of [Ca2+]i in cells treated with 3 μM histamine and the indicated concentrations of CQ. (B) Biphasic [Ca2+]i response to CQ in HASM concomitantly treated with 3 μM histamine. (C) Inhibitory limb of the [Ca2+] response to CQ in HASM concomitantly treated with 3 μM histamine. (D) Stimulation of [Ca2+] by CQ in HASM. There is no co-treatment with histamine in this experiment. Shown are representative results (mean ± SE of quadruplicates) of 3–5 independent experiments performed.
Inhibition of [Ca2+]i by TAS2R Agonists is Contingent Upon the Procontractile Receptor that Stimulates [Ca2+]i Mobilization
We next examined whether activation of TAS2R31 with AA would also inhibit [Ca2+]i elicited by histamine. Fig 2A shows that in contrast to CQ, AA failed to decrease [Ca2+]i evoked by histamine. The magnitude of the [Ca2+]i peaks remained unchanged up to ~200 μM AA, above which an increase in the signal was detected. Similar to AA, co-treatment of HASM cells with SAC, a bitter tastant that activates TAS2R31, did not inhibit [Ca2+]i stimulated by histamine (Fig 2B). The lack of inhibitory effect of AA and SAC on histamine-stimulated [Ca2+]i prompted us to investigate whether AA inhibits elevated [Ca2+]i induced by other GPCR agonists. Fig 3A shows that AA does indeed inhibit [Ca2+]i that is elevated by ET-1. The calculated IC50 of AA with HASM cells stimulated with 1 μM ET-1 was 18.1 ± 6.7 μM (N = 3). As expected, AA in the absence of ET-1 stimulates [Ca2+]i (Fig 3B), although this requires higher doses of AA to observe such stimulation. Remarkably, CQ, which was highly effective at inhibiting histamine-stimulated [Ca2+]i (Fig 1C) showed a minimal effect on ET-1 stimulated [Ca2+]i (Fig 3C). The inhibitory effect of the TAS2R agonists was not an artifact associated with drug-induced cell death (S1 Fig). Furthermore, this [Ca2+]i inhibitory effect was fully reversed when the cells were washed to remove TAS2R agonist and rechallenged with procontractile agonist (S2 Fig). The efficacy of CQ to inhibit [Ca2+]i elicited by histamine but not by ET-1, versus the efficacy of AA to inhibit [Ca2+]i elicited by ET-1 but not by histamine, strongly suggests that the potential for TAS2R agonists to inhibit [Ca2+]i is conditional upon the GPCR that stimulates [Ca2+]i elevation.
Fig 2. Effects of TAS2R31 agonists on histamine-stimulated [Ca2+] in HASM cells.

Effect of varying concentrations of AA (A) or SAC (B) on 3 μM histamine-mediated [Ca2+]i increase. Shown are representative results (mean ± SE of quadruplicates) of 3–5 independent experiments performed.
Fig 3. Inhibition of ET-1 stimulated [Ca2+]i in HASM cells differs based on TAS2R agonist.
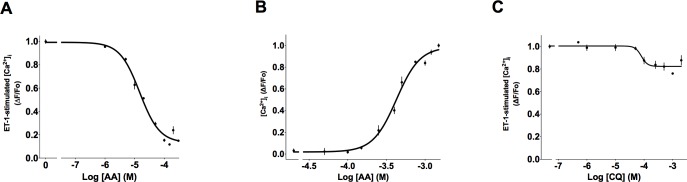
(A) Inhibition of ET-1 stimulated [Ca2+] by AA. (B) AA stimulates [Ca2+]i in the absence of ET-1. (C) CQ has a minimal inhibitory effect on ET-1 stimulated [Ca2+] (compared to histamine stimulated effect in Fig 1). Shown are representative results (mean ± SE of quadruplicates) of 3–5 independent experiments performed.
To better comprehend the extent of this selectivity, we utilized a battery of the aforementioned eight TAS2R agonists and five procontractile GPCR stimulators of [Ca2+]i: BK, ACh, Ang II, histamine, and ET-1. As introduced earlier, each of the TAS2R agonists stimulates [Ca2+]i in HASM cells with low potency (S1 Table), and at the 50 μM concentration, no agonist consistently increased [Ca2+]I levels above background. We therefore utilized 50 μM of TAS2R agonists in the screen. Because DPD is a histamine receptor antagonist, it was not studied in the context of histamine-stimulated [Ca2+]i (denoted as N/A in figures). Quantitative analysis revealed that CQ, STRY, YOH and QUI inhibited [Ca2+]i evoked by histamine while AA, FFA, and SAC had no effect (Fig 4A). On the other hand, AA and QUI suppressed [Ca2+]i elicited by ET-1 but CQ, STRY, YOH, DPD and FFA did not. FFA and AA inhibited [Ca2+]i stimulated by BK while AA, QUI, STRY, CQ, and YOH were without effect. Similar heterogeneity was observed when ACh and Ang II were utilized as the [Ca2+]i stimulant (Fig 4D and 4E).
Fig 4. Quantitative effect of TAS2R agonists on [Ca2+]i in HASM stimulated by five GPCR agonists.

(A-E) Cells were treated with the 3 μM histamine, 1 μM ET-1, 5 mM BK, 1 mM ACh, or 100 μM Ang II in the absence (buffer) or presence of 50 μM of the indicated TAS2R agonists. Statistical analysis was not performed on co-treatments that resulted in [Ca2+]i enhancement. Large positive values were truncated to allow visualization of the smaller values. *, P<0.05; # P<0.005 vs control (buffer). N = 5–8 experiments.
