

Abstract
Protein phosphatase 6 (PP6) is a ubiquitous Ser/Thr phosphatase involved in an array of cellular processes. To assess the potential of PP6 as a therapeutic target in liver disorders, we attenuated expression of the PP6 catalytic subunit in HepG2 cells using lentiviral-transduced shRNA. Two PP6 knock-down (PP6KD) cell lines (90% reduction of PP6-C protein content) were studied in depth. Both proliferated at a rate similar to control cells. However, flow cytometry indicated G2/M cell cycle arrest that was accounted for by a shift of the cells from a diploid to tetraploid state. PP6KD cells did not show an increase in apoptosis, nor did they exhibit reduced viability in the presence of bleomycin or taxol. Gene expression analysis by microarray showed attenuated anti-inflammatory signaling. Genes associated with DNA replication were downregulated. Mass spectrometry-based phosphoproteomic analysis yielded 80 phosphopeptides representing 56 proteins that were significantly affected by a stable reduction in PP6-C. Proteins involved in DNA replication, DNA damage repair and pre-mRNA splicing were overrepresented among these. PP6KD cells showed intact mTOR signaling. Our studies demonstrated involvement of PP6 in a diverse set of biological pathways and an adaptive response that may limit the effectiveness of targeting PP6 in liver disorders.
Keywords: Protein phosphatase 6, hepatic cells, signal transduction, protein phosphorylation, gene expression, DNA replication, DNA repair, polyploidy
Graphical Abstract
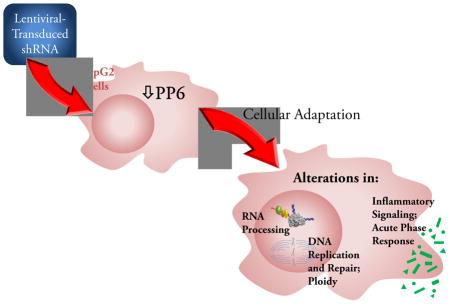
Introduction
The reversible phosphorylation of cellular proteins is controlled by the activities of both protein kinases and protein phosphatases. The protein serine/threonine phosphatases are subdivided into four broad classes: type1 (PP1), type 2A (PP2A), type 2B (PP2B) and type 2C (PP2C). All four classes play an important role in the regulation of a broad range of cellular proteins that include metabolic enzymes, ion channels, hormone receptors, cytostructural proteins and components of kinase cascades [1].
Protein phosphatase 6 (PP6) is classified as a PP2A-type phosphatase based on its sequence homology to PP2A and its sensitivity to protein phosphatase inhibitors such as okadaic acid, microcystin-LR, and calyculin A [2]. The PP6 holoenzyme is proposed to be a heterotrimer consisting of a catalytic subunit (PP6-C), a SAPS (Sit4 Associated Protein Subunit; SAPS1, SAPS2, or SAPS3), and an ankyrin repeat domain containing subunit (ANKRD28, ANKRD44, or ANKRD52) [3]. The SAPS have been shown to bind directly to PP6-C [4]. ANKRD28, the only ANKRD subunit to be studied to any degree, has been shown to bind to all of the SAPS [3]. In general, phosphatase regulatory subunits control enzyme activity, confer substrate specificity, and target the enzyme to specific cellular locations. While this is presumed to be the case for PP6, the precise role of its regulatory subunits is yet to be fully characterized.
PP6-C is the human homologue of the yeast phosphatase, Sit4, with which it shares 61% amino acid identity [4]. Studies in Saccharomyces cerevisiae have implicated Sit4 and its regulatory subunits SAP155, SAP185, SAP190, and SAP4 in G1 to S progression [5]. PP6 has been shown to function similarly in human cancer cells [6, 7]. Other studies in yeast have shown that PP6 contributes to the response to mitochondrial DNA damage [8]. In addition, PP6 in yeast plays a role in signaling by the target of rapamycin (TOR), a key nutrient-sensing kinase [9]. Activation of TOR is associated with the inhibition of Sit4 by its association with regulatory subunits, including TAP42, the mammalian homologue of which is α4, and the SAP proteins [10]. This may account for a mechanism by which TOR can enhance protein phosphorylation through inhibition of a phosphatase. PP6 has been shown to functionally substitute for Sit4 mutations in S. cerevisiae and the Sit4 homolog ppe1 in fission yeast [11]. Deletion of the SAP or Sit4 genes in S. cerevisiae results in increased sensitivity to rapamycin and defects in the expression of certain TOR-regulated genes [10]. It has further been reported that human SAPS, when expressed in S. cerevisiae, are able to physically interact with Sit4, and that the expression of either SAPS2 or SAPS3, but not SAPS1, can rescue the growth defect and rapamycin sensitivity of yeast cells lacking all four yeast SAPs. This effect was dependent on the presence of Sit4 [12].
Evidence for analogous involvement of PP6 in TOR signaling in mammalian cells has been lacking. However, Wengrod et al. [13] recently showed that PP6 is required for the pharmacological induction of autophagy by pharmacologic inhibition of mTORC1, an action that also required GCN2 (general control nonrepressed 2) and eukaryotic initiation factor 2α (eIF2α). As noted above, PP6 has been implicated in the regulation of cell growth and proliferation, which may reflect its involvement in signaling by the mammalian homolog of the Target of Rapamycin, mTOR. As reviewed by Douglas et al. [14], PP6 has an established role in the cell response to DNA damage [15]. This aspect of PP6 function likely reflects its ability to modulate signaling by DNA-dependent protein kinase [16] as well as its interactions with Aurora A kinase [17, 18]. More recent studies suggest a broader role for PP6 in endoplasmic reticulum-to-Golgi trafficking [19], pre-mRNA splicing [20], formation of adherens junctions through interaction with E-cadherin [21], control of apoptosis in immune cells [22], response to mitochondrial DNA damage [8], and modulation of signaling through the Hippo pathway [23]. With regard to its role in liver cells, PP6 has been identified as a tumor suppressor based on its ability to induce G1/S cell cycle arrest in two human hepatocellular carcinoma (HCC) cell lines [7]. Additional data on a hepatic role for PP6 are lacking.
In recent years, while protein kinases have been widely targeted in the development of drug therapies for a broad spectrum of disease entities, the therapeutic targeting of protein phosphatases has only begun to emerge [24]. Given the diversity of functions served by PP6, the present studies were undertaken to explore the role of PP6 in determination of cell phenotype, regulation of gene expression, and involvement in signal transduction pathways in liver cells. Our goal was to explore the potential for PP6 to serve as a therapeutic target in liver disorders. We chose to study HepG2 cells in which PP6-C expression was stably attenuated in an effort to mimic the effects of tonic administration of a specific phosphatase inhibitor.