Fig 5 summarizes the above results for inhibition of [Ca2+]i as a heat map, with TAS2R agonist subtype specificities noted for the different compounds. From this map, it does not appear that TAS2R inhibition of [Ca2+]i stimulation by the various GPCR agonists is TAS2R subtype-dependent (note the lack of a pattern in the columns). We considered that the heterogeneity in the inhibitory response could be due to whether [Ca2+]i was stimulated by a Gq—vs a Gi-coupled receptor. Of note, pertussis toxin treatment cannot be utilized for this differentiation, since it also inactivates the TAS2R G-protein gustducin [37]. We thus utilized subtype-specific agonists for the histamine HRH1 (Gq-coupled) and HRH3 (Gi-coupled) receptors in the absence or presence of CQ, and measured HASM [Ca2+]i mobilization (Fig 6A). N-Methyl-histaprodifen (NMH, a HRH1 agonist) stimulated [Ca2+]i was inhibited ~50% by CQ. Similarly, stimulation of HASM [Ca2+]i by another HRH1 agonist, 2-((3-Trifluoromethyl)phenyl)histamine (2,3 TFMP), was also inhibited by CQ. Importantly, the Gi-coupled HRH3 agonist immethridine-stimulated [Ca2+]i was inhibited by CQ to a similar extent. To further assess the possibility of selectivity for Gq- vs Gi-coupled receptors for TAS2R-mediated inhibition, we exposed cells to 400 μM somatostatin, which activates the four somatostatin subtypes, all of which couple to Gi but not to Gq. As shown in Fig 6B, [Ca2+]i stimulation by somatostatin was also inhibited by TAS2R agonists to ~50%. Taken together, these data indicate that regardless of whether the GPCR couples to Gq or Gi, TAS2R activation can reduce elevated [Ca2+]i, and thus the heterogeneity that we observe between contractile GPCR agonists and TAS2R agonists cannot be readily attributed to this mechanism.
Fig 5. Heat map of the relative effects of TAS2R agonists on [Ca2+] stimulated by the indicated procontractile CPCR agonists.

Data from the experiments in Fig 4 were normalized to maximal stimulation (red) or inhibition (blue). TAS2R agonists are on the top row and their subtype specificity shown. Procontractile GPCR agonists are listed in the first column. N/A, not applicable.
Fig 6. TAS2R inhibition of stimulated [Ca2+]i is not dependent on Gi or Gq coupled receptor stimulation.
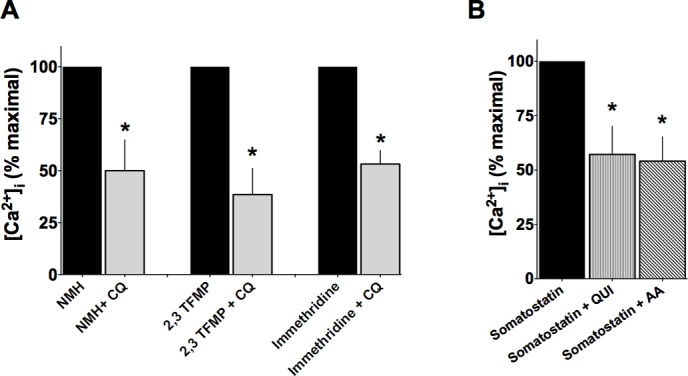
(A) HASM were co-treated with Gq-coupled HRH1 agonists NMH or 2,3 TFMP, or the Gi-coupled HRH3 agonist immethridine and 50 μM CQ. (B) Somatostatin (Gi-coupled) stimulated [Ca2+]i is inhibited by QUI and AA. *, P<0.05, N = 4 experiments.
Heterogeneity in TAS2R Responses of HASM Cell Membrane Potential
The increase in [Ca2+]i induced by procontractile agonists promotes cell membrane depolarization and actin-myosin activation, ultimately leading to contraction. Conversely, hyperpolarization of the cell membrane antagonizes contraction. One of the proposed mechanisms by which TAS2R activation induces smooth muscle relaxation is a [Ca2+]i-dependent decrease in plasma membrane potential (hyperpolarization). We therefore explored the changes in membrane potential that occurred upon stimulation of HASM cells with bronchoconstrictive agonists in the presence and absence of co-administration of TAS2R agonists. We used a validated assay in which an increase or decrease in probe fluorescence is indicative, respectively, of depolarization or hyperpolarization of the cell membrane. Exposure of HASM to 60 mM KCl resulted in the expected depolarization (Fig 7). Similarly, the bronchoconstrictive GPCR agonist histamine caused a sustained depolarization. However, in the presence of 50 μM CQ, histamine-mediated membrane depolarization was markedly inhibited and indeed resulted in hyperpolarization. In contrast, SAC had no such effect. These results are consistent with the [Ca2+]i inhibition studies, where CQ decreased histamine-stimulated [Ca2+]i while SAC was ineffective (see Fig 4A). ET-1 also evoked depolarization, although it displayed an early peak with a lower-magnitude depolarization thereafter. Concomitant treatment with 50 μM AA blocked ET-1 depolarization (Fig 7), also consistent with the AA effect on lowering ET-1 stimulated [Ca2+]i (see Fig 4B).
Fig 7. Heterogeneity of HASM membrane potential responses evoked by procontractile agonists co-treated with TAS2R agonists.

Cultured HASM cells were studied using a fluorescence-based membrane potential dye. The indicated agents were added singly, or in combination, at the 16 sec time point. The final concentrations were: Hist 3 μM, CQ 50 μM, SAC 50 μM, ET-1 1 μM, AA 50 μM. Results are mean ± SE of triplicate determinations from a single representative experiment of 2–5 performed.