Material and Methods
Cell Culture
HepG2 cells were obtained from the American Type Culture Collection. Low passage cells were maintained in minimal essential medium with Earle’s salts, L-glutamine and non-essential amino acids (Invitrogen, Carlsbad, CA) supplemented with 1 mM sodium pyruvate, 10% fetal bovine serum, 50 units/ml penicillin and 50 μg/ml streptomycin.
To generate cells lines in which PP6-C was reduced in expression (PP6-C knock-down; PP6KD), HepG2 cells were seeded into 96-well plates and transduced with lentiviral particles (multiplicity of infection of 5) corresponding to different short hairpin RNA (shRNA) constructs targeted to the PP6-C gene (Mission shRNA; Sigma-Aldrich, St. Louis, MO). Procedures followed the manufacturer’s protocol. Cell lines were selected in 2 μg/ml puromycin and surviving colonies of cells were expanded as mass cultures in puromycin. Five individual shRNA lentiviral particle constructs were tested. The Mission lentiviral designations were as follows: Cell line 16.5, TRCN0000279890; 17.5, TRCN0000279949; 18.5 TRCN0000297274; 19.5, TRCN0000379835; 20.5, TRCN0000379918). Except where noted, all studies were carried out on cell lines 18.5 and 19.5. Mission TRC2 non-target shRNA control transduction particles were used for control transduction.
Western immunoblotting of phosphatase subunits and other signaling proteins
Cell lines were plated at 1×106 cells per 10 cm tissue culture plate and allowed to proliferate for 48 hours. Samples were prepared using 0.5 ml per plate of a lysis buffer containing 10 mM Tris-base, pH 7.6, 5 mM EDTA, 50 mM NaCl, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 100 μM sodium orthovanadate, 1% Triton X-100, freshly added protease inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, and 34.4 μg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, and the phosphatase inhibitor microcystin-LR (500 nM). Samples were allowed to incubate on ice for 30 min followed by centrifugation at 16,000g for 10 min at 4°C. Protein concentration of the lysates was measured using the Pierce BCA Protein Assay (Thermo Scientific, Rockford, IL).
Western immunoblotting, image acquisition and quantification of results were carried out as described previously [25]. Primary antibodies were obtained from the following sources: PP6-C, Millipore, Billerica, MA; SAPS1, 4E-BP1 and p-PKCα(Ser657), Santa Cruz Biotechnology, Inc., Santa Cruz, CA; SAPS2 and SAPS3, Bethyl Laboratories, Inc., Montgomery, TX; p-S6(Ser235/236) and p-NDRG1(Thr346), Cell Signaling Technology, Danvers, MA. Immunoblot detection was by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
Cell analyses and imaging
For phalloidin staining, cells were plated on a 6-well μSlideVI, 0.4, tissue culture treated slide (Ibidi LLC, Verona, WI) at 4.5×103 cells per well. After two days in culture, cells were fixed using 4% paraformaldehyde, permeabilized with 0.5% Triton-X100, and stained with rhodamine phalloidin (Cytoskeleton Inc., Denver, CO) according to manufacturer’s protocol. Nuclei were stained with Hoechst 33342 (Life Technologies, Grand Island, NY). Confocal images were acquired with a Nikon C1si confocal microscope (Nikon Inc. Melville, NY) using diode lasers 402 and 561. Wavelengths were collected separately by invoking frame lambda. Serial optical sections were performed with EZ-C1 computer software (Nikon Inc.). Z series sections were collected at 0.5μm with a 20x Plan Apo lens and scan zoom of 2. Deconvolution and projections were performed in Elements version 3.1 (Nikon Inc.) computer software.
Fluorescence in situ suppression hybridization was performed on fixed cells using chromosome enumeration probes (centromeric regions) of chromosomes X, Y and 18 (Abbott Molecular, Des Plaines, IL) following the manufacturer’s suggested protocol. Signals were quantified under a fluorescent microscope using appropriate emission and excitation filters.
Cell migration was assessed with the OrisTM Cell Migration Assay (Platypus Technologies, LLC, Madison, WI) using the Collagen 1 coated 96-well plate. Cells were plated at 5×104 cells/well in 100 μl of culture media containing 2 μg/ml puromycin. After overnight incubation, the stoppers were removed from the experimental wells. After a second overnight incubation, the control stoppers were removed and all wells were washed gently with 100 μl of phosphate buffered saline. Cells were fixed with methanol followed by staining with 4′,6-diamidino-2-phenylindole (DAPI). Images were obtained using a Zeiss Axiovert 200M fluorescent microscope with a Roper CoolSnap CF color camera controlled by Metaporph 6.0 software at 5x magnification. Data were acquired using NIH ImageJ software.
To assess the sensitivity of the three cell lines to taxol and bleomycin, cells were treated with either Paclitaxol (Sigma-Aldrich) dissolved in DMSO at a final concentration of 8 ng/ml, or bleomycin sulfate (Enzo Life Sciences, Farmingdale, NY) dissolved in cell media at a final concentration of 0.3125 μg/ml. DMSO alone was used as a control for the former. At 20, 45, and 69 hours, the cells were stained with the Live/Dead Reduced Biohazard Cell Viability Kit #1, green & red fluorescence (Life Technologies). Images were obtained using a Nikon Eclipse Ti-S Microscope with a Nikon DS-Fi2 camera attached. Images captured with NIS-Elements 4.0 software (Nikon Inc.) were quantified using NIH Image J.
For flow cytometry, cells were plated at 106 cells per 100 mm plate. After 2 days, cells were detached, fixed in ethanol and stained with propidium iodide. Flow cytometry was performed using a Becton Dickinson FACSort flow cytometer (BD Biosciences, San Jose, CA) equipped with an argon ion laser with an excitation wavelength of 488 nm. Analysis was done using ModFitLT software (Verity Software House, Inc., Topsham, ME).
Cell size determination, and DNA, RNA and protein assays
Viable cell counting and determination of their mean diameter was done using the CountessTM automated cell counter (Life Technologies). To determine DNA and RNA content, the MasterPure Complete DNA and RNA Purification Kit (Epicentre Biotechnologies, Madison, WI) was used to prepare samples. A second set of samples was processed for protein using the lysis buffer described above. DNA content was measured using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). rRNA was visualized with ethidium bromide and band intensity quantified using the Bio Doc-iT System and quantified with Labworks 4.5 Image Acquisition and Analysis Software (UVP, Inc., Upland, CA). Protein quantification was accomplished using the Pierce BCA Protein Assay (Thermo Scientific).
RNA isolation, PCR, and microarray hybridization
Total RNA was prepared from triplicate plates of cells using TRIzol reagent (Invitrogen Corporation) following the manufacturer’s directions. RNA was analyzed by RT-PCR for the abundance of PP6-C (5′-GGCTAAATGGCCTGATCGTA-3′, 5′-AAGGAAATATGGCGTTGTCG-3′), SAPS1 (5′-AAGGCTACATGGGTCACCTG-3′, 5′-TTCGTCCTCACTGTCTGTGC-3′), SAPS2 (5′-CCACACAGTCCTGCGACTCC-3′, 5′-TTCTTGGACCATGAGCCGCC-3′), and SAPS3 (5′-ACACGACGACCAACATTTGA-3′, 5′-TCATTCATTTCCCAGGCTTC-3′) mRNA. RT-PCR for 18S rRNA (Applied Biosystems, Foster City, CA) was used as a control. PCR primers were designed using the MIT Primers3 design program (http://frodo.wi.mit.edu/primer3/) and human sequence data.
Gene expression analysis using the Affymetrix GeneChip Human Gene 1.0 ST Array (Affymetrix, Santa Clara, CA) was carried out at the Brown University Center for Genomics and Proteomics Core Facility. Sample quality control was done using Affymetrix Expression Console and the differential gene expression analysis was performed using the Partek Genomic Suite v 6.5 (Partek Inc., St. Louis, MO). Heat map generation and hierarchical clustering were performed using the Gene Pattern analysis platform [26]. We performed further analyses using the Ingenuity Pathway Analysis (IPA®) platform (Ingenuity Systems, www.ingenuity.com), A control dataset was generated and analyzed as previously described [27] to determine the significance of IPA categories. Gene Set Enrichment Analysis (GSEA) utilized the Molecular Signature Database (MSigDB) [28].
Microarray data have been deposited in GEO (accession number GSE67891; reviewer link http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wpkriicifpanpsv&acc=GSE67891).
Phosphoproteomic analyses
Control and PP6KO cell lines were plated at 106 cells per 100 mm plate and maintained under the culture conditions described above for 72 hours. At the end of this incubation period, the cells were washed twice with 10 ml ice cold PBS. Cells were scraped into 0.5 ml of PBS, pooling two plates per sample. The cells were collected by centrifugation at 100g for 5 min. The PBS was removed and the cell pellets were frozen at −70°C until use.
Cell pellets were sonicated in 1.2 ml of a lysis buffer containing 50 mM Tris, pH 8.1, 75 mM NaCl, 6 M Urea, 10 mM sodium pyrophosphate, 1 mM NaF, 1 mM β-glycerophosphate, and 1 mM sodium orthovanadate. Lysates were centrifuged at 14,300xg for 15 min. Equal amounts of protein, as determined with a Bradford protein assay, were reduced, alkylated and trypsinized overnight (ratio of 1:100 of trypsin:protein by weight) at 37°C. The resulting samples were desalted using Oasis HLB 6 ml cartridges (Waters, Milford, MA). From these preparations, phosphopeptides were enriched using Titansphere Phos-TiO reagents (GL Sciences, Torrance, CA) as described previously [29].
The resulting samples were analyzed through the Rhode Island Hospital/Lifespan proteomics core facility using a Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA). Using the Mascot algorithm version 2.4 from Matrix Science [30], MS/MS spectra were searched against the non-redundant human UniProt complete proteome set database containing 72,078 forward (Uniprot database released 2013.2.01) and an equal number of reversed decoy protein entries. Peak lists were generated with msconvert from ProteoWizard (3.0.4888) using default parameters with the MS2Deisotope filter. The Mascot database search was performed with the following parameters: trypsin enzyme cleavage specificity, 2 possible missed cleavages, 10 ppm mass tolerance for precursor ions, 50 mmu mass tolerance for fragment ions. Search parameters specified a differential modification of phosphorylation (+79.9663 Da) on serine, threonine, and tyrosine residues, a dynamic modification of methionine oxidation (+15.9949 Da), and a static modification of carbamidomethylation (+57.0215 Da) on cysteine. The resulting unique peptide assignments were filtered down to 1% false discovery rate (FDR) by a logistic spectral score filter [31]. FDR was estimated with the decoy database approach after final assembly of non-redundant data into heatmaps [32]. To validate the position of the phosphorylation sites, the Ascore algorithm [33] was applied to all data. The reported phosphorylation site position reflected the top Ascore prediction.
Label-free comparison of phosphopeptide abundance was accomplished as described previously [34], as was retention time alignment of individual replicate analyses [29]. Peak areas were calculated by inspection of select ion chromatograms (SICs) using in-house software programmed in Microsoft Visual Basic 6.0 based on Xcalibur Development kit 2.1 (Thermo Fisher Scientific). This approach used the ICIS algorithm available in the Xcalibur XDK with the following parameters: multiple resolutions of 8, noise tolerance of 0.1, noise window of 40, scans in baseline of 5, and inclusion of refexc peaks parameter value, which is false. SIC peak areas were determined for every phosphopeptide that was identified by MS/MS. In the case of a missing MS/MS for a particular peptide, in a particular replicate, peak areas were calculated according to the peptide’s isolated mass and the retention time calculated from retention time alignment. A minimum SIC peak area equivalent to the typical spectral noise level of 300 was required of all data reported for label-free quantitation.
A label-free data heatmap was generated for comparison of phosphopeptides [35]. The coefficient of variation was calculated for each heatmap square. Label-free P-values were calculated from the replicate data for each cell line compared to the cell line with the minimum average peak area for that phosphopeptide. Q values for multiple hypothesis tests were also calculated for each time point based on the determined P-values using the QVALUE package in R [36].
Additional statistical analyses
All data are provided as mean and standard deviation. Multiple comparisons were made using ANOVA with a Tukey post hoc test.
Results
The phenotype of hepatic cells with reduced PP6-C expression
For the present studies, we utilized HepG2 cells, a well-characterized liver HCC cell line that is not tumorigenic and retains differentiated liver cell functions [37, 38]. Using lentiviral-mediated introduction of shRNA constructs targeting the PP6-C gene, we generated and analyzed five HepG2 clones. Relative to control (non-target) shRNA lentiviral-transduced HepG2 cells, we achieved stable reductions of PP6-C at the protein level ranging from less than 20% to approximately 90% in the five resulting cell lines (Fig. 1A). Two PP6KD cell lines, designated 18.5 and 19.5, showed a 90% reduction in PP6-C protein content. These two lines were chosen for further study. PP6-C mRNA levels, as determined by RT-PCR (Fig. 1B and 1C) were similarly reduced in these two cell lines by about 90%.