The Dichotomy of CQ and AA Inhibition of [Ca2+]i is Recapitulated in HASM Physiologic Responses
The above results suggest that HASM relaxation by a given TAS2R agonist would be dependent upon which GPCR is acting to contract the muscle. To test this, we utilized MTC, a sensitive method that detects changes in stiffness in single cells and is considered a surrogate for ASM contraction [34]. As shown in Fig 4A and 4B, a clear dichotomy in TAS2R responses is observed between histamine and ET-1 stimulated [Ca2+]i. CQ inhibits the former but not the latter, which is in contrast to that of AA, which inhibits ET-1-but not histamine-stimulated [Ca2+]i. To ascertain if these biochemical findings correlate with the expected physiologic effects, HASM cells were treated with 3 μM histamine alone, or in combination with 50 μM of either AA or CQ. Similarly, HASM cells were treated with 1 μM ET-1 alone, or in combination with 50 μM of either AA or CQ. HASM cell stiffness was measured for 60 sec and normalized to baseline (absence of any drugs). As shown in Fig 8A, histamine caused the expected increase in cell stiffness. Co-administration with 50 μM CQ, which inhibited histamine-evoked [Ca2+]i by ~70%, fully blocked the histamine-mediated increase in cell stiffness (Fig 8A). In contrast, and consistent with the [Ca2+]i results of Fig 4A, AA had no effect on histamine-induced cell stiffening. For ET-1 evoked stiffness (Fig 8B), AA caused an attenuation of the stiffness response, amounting to a ~60% reduction. The magnitude of this response is nearly identical to the reduction in [Ca2+]i shown in Fig 4B. Consistent with the lack of an inhibition of the [Ca2+]i response to ET-1, CQ had no physiological effect on the HASM stiffness response to ET-1 (Fig 8B).
Fig 8. The heterogeneity of TAS2R inhibition of [Ca2+]i is recapitulated in HASM physiological responses.
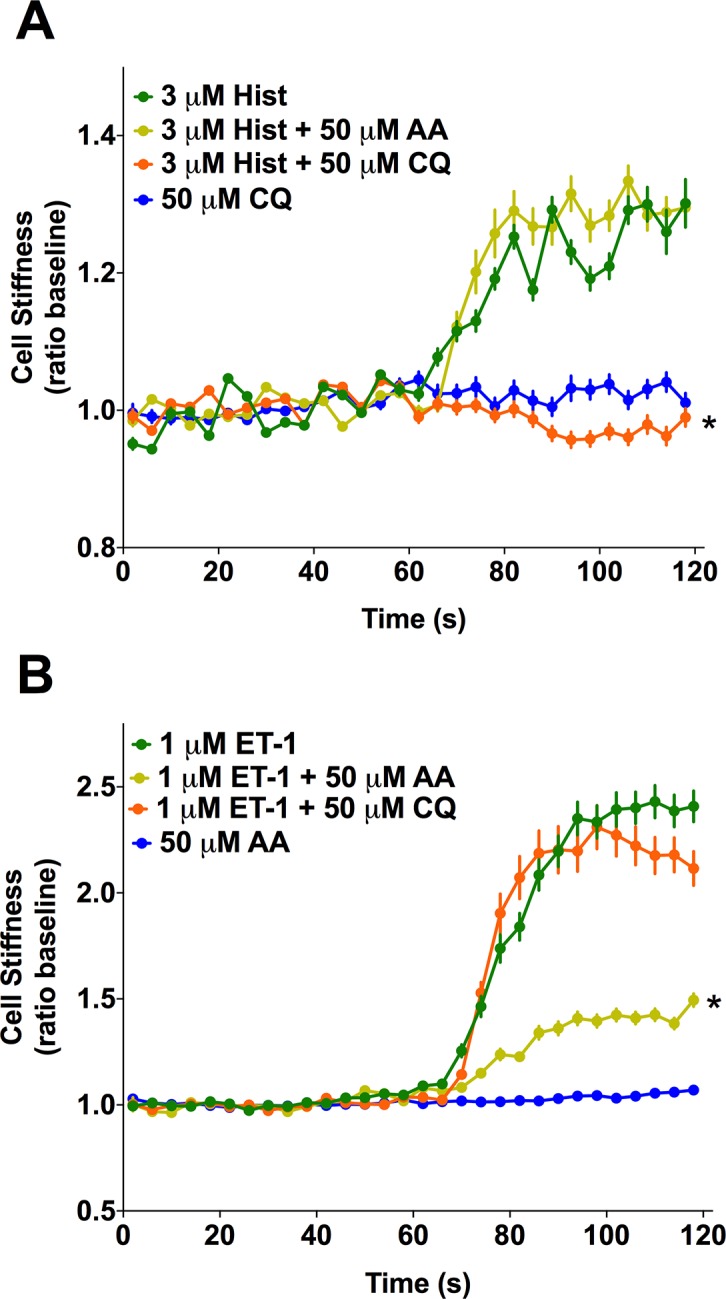
(A, B) Isolated HASM in culture were studied using magnetic twisting cytometry. Cells were treated with 3 μM histamine or 1 μM ET-1 alone or together with the indicated TAS2R agonists (50 μM). * P<0.01 vs control (histamine or ET-1 with buffer). N = 303–400 cells per condition.