Figure 1. PP6-C, SAPS1, SAPS2, and SAPS3 mRNA and protein content in control and PP6KD cells.

A. Cell lysates were prepared from control transduced cells and five cell lines that were transduced with shRNA targeting PP6-C. Western immunoblotting performed on triplicate samples was used to calculate the mean and standard deviation for the reduction in PP6-C protein content. B. Triplicate total RNA samples from control transduced and the 18.5 and 19.5 PP6KD HepG2 cell lines were analyzed by RT-PCR for PP6-C, the three SAPS and 18s ribosomal RNA. The resulting ethidium bromide-stained gels are shown. C. The results for PP6-C and 18s were quantified by densitometry and expressed as the ratio of PP6-C to 18s (mean and standard deviation of the triplicate analyses). D. Triplicate lysates from control transduced and the 18.5 and 19.5 PP6KD cell lines were analyzed by immunoblotting for PP6-C and the three SAPS. E. The gels shown in Panel D were analyzed by densitometry. Results were normalized to the mean intensity for the triplicate controls and expressed as the mean and standard deviation. Black bars, control cells; white bars, 18.5; gray bars, 19.5.
mRNA expression of SAPS1, SAPS2 and SAPS3 in the 18.5 and 19.5 cell lines was unchanged relative to levels seen in control cells (Fig. 1B). However, SAPS1 and SAPS2 showed a decrease at the protein level of about 50% (Fig. 1D and 1E). SAPS3 protein content was similarly reduced in the 19.5 cell line but only marginally reduced in the 18.5 cell line. In preliminary experiments, we found that Western immunoblotting with available antibodies to determine the level of ANKRD proteins did not yield interpretable results (data not shown). However, ANKRD expression, as determined through microarray analyses, (supplemental Table 1) was similar in both PP6KD cell lines to the control cells. As was the case for PP6-C and all three SAPS, ANKRD 28 and ANKRD52 expression levels were in the top quartile for expression level in all three cell lines. In contrast, ANKRD44 expression was in the lowest quartile in all three cell lines.
The appearance of the 18.5 and 19.5 cells was similar to control-transduced cells (phase contrast microscopy; data not shown). On examination of dual staining using phalloidin and Hoechst 33342 (Fig. 2A), both the 18.5 and 19.5 PP6KD cells appeared larger than the control cells relative to the number of nuclei visible in a given field or cluster of cells. In fact, a direct measure of cell size (Fig. 2B) showed that both of the PP6KD cells were slightly larger than the control cells. To pursue this observation, we measured RNA, protein and DNA content. Results (Fig. 2C) showed a 2-fold lower ratio of both RNA:DNA and protein:DNA in the two PP6KD cell lines. There were no differences in the protein:RNA ratio (Fig. 2C).
Figure 2. Phenotypic characterization of control and PP6KD cells.

A. Control and PP6KD cells were stained with phalloidin (red) and Hoechst 33342 (blue). Representative images were acquired at 20x magnification. B. Cell diameter was determined for the control and PP6KD cell lines using the CountessTM automated cell counter. Results are shown as the mean and standard deviation of triplicate analyses * P < 0.01 by ANOVA and Tukey post-hoc test. C. Protein, DNA and RNA were extracted from HepG2 control transduced (black bars) and PP6KD cells (18.5, white bars; 19.5, grey bars). The graphs show RNA and protein content relative to DNA (left) and the protein:RNA ratio (right). Data are expressed as mean and standard deviation for triplicate analyses. * P < 0.02 by ANOVA and Tukey post-hoc test.
Reasoning that these results were consistent with cell cycle arrest in G2/M, we characterized the cell cycle kinetics of the two PP6KD cell lines and the control-transduced cell line. Although all three cell lines grew at similar rates (Fig. 3A), cell cycle analysis by flow cytometry showed marked differences (Fig. 3B and 3C). There was no evidence for an increase in apoptotic cells (sub-G1 peak) in either the 18.5 and 19.5 cell lines. However, both showed an accumulation of cells in a peak consistent with diploid G2/M. The diploid G1 peak was markedly reduced. There was also a shift in fluorescence consistent with the presence of a tetraploid G2/M peak. In order to assess this directly, we performed fluorescent in situ suppression hybridization for the X and Y chromosomes and for chromosome 18. We included all of the PP6KD cell lines in our analysis. Results (Fig. 3D) indicated that both the 18.5 and 19.5 cell lines were tetraploid. The 16.5 and 18.5 cell lines showed an intermediate degree of tetraploidy, coincident with an intermediate level of reduction in PP6-C. The 17.5 cell line was an outlier in that results on this cell line were consistent with a high degree of tetraploidy, similar to the 18.5 cell line, even though the former seemed to exhibit only a slight reduction in PP6-C content.
Figure 3. Proliferation and cell cycle kinetics of control and PP6KD cells.

A. Growth rate was measured for the control and two PP6KD cell lines. Data are given as mean±standard deviation of 6 replicate determinations. B. The percent of cells in each of three cell cycle phases, G0/G1, S and G2/M, was determined using flow cytometry. Triplicate analyses were performed using control cells (black bars) and the two PP6KD cell lines (18.5, white bars; 19.5, gray bars). Data are shown as the mean plus standard deviation. C. Representative flow cytometry profiles are shown for each of the three cell lines. D. Fluorescent in situ hybridization was used to determine the copy number in each individual cell for the X and Y chromosomes and chromosome 18. Data are shown for the control cells and the five PP6KD cell lines. The percent reduction in PP6-C, taken from the experiment in Figure 1, is given next to the cell line identification in each graph. The numbers below the bars represent the mean number of copies of each chromosome in the particular cell line.
Gene expression in the PP6KD cells
Total RNA preparations from control-transduced and PP6KD cell lines were analyzed by microarray (supplemental Table 1). Hierarchical clustering using the 182 genes that were significantly different between any two cell lines (FDR<0.05 with a fold-difference of at least 1.25) showed clustering of the 18.5 and 19.5 cells (Fig. 4A and supplemental Table 2A).
Figure 4. Gene expression in control and PP6KD cells.