To confirm these results from isolated cell mechanics experiments, we studied human bronchi in the ex vivo setting, measuring contraction and relaxation of airway rings in a lateral myograph. In this system, the coordinated effect of the ASM cell phenotypes can be ascertained in the context of the intact airway. Mean data from measurements using 10–15 rings derived from three independent donors are shown in Fig 9. When airways were contracted with histamine, CQ evoked ~90% relaxation. However, AA had no effect on histamine-mediated tension (Fig 9A). In contrast, when airways were contracted with ET-1, AA caused ~75% relaxation while CQ had a non-significant effect (Fig 9B). These results are fully consistent with the results from the MTC experiments with isolated ASM cells, as well as the membrane potential and [Ca2+]i inhibition results.
Fig 9. TAS2R inhibition of contracted human bronchi.
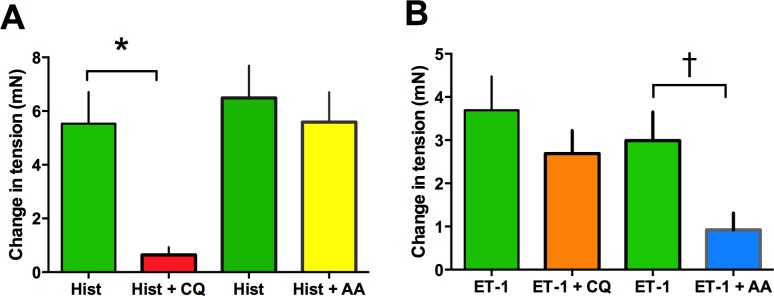
Results are from experiments performed on 10–15 airway rings from three different donated lungs. The concentrations of drugs were: histamine 10 μM, ET-1 1 μM, AA 100 μM, CQ 100 μM. *, P<0.001; † P<0.05, compared to histamine or ET-1 alone.
Discussion
As introduced earlier, TAS2Rs expressed on HASM cells represent a novel target for a new class of direct bronchodilators for the treatment of obstructive lung diseases such as asthma and chronic obstructive lung disease. Here we report the function of eight TAS2R agonists acting on HASM under physiologically relevant conditions of exposure to procontractile GPCR agonists. We hypothesized that TAS2R activation decrease [Ca2+]i stimulated by these procontractile GPCR agonists, and, that this inhibitory effect occurs with lower concentrations of TAS2R agonists than those that promote the TAS2R-mediated [Ca2+]i stimulatory pathway. We found that the efficacy of TAS2R agonists to oppose the increase in [Ca2+]I evoked by other GPCRs is contingent upon which procontractile GPCR is being activated and that both Gi and Gq coupled receptors are subject to TAS2R inhibition. Furthermore, the decrease in [Ca2+]i by TAS2R agonists was associated with a decrease in cell membrane depolarization. When a given TAS2R agonist-GPCR procontractile agonist pair did not reveal a TAS2R-mediated inhibition of [Ca2+], there was consistently no effect on membrane potential. The physiological consequences of these TAS2R-mediated events were ascertained by measuring HASM cell mechanics (which examines the effect on isolated smooth muscle cells) and by measuring force in intact human bronchi (which ascertains the effect on coordinated smooth muscle function of the intact airway). The heterogeneity observed for certain pairs for inhibiting [Ca2+]i was indeed recapitulated in measurements of stiffness in single cells. Cell stiffness, which was increased by the procontractile agonists was antagonized only by TAS2R agonists that decreased [Ca2+] (and opposed membrane depolarization) under the same conditions. Finally, intact human airway responses were consistent with the results from the [Ca2+]i, membrane potential, and isolated cell mechanics studies. Taken together, these findings demonstrate the relevance of this TAS2R pathway to human physiologic responses.
Our initial identification and characterization of TAS2Rs on mouse and human ASM centered around the increase in [Ca2+]i as a key intracellular event leading to ASM relaxation [11]. Indeed, agonists for these receptors caused robust increases in [Ca2+]i in isolated HASM cells, that was blocked by Gβγ and PLC inhibitors, and substantially attenuated by an IP3 receptor antagonist. In addition, human and mouse ASM have been shown to express gustducin [11,15]. This stimulatory pathway pointed towards a taste cell-like response, where the Gβγ activation of PLC caused IP3 production which activated the endoplasmic reticulum IP3 receptor, releasing [Ca2+] from these stores into an intracellular space. However, we found that the temporal and spatial distribution of this Ca2+ in HASM cells was suggestive of the activation of one or more cell surface channels such as the large conductance calcium dependent K+ channel (BKCa), which leads to ASM cell hyperpolarization. There appears to be additional mechanisms by which this specialized pool of [Ca2+]i evoked from TAS2R activation leads to relaxation [15]. Regardless of potential mechanism, TAS2R agonists that fail to increase HASM [Ca2+]i also fail to relax [11]. This stimulatory pathway appears to be somewhat inefficient, in that most TAS2R agonists activate [Ca2+]i with EC50 values in the high μM to mM range in HASM (S1 Table) and in transfected cell lines [17]. With this current report and studies by others using different cells [15], it is now apparent that TAS2Rs acting through a distinct pathway can also decrease [Ca2+]i in HASM cells that have been stimulated by other means. This decrease in [Ca2+]i would be expected to cause relaxation since it antagonizes the procontractile GPCR-mediated elevated [Ca2+]i, and thus supports the concept that TAS2Rs in ASM cells initiate two signaling events. The lower efficiency transduction pathway results in an increase in [Ca2+]i and subsequent membrane hyperpolarization and relaxation. The more efficient process, instead, acts to functionally compete with a stimulated increase in [Ca2+]i and dampen, or prevent, depolarization. The physiologic response, then, is relaxation from the contracted state. We also show that there is heterogeneity of the response that hinges upon which procontractile GPCR is being stimulated.
We propose the operational model depicted in Fig 10, where three [Ca2+]i pools are indicated. GPCR-A is a bronchoconstrictive receptor whose activation elevates [Ca2+]i leading to contraction. Because there is no interaction with TAS2R-X signaling, agonists for this TAS2R do not inhibit [Ca2+]i or reverse depolarization from this bronchoconstrictive receptor and thus we would expect no TAS2R physiological effect. An example of this signaling is histamine (representing GPCR-A) in the presence of AA, which causes no change in [Ca2+]i or relaxation (Figs 4A, 8A and 9, respectively). In contrast, the signaling of GPCR-B to elevate [Ca2+]i interacts with the signaling of TAS2R-X. This could occur in a very early event (Fig 10, i) such as through βγ as has been suggested [15], or at later points (Fig 10, ii) such as between [Ca2+]i pools. In this case, TAS2R-X inhibits the [Ca2+]i elevated by GPCR-B, leading to reversal of depolarization and relaxation. An example of this scenario is AA acting at TAS2R31 to inhibit endothelin receptor-stimulated [Ca2+] (Fig 4A), reverse depolarization (Fig 7), decrease cell stiffness (Fig 8A), and relax intact airways (Fig 9B). A third scenario in the model is the action of TAS2R-Y in the absence of [Ca2+] stimulation by Gq- or Gi-coupled GPCRs. Here, there is no interaction between the signaling of the TAS2R and the bronchoconstrictive GPCR. In this instance, TAS2R-Y relaxes the ASM cell by a [Ca2+]i-dependent cell surface transducer, which hyperpolarizes the membrane when [Ca2+]i is increased by TAS2R-Y agonists, acting to decrease ASM tone. This represents the less efficient pathway, based on EC50 values being higher than those for [Ca2+]i inhibition. An example of this scenario is SAC, which clearly stimulates [Ca2+]i, hyperpolarizes the HASM cell membrane, and relaxes HASM in the absence of a contractile stimulus [11].
Fig 10. Model of TAS2R signaling to relaxation of HASM.

Stimulation of GPCR-A results in an increase in [Ca2+]i, depolarization, and contraction (red). GPCR-A is not influenced by TAS2R interaction, thus [Ca2+]i mobilization and ultimately contraction is not affected. Activation of certain GPCRs (GPCR-B) also promote contraction by increasing [Ca2+]i, but are influenced (green dotted lines) by TAS2R agonists (acting at TAS2R-X). This results in a decrease in [Ca2+]i, which opposes depolarization and relaxes HASM (green). This GPCR-B/TAS2R-X interaction could be in proximal (i) components or other downstream components (ii). TAS2R-Y does not specifically interact with a procontractile GPCR, but relaxes HASM by a calcium-dependent transducer, such as BKCa, that hyperpolarizes the membrane (blue).
In humans, there are 25 TAS2R subtypes expressed on taste buds, which presumably evolved to trigger avoidance of ingestion of toxic bitter plants [17,18]. Most TAS2R subtypes are activated by large numbers of naturally occurring bitter substances, and given that these substances come into direct contact with the tongue, probably evolved towards low affinity and broad ligand recognition to accomplish this function. Recently, TAS2Rs have been identified on other cell types and regions of the body, including the nose, gastrointestinal tract, thyroid, lung, heart, lymphocyte, brain and testes. This suggests a previously unrecognized chemosensory system in the body that may respond to exogenous substances ingested in food or endogenously produced substances such as from resident bacteria [38,39]. These extraoral receptors may represent targets for new therapeutics, such as the TAS2R subtypes expressed on ASM which act to markedly relax the muscle resulting in bronchodilation. The dual pathway that we show for TAS2Rs in HASM cells offers intriguing possibilities for drug discovery and design. In the initial phase, measurement of fluorescent-based [Ca2+] measurements using cells transfected with the cDNA for a specific TAS2R subtype will provide a platform for compound screening. Typically, the cDNA for a Ggust/G44 chimeric G-protein is also transfected, which directs signaling to PLC activation and an increase in [Ca2+]i [17]. In this system, [Ca2+]i acts as a readily acquired indicator of receptor activation, but is not necessarily the physiologically relevant signal. The “inhibition pathway”, therefore, is not revealed in this screening approach. In the second phase, moving lead compounds from the transfected cell studies to HASM cell-based assays would be appropriate, measuring both TAS2R-stimulation of [Ca2+]i as well as TAS2R mediated inhibition of [Ca2+]i stimulated by a procontractile GPCR agonist. Our findings caution against excluding compounds based on the lack of inhibition of [Ca2+]i elevated by a single GPCR procontractile agonist (Fig 4). Rather, multiple such contractile agonists would need to be explored. This would also be the case for membrane potential assays as well as physiological assays.
There is considerable precedence for receptors within the GPCR superfamily (of which the TAS2R family is a member) coupling to multiple pathways, including those with competing or opposing effects. For example, the α2ARs couple to Gαi (inhibiting cAMP) and to Gαs (stimulating cAMP) [40]. The latter function requires higher doses of agonist compared to the inhibitory function due to the lower efficiency of coupling [40,41]. Specific regions within the intracellular loops of the α2AAR have been identified which direct coupling to Gαs or Gαi [42,43], thus showing receptor structure as the basis for these multifunctional events. In this instance one receptor couples to two G-proteins with opposing actions on the effector adenylyl cyclase. Multifunctional signaling can also be from non-G-protein interactions. For example, activation of the angiotensin II type 1A receptor couples to PLC activation via Gq, but also activates c-Jun amino-terminal Kinase 3 (JNK3) by βarrestin-2 binding to the receptor, which provides a scaffold for JNK3 activation [44]. GPCR signaling can also be directed based on spatial distribution of the receptor, G-protein, or effector within the cell, thereby resulting in specialized pools of second messenger [11,45,46]. These and other mechanisms of multifunctional GPCR signaling have been reviewed elsewhere [47–49]. The mechanisms by which TAS2Rs couple to both [Ca2+]i stimulation and inhibition are not readily apparent. This phenomenon is made even more complex by the heterogeneity of the inhibitory response, which is dependent upon which GPCR is providing the [Ca2+]i stimulation.