RNA prepared from triplicate control transduced and PP6KD cells was analyzed by microarray. A. Results are shown in the form of a heat map for all genes that differed significantly (FDR<0.05, and fold change ≥ 1.25) between any two of the three cell lines studied. Red and blue reflect high and low gene expression, respectively. A dendrogram of the cluster analysis is shown above the heat map. B. Ninety-two genes that were differentially expressed in both PP6KD cell lines (FDR<0.05; direction of change concordant in 18.5 and 19.5) were subjected to Ingenuity Pathways Analysis (IPA). The graph shows the P-values obtained for specific IPA results in four categories: purple, canonical pathways; blue, upstream regulators; red, diseases and functions; orange, toxicological functions. In all cases, the P-values for the categories shown were less than the lowest P-value obtained in five control analyses.
For IPA, results were further filtered to include only those significant genes for which the PP6KD:Control ratios for both PP6KD cell lines were >1.25-fold (supplemental Table 2B). The resulting 92 genes yielded a range of gene ontology categories (Fig. 4C and supplemental Tables 2C through 2F). We considered IPA categories to be significant only if their P-value was less than the lowest P-value obtained for five control analyses (supplemental Table 2G). Based on this criterion, one canonical pathway category, acute phase response signaling, was highly significant at P = 4.27×10−8. Results of the analysis for upstream regulators yielded a corresponding result. Among the most highly significant regulators were interleukin 1B (IL1B), interleukin 6 (IL6) and tumor necrosis factor (TNF). The genes that contributed to the identification of these regulators (supplemental Table 2H) all showed increased expression in the two PP6KD cell lines with the exception of one, glutathione S-transferase α1 (GSTA1). Other upregulated genes included ceruloplasmin, fibrinogen β chain, plasminogen and three serpin peptidase inhibitors. A number of “Diseases and Function” and “Toxicological Function” categories were also significant (Fig. 4B). Identification of these categories was largely dependent on the same genes that accounted for the acute phase response and upstream regulators (supplemental Table 2H). We interpreted the IPA results as indicating that PP6KD cells exhibit an increase in acute phase reactants through an upregulation of cytokine signaling.
GSEA (supplemental Table 3A through 3I) yielded numerous results that were borderline significant and/or were inconsistent between the two PP6KD cell lines. However, two results were highly significant in both cell lines. The KEGG gene set entitled “Complement and Coagulation Cascades” was enriched in the PP6KD cells (Fig. 5A). An examination of the genes that contributed to this result (supplemental Table 3J) revealed overlap with the acute phase reactants identified by IPA as well as a spectrum of other acute phase reactant genes. The KEGG “DNA Replication” gene set was enriched in the control cells relative to PP6KD cells. The genes accounting for this finding (supplemental Table 3K) were largely accounted for by DNA polymerases and replication factors.
Figure 5. Gene set enrichment analysis of gene expression in the control and PP6KD cell lines.
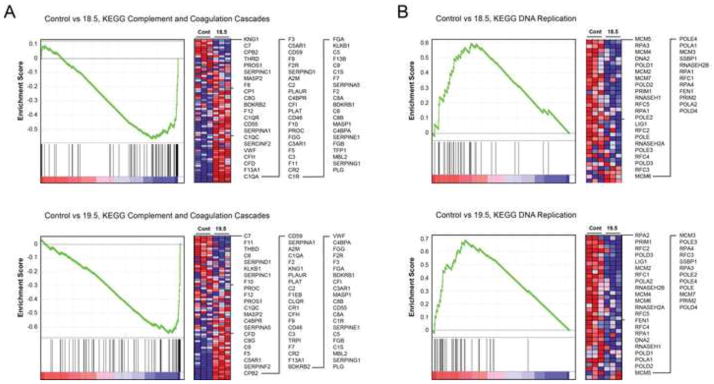
Results are shown for two gene sets, Complement and Coagulation Cascades (A) and DNA replication (B). The graphs show the hierarchical ordering of enrichment scores. The intensity of the genes accounting for these data is depicted in the heat maps. Red and blue reflect high and low gene expression, respectively. Individual genes corresponding to rows in the heat map are shown to the right in each panel.
Our gene expression results did not indicate an effect on the gene expression categories that we have found previously to be associated with reduced mTORC1 [39, 40] or mTORC2 signaling [27] in hepatic cells and liver. Among these unrepresented categories were metabolic enzymes, translation factors, ribosomal proteins or proteosome constituents.
The effect of reduced PP6-C expression on the phosphoproteome
We performed mass spectrometry-based phosphoproteomic analysis with the aim of identifying protein phosphorylation sites that were sensitive to a reduction in PP6 catalytic subunit content. Unlabeled quantitation was performed on four replicate samples taken from the control and PP6KD cells. Following the generation of raw MS phosphopeptide identifications, high quality sequence assignments were made using the stringent criteria defined above. A complete list of phosphopeptides with MOWSE score >20 and mass error <2 ppm, including reversed database hits, is provided in supporting information (supplemental Table 4). Complete sequence and quantitation data, identifications derived from either MS/MS or accurate mass/aligned retention time, and results of statistical analyses are also provided (supplemental Table 5A). Results showed that we identified 6,894 unique phosphopeptides representing 1,568 unique proteins. Of these, 6,614 phosphopeptides on 1,538 proteins were quantified in 12 of 12 samples. An additional 252 unique phosphopeptides were quantified in 11 of the 12 samples.
Data analysis focused on the ratios of phosphopeptide abundance, 18.5/Control and 19.5/Control. The analysis used the mean ratios for both PP6KD cell lines. A comparison of the two (Fig. 6A, supplemental Table 5B and 5C) showed a large number of phosphosites that were minimally affected by a reduction in PP6-C content. Among all of the phosphopeptides that were quantified, the ratios for 819 were significant based on a FDR Q-value <0.05. However, only 231 of these sites showed an effect of PP6-C expression that was concordant for the two clones (increased or decreased phosphopeptide abundance in both PP6KD cell lines relative to control). We considered the changes in the remaining 588 phosphopeptides as representing off-target effects. Of the 231 concordant phosphopeptides, 134 were increased in abundance in the PP6KD cells. The remaining 97 were decreased in abundance.
Figure 6. Results of phosphoproteomic analyses.
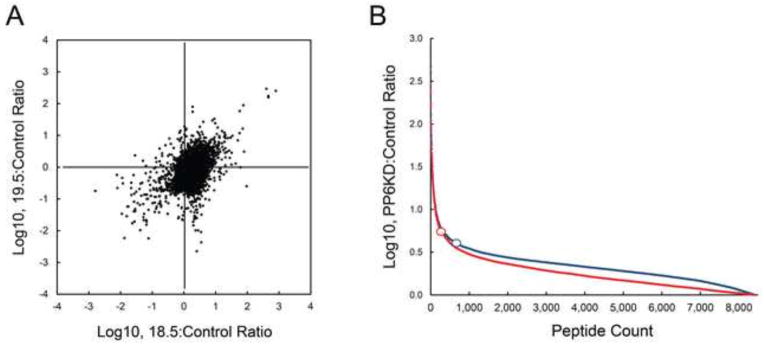
Mean abundance data for each phosphopeptide was used to generate a ratio of PP6KD to control. A. The relationship between the ratios for the two PP6KD cell lines is shown as a scattergram. B. The same data are shown for the two cell lines (18.5, blue; 19.5, red) as a hierarchical ordering from highest to lowest. The open circles represent the inflection point for each curve.
Based on the distribution of the PP6KD:Control ratios for all phosphopeptides, an inflection point for this distribution [29] was calculated for each cell line (Fig. 6B, supplemental Table 5D). Using this statistic, we generated a list of significant phosphopeptides based on three criteria: a mean PP6KD:Control ratio beyond the inflection point for each particular cell line, concordance in both cell lines for the direction of change, and a Q-value <0.05. The result of this highly stringent analysis (supplemental Table 5E) included 88 quantified phosphopeptides of which 80 were non-redundant. The 80 unique phosphopeptides were on 56 unique proteins. Of these 80 phosphopeptides, 27 were increased in abundance in the PP6KD cells while the remaining 53 were decreased in abundance.
We examined all of the significantly altered phosphosites using two annotated resources, PhosphoSitePlus [41] and the Human Protein Reference Database (HPRD) [42]. Among all of the significant phosphosites, only one had a well-defined regulatory role. The Ser729 site on B-Raf has been reported to be a substrate of AMP-activated protein kinase (AMPK) that, when phosphorylated attenuates signaling through the ERK pathway [43]. This site was downregulated in the PP6KD cells.
Although we did not identify specific phosphosites that could account for the phenotype of the PP6KD cells, we identified phosphorylation changes in a number of proteins with defined functions that related to the cells’ phenotype. Three of the 56 significant proteins have been shown to be involved in DNA replication and repair. Chromobox Protein Homolog 3 (CBX3) is a DNA binding protein and heterochromatin component that has been shown to be recruited to sites of DNA damage and double-strand breaks. Chromodomain helicase DNA binding protein 2 (CHD2) is also involved in modification of chromatin structure. Replication factor C1 (RFC1) a large subunit of replication factor C, is a DNA-dependent ATPase. Matrin 3 (MATR3), an RNA-binding nuclear matrix protein, has also been implicated in the DNA damage response [44]. The multiple phosphorylation sites we identified in this protein, all of which were upregulated in the PP6KD cells, have not been assigned functional significance. In fact, the parallel upregulation of multiple sites raises the possibility of a change in the abundance of the protein rather than a change in the stoichiometry of phosphorylation. In contrast, the multiple sites in nucleolin were not all concordant for the direction of change in the PP6KD cell lines; one multiply phosphorylated peptide that contained Ser28, Ser34, Ser41 and Ser42, was upregulated in the PP6KD cells. Ser67 and Ser69 were downregulated. Nucleolin has also been implicated in the DNA damage response and in the Aurora-B kinase signaling network [45].
In addition to MATR3, 13 other significant proteins had documented or inferred RNA binding activity. Several (TRA2B, DDX46, SRRM1, SFRS2, SRRM2, BUD13) have purported roles in mRNA splicing. This number was disproportionate relative to the number of mRNA binding proteins involved in mRNA splicing in the proteome.
Our method for data analysis incorporates information regarding gene ontology (GO). We performed a comparison of the GO categories using a set of 135 non-redundant proteins for which the mean PP6KD:Control ratios were significant (FDR<0.05) and altered in a concordant direction in both PP6KD controls (no filtering for fold-difference). A control set of 1,527 non-redundant proteins was generated based on PP6KD:Control ratios that were not significant and/or non-concordant. The results of this analysis yielded several significant results based on the Fischer’s Exact Test and a correction for multiple comparisons. Only the GO categories that were represented at least three times among the test sample were included.
Using this approach, three GO Location categories, membrane, microtubule and nucleus, were significant (P-values of 0.008, 0.001 and 0.003, respectively). Two GO Biological Processes categories (cortical actin cytoskeleton organization, P = 0.014; ubiquitin-dependent protein catabolism, P = 0.022) were significantly over-represented in the PP6KD cells.
Further phenotypic characterization of the PP6KD cell lines
The analyses described above did not address several characteristics of the PP6KD cell lines that warranted investigation based on published data. Given our results showing accumulation of PP6KD cells in diploid G2/M, published data on the role of PP6 in DNA repair, and our own gene expression data, we performed experiments aimed at directly assessing the sensitivity of the PP6KD cell lines to two pharmacologic agents, bleomycin and taxol. In a preliminary experiment, we identified concentrations of these two agents and durations of exposure that resulted in subtle loss of viability. However, neither PP6KD cell line showed heightened sensitivity to either agent under the conditions defined by the preliminary experiment (data not shown).
Based on published data demonstrating a relationship between PP6 and E-cadherin [21], and the known role of E-cadherin in cell migration [46, 47], we performed cell migration studies. Results (Fig. 7A) showed a modest reduction in migration of the 18.5 cells that was not replicated in the 19.5 cells. We interpreted these results as indicating that the change in the 18.5 cell line represented an off-target effect and that altered cell migration was not associated with a stable reduction in PP6-C content.
Figure 7. Further characterization of the PP6KD cell lines.

A. Cell migration was measured as the number of cells that moved to the void area of a migration chamber over a period of 48 hr. Control measurements (white bars) and experimental results (black bars) are shown for each of the three cell lines as the mean plus standard deviation for triplicate determinations. B. Control and PP6KD cells, as indicated at the top of the panel, were exposed to 100 nM rapamycin for 1 hr prior to preparation of cell lysates, 100 nM insulin for 30 min, neither, or both. Samples were prepared in triplicate and analyzed by Western immunoblotting. One set of samples that is representative is shown. The α, β and γ designations to the right of the 4EBP1 blot represent the positions of the hypo-, intermediate and hyperphosphorylated forms of the protein, respectively.