In conclusion, TAS2Rs on HASM are recognized to stimulate [Ca2+] with high agonist concentrations which results in membrane hyperpolarization and HASM relaxation. In addition, when TAS2Rs are activated by lower concentrations of agonist under conditions of elevated [Ca2+] by various bronchoconstrictors, they inhibit this [Ca2+]i increase, oppose membrane depolarization, and thus relax precontracted HASM. The inhibitory response is dependent on which GPCR is acting to stimulate [Ca2+] mobilization. This suggests a compartmentalization of [Ca2+]i signals in HASM, of which some are accessible to TAS2Rs, or, other forms of signaling interactions between TAS2Rs and bronchoconstrictive receptors.
Supporting Information
(PDF)
A) Cell death was determined using the Vybrant assay (Life Technologies), which quantitates the formation of reduced red fluorescent resazurin from a coupled enzymatic reaction in which NADPH is generated from the activity of glucose-6-phosphate dehydrogenase released from dying cells. 40,000 HASM cells/well were treated with buffer or buffer with 50 μM or 1 mM of the indicated TAS2R agonists for 5 min. As a positive control, cells were treated with lysis buffer. Data is from 4–7 experiments performed in triplicate. P>0.05 for all agonists compared to buffer, indicating no significant cell death. B) The proportion of live HASM cells was determined with the LIVE assay (Life Technologies) which measures intracellular esterase activity on calcein-AM which fluoresces green when hydrolyzed. Cells were plated at 40,000/well and treatments were with buffer alone or buffer with 50 μM of the indicted TAS2R agonists for 5 min. No agonist caused a decrease in viable cells. Data is from 4–6 experiments performed in triplicate. P = 0.04 for YOH which was greater than control (buffer).
(TIFF)
40,000 HASM cells/well were exposed to buffer alone (representing “untreated”) or buffer with 50 μM CQ or AA for 5 min. Cells were washed twice with PBS, and then [Ca2+]i mobilization measured in response to 3 μM histamine, 1 μM ET-1, or 1 μM ionomycin. The responses to histamine and ET-1 (as well as ionomycin) were no different in cells pretreated with CQ or AA, compared to pretreatment with buffer alone, indicated a reversal of TAS2R agonist effect. Data is from 4–5 experiments performed in triplicate.
(TIFF)
Acknowledgments
We acknowledge the technical assistance of David J. Disimile, Madison A. Dixon, Jacqueline Carson, Shariff Bushra, Atif Khan and manuscript preparation by Charmaine Disimile.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by the National Institutes of Health, http://www.nhlbi.nih.gov: P01HL114471 to SBL SSA, R01HL107361 to SSA, K01HL092588 to BC-M, and NIH R01HL045967 to SBL. American Asthma Foundation to DAD. Cracchiolo Family Foundation to SBL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Green SA, Liggett SB (1996) G-protein-coupled receptor signaling in the lung In: Liggett SB, Meyers DA, editors. The Genetics of Asthma. New York: Marcel Dekker, Inc; pp. 67–90. [Google Scholar]
- 2. Barnes PJ, Chung KF, Page CP (1998) Inflammatory mediators of asthma: an update. Pharmacological Reviews 50: 515–596. [PubMed] [Google Scholar]
- 3. Lipworth BJ (1997) Airway subsensitivity with long-acting beta 2-agonists. Is there cause for concern? Drug Saf 16: 295–308. [DOI] [PubMed] [Google Scholar]
- 4. Kraan J, Koeter GH, van der Mark TW, Sluiter HJ, De Vries K (1985) Changes in bronchial hyperreactivity induced by 4 weeks of treatment with antiasthmatic drugs in patients with allergic asthma: a comparison between budesonide and terbutaline. J Allergy Clin Immunol 76: 628–636. [DOI] [PubMed] [Google Scholar]
- 5. Cheung D, Timmers MC, Zwinderman AH, Bel EH, Dijkman JH, Sterk PJ (1992) Long-term effects of a long acting β2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N Engl J Med 327: 1198–1203. [DOI] [PubMed] [Google Scholar]
- 6. Drazen JM, Silverman EK, Lee TH (2000) Heterogeneity of therapeutic responses in asthma. Br Med Bull 56: 1054–1070. [DOI] [PubMed] [Google Scholar]
- 7. Beasley R, Pearce N, Crane J, Burgess C (1999) Beta-agonists: what is the evidence that their use increases the risk of asthma morbidity and mortality? J Allergy Clin Immunol 103: S18–S30. [DOI] [PubMed] [Google Scholar]
- 8. Sears MR, Taylor DR (1994) The β2-agonist controversy: Observations, explanations and relationship to asthma epidemiology. Drug Safety 11: 259–283. [DOI] [PubMed] [Google Scholar]
- 9. Salpeter SR, Wall AJ, Buckley NS (2010) Long-acting beta-agonists with and without inhaled corticosteroids and catastrophic asthma events. Am J Med 123: 322–328. 10.1016/j.amjmed.2009.07.035 [DOI] [PubMed] [Google Scholar]