Our phosphoproteomic results did not indicate a relationship between PP6 and mTOR signaling, an association that has been inferred from yeast studies [9, 10]. To further assess the effect of reduced PP6-C content on signaling by mTOR Complex 1 (mTORC1), we performed immunoblotting for the regulator of eIF4E, 4EBP1, and for the phosphorylation state of ribosomal protein S6. We similarly examined mTORC2 signaling in the PP6KD cells through immunoblotting for phospho-PKCα (Ser657) and phospho-NDRG (Thr346). Results (Fig. 7) did not indicate altered basal or insulin-activated mTOR signaling in PP6KD cells, nor was there an apparent effect on the potency with which rapamycin inhibited the phosphorylation of mTORC1 targets.
Discussion
Relative to protein kinases, the assignment of function to Ser/Thr protein phosphatases has proved challenging [1]. While phosphatase catalytic subunits can be assigned to several superfamilies, regulation and specificity of phosphatase holoenzymes is dependent on a large, diverse array of regulatory subunits. Therefore, it is not surprising that phosphatase inhibitors, such as okadaic acid [48], have low specificity, even crossing superfamilies to inhibit a broad range of phosphatases. It is the Ser/Thr phosphatase regulatory subunits that are responsible for conferring the specificity and regulation of the hundreds of holoenzymes in which they participate.
PP6 is a case in point. Although the yeast homolog of this phosphatase was identified twenty-five years ago [49], new information continues to emerge on its biological role. Studies in yeast have long pointed to involvement of PP6 in cell cycle regulation [50]. In human cancer cells, PP6 is reported as being involved in G1-to-S transition [6]. More recently, it was demonstrated that PP6-C is a target of microRNA-373, which is overexpressed in HCC [7], and that microRNA-373 has oncogenic activity dependent on regulation of PP6-C. These observations were consistent with the designation of PP6 as a tumor suppressor. However, PP6 has also been identified as oncogenic in human mesothelioma cells [51], making the contribution of this phosphatase to cell transformation and cancer pathogenesis uncertain. In other studies, PP6 was identified as a target that could serve to sensitize cells to radiation [52], an effect that involved the SAPS1-dependent association of PP6-C with DNA-dependent protein kinase (DNA-PK) [53].
Given our longstanding interest in mTOR signaling and cell cycle control in liver development, carcinogenesis and response to injury [40, 54, 55], we undertook a broad assessment of the role of PP6 in hepatic cells. In contrast to studies in which PP6 was acutely downregulated or overexpressed, we took a somewhat less conventional approach. We generated two cell lines in which PP6-C content was stably reduced by about 90%, a condition similar to that which would pertain if PP6 were a targeted therapy. To broadly characterize the phenotype of these cells, we performed genomic and proteomic profiling in addition to more conventional characterization of the cells.
Among our observations, the most unexpected may be the stable transition of PP6KD HepG2 cells from diploid to tetraploid. This occurred to varying degrees with all degrees of reduction in PP6-C. The induction of tetraploidy occurred without a change in proliferation rate relative to the diploid, control-transduced HepG2 cells. This phenotype is reminiscent of normal, mature hepatocytes, which retain an ability to proliferate in the polyploid state [56, 57]. In general, polyploidization can occur through cell fusion or abnormal cell division, the latter involving endoreplication, mitotic slippage or failure of cytokinesis [56]. There was no indication in our data that mechanisms involved in cell fusion resulted from reduced PP6-C expression. However, our gene expression profiling was consistent with the emergence of tetraploid cells through endoreplication. This mechanism, in which G and S phase occur without mitosis, may result from downregulation of mitotic regulators and mediators [58], and it is consistent with the reduced expression of genes involved in DNA replication that we observed in both PP6KD cell lines. Among these were members of the minichromosome maintenance (MCM) complex, and the epsilon and delta DNA polymerases.
The concurrence of tetraploidy and a normal rate of proliferation may also relate to changes in the phosphorylation state of a number of proteins involved in DNA replication and the DNA damage response that we observed. This conclusion is consistent with published data indicating involvement of PP6 in homology directed repair of DNA double strand breaks [59] and dephosphorylation of the mitotic kinase, Aurora A [17]. Our results are also consistent with the recently assigned role of PP6 in regulating DNA-PK, the depletion of which results in mitotic defects [60].
The results of our genomic analyses indicated that PP6 functions to suppress pathways downstream from inflammatory mediators. This conclusion, based on a large number of small changes in gene expression, was supported by two distinct analytical approaches, the identification of pathways and upstream regulators using IPA, and Gene Set Enrichment Analysis. The increased expression of acute phase reactants and inflammation-associated genes with stable reduction of PP6-C content is consistent with a recently proposed mechanism [61, 62] in which deactivation of TAK1 by PP6 prevents prolonged inflammatory response. The synthesis of acute phase proteins by liver parenchymal cells represents a highly regulated part of a more generalized systemic response to cytokine release that is reflected in an altered serum proteome [63]. Acute phase signaling in liver is a response to systemic pathogens (e.g., sepsis), injury, and many forms of physiologic stress.
GSEA results were also indicative of impaired DNA replication in the PP6KD cells. We interpreted this result as consistent with the mitotic defect described above. Examination of our phosphoproteomic results supported a role for PP6 in DNA replication and the DNA damage response.
Our phosphoproteomic analyses used stringent criteria for reproducibility and significance of effects. Using these criteria, fewer than a hundred phosphorylation sites were identified. Of these sites, the reduced phosphorylation of B-Raf at Ser729 may be relevant to the previously observed cooperativity between PP6 and B-Raf in skin carcinogenesis [64]. Among the other phosphosites that were altered in the PP6KD cells, there was consistency in the categories of proteins that showed significant changes in phosphorylation. Of note, more than two thirds of the significantly altered sites were decreased in abundance in the PP6KD cells, not increased. We speculate that this relates to an adaptive response of the cells to the persistent reduction in PP6-C content, one that likely involves modulation of kinase signaling. We choose to be conservative in the interpretation of the phosphoproteomic data with regard to the identification of direct PP6 targets. While the phosphosites that were upregulated in the PP6KD cells are candidates, we consider it likely that they also represent changes that are part of an adaptive response to the stable reduction in PP6-C content.
In addition to DNA replication and repair, and inflammatory cytokine signaling, our results also support another functional role for PP6 that has been previously described, pre-mRNA splicing [20]. Although PP6 has an established role in TOR signaling in yeast [10], data confirming an analogous role in mammals has been scant. A recent report has linked PP6 to mTORC1 in the regulation of autophagy [13]. However, like our own results, these authors did not show that PP6 is directly responsible for the dephosphorylation of mTOR targets. Considering our data in the aggregate, we found no evidence for involvement of PP6 in mTOR signaling, nor was there any evidence that normal levels of PP6-C are required for cell survival, as has been previously suggested [65]. However, there is a caveat in the interpretation of these results; we achieved a marked reduction of PP6-C, but not a complete elimination of the phosphatase. With regard to mTOR signaling and the other observations we have made, it is certainly possible that the small amount of remaining PP6-C was sufficient to retain PP6 function.