- 10. Einstein R, Jordan H, Zhou W, Brenner M, Moses EG, Liggett SB (2008) Alternative splicing of the G protein-coupled receptor superfamily in human airway smooth muscle diversifies the complement of receptors. Proc Natl Acad Sci U S A 105: 5230–5235. 10.1073/pnas.0801319105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, et al. (2010) Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nature Medicine 16: 1299–1304. 10.1038/nm.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deshpande DA, Robinett KS, Wang WC, Sham JS, An SS, Liggett SB (2011) Bronchodilator activity of bitter tastants in human tissue. Nature Medicine 17: 776–778. 10.1038/nm0711-776a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belvisi MG, Dale N, Birrell MA, Canning BJ (2011) Bronchodilator activity of bitter tastants in human tissue. Nature Medicine 17: 776 10.1038/nm0711-776a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pulkkinen V, Manson ML, Safholm J, Adner M, Dahlen SE (2012) The bitter taste receptor (TAS2R) agonists denatonium and chloroquine display distinct patterns of relaxation of the guinea pig trachea. Am J Physiol Lung Cell Mol Physiol 303: L956–966. 10.1152/ajplung.00205.2012 [DOI] [PubMed] [Google Scholar]
- 15. Zhang CH, Lifshitz LM, Uy KF, Ikebe M, Fogarty KE, ZhuGe R (2013) The cellular and molecular basis of bitter tastant-induced bronchodilation. PLoS Biology 11: e1001501 10.1371/journal.pbio.1001501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grassin-Delyle S, Abrial C, Fayad-Kobeissi S, Brollo M, Faisy C, Alvarez JC, et al. (2013) The expression and relaxant effect of bitter taste receptors in human bronchi. Respir Res 14: 134 10.1186/1465-9921-14-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyerhof W, Batram C, Kuhn C, Brockhoff A, Chudoba E, Bufe B, et al. (2010) The molecular receptive ranges of human TAS2R bitter taste receptors. ChemSenses 35: 157–170. [DOI] [PubMed] [Google Scholar]
- 18. Meyerhof W (2005) Elucidation of mammalian bitter taste. Rev Physiol Biochem Pharmacol 154: 37–72. [DOI] [PubMed] [Google Scholar]
- 19. Liggett SB (2014) Bitter taste receptors in the wrong place: novel airway smooth muscle targets for treating asthma. Trans Am Clin Climatol Assoc 125: 64–74; discussion 74–65. [PMC free article] [PubMed] [Google Scholar]
- 20. Liggett SB (2013) Bitter taste receptors on airway smooth muscle as targets for novel bronchodilators. Expert Opin Ther Targets 17: 721–731. 10.1517/14728222.2013.782395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. An SS, Robinett KS, Deshpande DA, Wang WC, Liggett SB (2012) Reply to: Activation of BK channels may not be required for bitter tastant-induced bronchodilation. Nature Medicine 18: 650–651. 10.1038/nm.2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. An SS, Wang WC, Koziol-White CJ, Ahn K, Lee DY, Kurten RC, et al. (2012) TAS2R activation promotes airway smooth muscle relaxation despite beta(2)-adrenergic receptor tachyphylaxis. Am J Physiol Lung Cell Mol Physiol 303: L304–311. 10.1152/ajplung.00126.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinett KS, Koziol-White CJ, Akoluk A, An SS, Panettieri RA Jr, Liggett SB (2014) Bitter taste receptor function in asthmatic and nonasthmatic human airway smooth muscle cells. Am J Respir Cell Mol Biol 50: 678–683. 10.1165/rcmb.2013-0439RC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinett KS, Deshpande DA, Malone MM, Liggett SB (2011) Agonist-promoted homologous desensitization of human airway smooth muscle bitter taste receptors. Am J Respir Cell Mol Biol 45: 1069–1074. 10.1165/rcmb.2011-0061OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paul RJ, de Lanerolle P (1996) Regulation of Smooth Muscle Contractility In: Liggett SB, Meyers DA, editors. The Genetics of Asthma. New York: Marcel Dekker, Inc; pp. 91–117. [Google Scholar]
- 26. Syyong HT, Raqeeb A, Pare PD, Seow CY (2011) Time course of isotonic shortening and the underlying contraction mechanism in airway smooth muscle. J Appl Physiol (1985) 111: 642–656. 10.1152/japplphysiol.00085.2011 [DOI] [PubMed] [Google Scholar]
- 27. McGraw DW, Forbes SL, Witte DP, Fortner CN, Paul RJ, Liggett SB (1999) Transgenic overexpression of β2-adrenergic receptors in airway smooth muscle alters myocyte function and ablates bronchial hyperreactivity. J Biol Chem 274: 32241–32247. [DOI] [PubMed] [Google Scholar]
- 28. Panebra A, Schwarb MR, Glinka CB, Liggett SB (2007) Heterogeneity of transcription factor expression and regulation in human airway epithelial and smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 293: L453–L462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carr R 3rd, Du Y, Quoyer J, Panettieri RA Jr, Janz JM, Bouvier M, et al. (2014) Development and characterization of pepducins as Gs-biased allosteric agonists. Journal of Biological Chemistry 289: 35668–35684. 10.1074/jbc.M114.618819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Comer BS, Camoretti-Mercado B, Kogut PC, Halayko AJ, Solway J, Gerthoffer WT, et al. (2014) MicroRNA-146a and microRNA-146b expression and anti-inflammatory function in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L727–734. 10.1152/ajplung.00174.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fairless R, Beck A, Kravchenko M, Williams SK, Wissenbach U, Diem R, et al. (2013) Membrane potential measurements of isolated neurons using a voltage-sensitive dye. PLoS ONE 8: e58260 10.1371/journal.pone.0058260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fitch RW, Xiao Y, Kellar KJ, Daly JW (2003) Membrane potential fluorescence: a rapid and highly sensitive assay for nicotinic receptor channel function. Proc Natl Acad Sci U S A 100: 4909–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whiteaker KL, Gopalakrishnan SM, Groebe D, Shieh CC, Warrior U, Burns DJ, et al. (2001) Validation of FLIPR membrane potential dye for high throughput screening of potassium channel modulators. Journal of Biomolecular Screening 6: 305–312. [DOI] [PubMed] [Google Scholar]
- 34. An SS, Fabry B, Trepat X, Wang N, Fredberg JJ (2006) Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am J Respir Cell Mol Biol 35: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krzywinski M, Altman N, Blainey P (2014) Points of significance: nested designs. For studies with hierarchical noise sources, use a nested analysis of variance approach. Nature Methods 11: 977–978. [DOI] [PubMed] [Google Scholar]
- 36. Chandrashekar J, Hoon MA, Ryba NJ, Zuker CS (2006) The receptors and cells for mammalian taste. Nature 444: 288–294. [DOI] [PubMed] [Google Scholar]
- 37. Van Dop C, Yamanaka G, Steinberg F, Sekura RD, Manclark CR, Stryer L, et al. (1984) ADP-ribosylation of transducin by pertussis toxin blocks the light-stimulated hydrolysis of GTP and cGMP in retinal photoreceptors. Journal of Biological Chemistry 259: 23–26. [PubMed] [Google Scholar]
- 38. Brockhoff A, Behrens M, Massarotti A, Appendino G, Meyerhof W (2007) Broad tuning of the human bitter taste receptor hTAS2R46 to various sesquiterpene lactones, clerodane and labdane diterpenoids, strychnine, and denatonium. J Agric Food Chem 55: 6236–6243. [DOI] [PubMed] [Google Scholar]
- 39. Sbarbati A, Tizzano M, Merigo F, Benati D, Nicolato E, Boschi F, et al. (2009) Acyl homoserine lactones induce early response in the airway. Anat Rec (Hoboken) 292: 439–448. [DOI] [PubMed] [Google Scholar]
- 40. Eason MG, Kurose H, Holt BD, Raymond JR, Liggett SB (1992) Simultaneous coupling of α2-adrenergic receptors to two G-proteins with opposing effects: Subtype-selective coupling of α2C10, α2C4 and α2C2 adrenergic receptors to Gi and Gs. J Biol Chem 267: 15795–15801. [PubMed] [Google Scholar]
- 41. Eason MG, Jacinto MT, Liggett SB (1994) Contribution of ligand structure to activation of α2AR subtype coupling to Gs . Mol Pharmacol 45: 696–702. [PubMed] [Google Scholar]
- 42. Eason MG, Liggett SB (1995) Identification of a Gs coupling domain in the amino-terminus of the third intracellular loop of the α2A-adrenergic receptor: Evidence for distinct structural determinants that confer Gs versus Gi coupling. J Biol Chem 270: 24753–24760. [DOI] [PubMed] [Google Scholar]
- 43. Eason MG, Liggett SB (1996) Chimeric mutagenesis of putative G-protein coupling domains of the α2A-adrenergic receptor. J Biol Chem 271: 12826–12832. [DOI] [PubMed] [Google Scholar]
- 44. McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, et al. (2000) Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290: 1574–1577. [DOI] [PubMed] [Google Scholar]
- 45. Hohendanner F, McCulloch AD, Blatter LA, Michailova AP (2014) Calcium and IP3 dynamics in cardiac myocytes: experimental and computational perspectives and approaches. Front Pharmacol 5: 35 10.3389/fphar.2014.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298: 834–836. [DOI] [PubMed] [Google Scholar]
- 47. Liggett SB (2006) Cardiac 7-transmembrane-spanning domain receptor portfolios: diversify, diversify, diversify. J Clin Invest 116: 875–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whalen EJ, Rajagopal S, Lefkowitz RJ (2011) Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends in Molecular Medicine 17: 126–139. 10.1016/j.molmed.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terrillon S, Bouvier M (2004) Roles of G-protein-coupled receptor dimerization. EMBO Rep 5: 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
A) Cell death was determined using the Vybrant assay (Life Technologies), which quantitates the formation of reduced red fluorescent resazurin from a coupled enzymatic reaction in which NADPH is generated from the activity of glucose-6-phosphate dehydrogenase released from dying cells. 40,000 HASM cells/well were treated with buffer or buffer with 50 μM or 1 mM of the indicated TAS2R agonists for 5 min. As a positive control, cells were treated with lysis buffer. Data is from 4–7 experiments performed in triplicate. P>0.05 for all agonists compared to buffer, indicating no significant cell death. B) The proportion of live HASM cells was determined with the LIVE assay (Life Technologies) which measures intracellular esterase activity on calcein-AM which fluoresces green when hydrolyzed. Cells were plated at 40,000/well and treatments were with buffer alone or buffer with 50 μM of the indicted TAS2R agonists for 5 min. No agonist caused a decrease in viable cells. Data is from 4–6 experiments performed in triplicate. P = 0.04 for YOH which was greater than control (buffer).
(TIFF)
40,000 HASM cells/well were exposed to buffer alone (representing “untreated”) or buffer with 50 μM CQ or AA for 5 min. Cells were washed twice with PBS, and then [Ca2+]i mobilization measured in response to 3 μM histamine, 1 μM ET-1, or 1 μM ionomycin. The responses to histamine and ET-1 (as well as ionomycin) were no different in cells pretreated with CQ or AA, compared to pretreatment with buffer alone, indicated a reversal of TAS2R agonist effect. Data is from 4–5 experiments performed in triplicate.
(TIFF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.