In a 2009 review of the therapeutic targeting of protein phosphatases [24], PP6 was not mentioned. The same is true for a more recent review of the regulatory roles of phosphatases in cancer [66]. The present report represents the first broad characterization of the role of PP6 in hepatic cells aimed at assessing its potential as a therapeutic target. A novel aspect of our studies is examination of the effects of a stable reduction in PP6-C content, a condition relevant to the therapeutic targeting of PP6 through inhibition of its activity or disruption of PP6 holoenzymes. Most unexpected was the observation that a stable reduction in PP6-C content is associated with a change in ploidy. This is particularly striking given the absence of an effect on rate of proliferation or survival. On the other hand, a number of our results are consistent with previously assigned roles for PP6. The subtle nature of the changes in gene expression and effect on the phosphoproteome suggest an adaptive response that abrogated phenotypic changes. Our findings illustrate the challenge of targeting PP6 given the extent to which it is promiscuous in its substrate interactions and functional roles. More specifically, we found no evidence that the PP6 catalytic subunit functions as an oncogenic protein in hepatic cells. Rather, it is involved in the modulation of a number of normal hepatic functions. It appears that detailed characterization of specific PP6 holoenzymes will be a necessary next step in targeting PP6 for the purpose of modifying liver pathophysiology.
Supplementary Material
Table S1A contains the full data set derived from microarray analysis. Triplicate RNA samples were prepared from control transduced cells and the two PP6KD cell lines, and analyzed as described in Materials and Methods. Table S1B contains the microarray data filtered to remove any probes that are not annotated. Probes for which all individual results were below the detection threshold of 50 were also removed. The mean control intensity was calculated, and results were ordered based on average intensity. Table S1C contains microarray data for PP6 catalytic subunits and regulatory components. The insert to the right shows the position of PP6 components in the hierarchy of control intensities.
Table S2A shows microarray results filtered for genes that were significant at FDR<0.05 for a fold-difference from the control group of >1.25. Ratios are shown for clones 18.5 and 19.5 as indicated in the first column. Table S2B contains the IPA input generated from the genes shown in Supplemental Table 1A. The expression ratios (PP6KD:Control) represent the mean of the ratios for clones 18.5 and 19.5. Tables S2C through S2F contain IPA results for individual gene ontology categories. Highly significant results are shown in dark blue; borderline significant results are shown in light blue. Level of significance was determined as the lower end of the range of P-values for 5 sets of control genes, which are shown in Table S1G. Control P-values were generated using randomly selected genes for which the fold-difference between PP6KD clones and control cells was between 0.95 and 1.05. Five sets of 93 randomly selected genes were used to generate these data. Table S2H shows the genes that contributed to the identification of selected, significant IPA categories. Red cells represent genes that were upregulated in the PP6KD cells; green represents downregulated genes.
Tables S3A through S3I show GSEA results for the major collections within the Molecular Signatures Database (MSigDB). Colored cells indicate genes that are common to both the 18.5 and 19.5 cell lines. Tables S3J and S3K show the gene enrichment data for KEGG “complement and coagulation cascade” and “DNA replication” categories, respectively.
Each tab represents the data for a biological replicate generated from the mock-transduced control cells, the 18.5 cell line, or the 19.5 cell line.
Table S5A contains the information used for data interpretation for all quantified phosphopeptides. Table S5B shows the correlation of results in the two PP6KD cell lines. Table S5C provides the distribution of ratios (PP6KD-to-control) for all peptides detected and quantified in both cell lines. These data were used to determine the inflection point, the calculation of which is shown in Table S5D. Table S5E contains data for the phosphopeptides for which the difference in abundance between the PP6KD cell lines and the control cell line was significant (FDR<0.05), beyond the inflection point, and concordant with regard to direction of change in the two PP6KD cell lines. Table S5F contains data for control phosphopeptides, which were identified based on a non-significant FDR and fold-change less than the inflection point. Table S5G contains data, including gene ontology categories for phosphopeptides deemed significant (FDR<0.05, direction of change concordant in both cell lines). Table S5H shows the results of the gene ontology analysis. Numbers under “Cont” and “KD” represent the number of phosphopeptides in each category for the control or PP6KD cell lines. The “Cont%” and “KD%” columns give the percentage of the total number of phosphopeptides in Tables S5F and S5G that are represented in the previous two columns in Table S5H, thus indicating the proportion of phosphopeptides representing each gene ontology category. These data were analyzed using a Fisher’s exact test to calculate the P value for each category.
Highlights.
Lentiviral-transduced shRNA was used to generate a stable knockdown of PP6 in HepG2 cells.
Cells adapted to reduced PP6; cell proliferation was unaffected, and cell survival was normal.
However, PP6 knockdown was associated with a transition to a tetraploid state.
Genomic profiling showed downregulated anti-inflammatory signaling and DNA replication.
Phosphoproteomic profiling showed changes in proteins associated with DNA replication and repair.
Acknowledgments
We thank Drs. Anatoli Zhitkovich, and Jennifer Sanders for their helpful discussions. We also appreciate Dr. Sanders’ careful reading of our manuscript. This work was supported by R01HD024455, and by P30GM103410, which provides support to the Brown University Genomics Core Facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brautigan DL. Protein Ser/Thr phosphatases--the ugly ducklings of cell signalling. FEBS J. 2013;280:324–345. doi: 10.1111/j.1742-4658.2012.08609.x. [DOI] [PubMed] [Google Scholar]
- 2.Prickett TD, Brautigan DL. The alpha4 regulatory subunit exerts opposing allosteric effects on protein phosphatases PP6 and PP2A. J Biol Chem. 2006;281:30503–30511. doi: 10.1074/jbc.M601054200. [DOI] [PubMed] [Google Scholar]
- 3.Stefansson B, Ohama T, Daugherty AE, Brautigan DL. Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry. 2008;47:1442–1451. doi: 10.1021/bi7022877. [DOI] [PubMed] [Google Scholar]
- 4.Stefansson B, Brautigan DL. Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IkappaBepsilon. J Biol Chem. 2006;281:22624–22634. doi: 10.1074/jbc.M601772200. [DOI] [PubMed] [Google Scholar]
- 5.Luke MM, et al. The SAP, a new family of proteins, associate and function positively with the SIT4 phosphatase. Mol Cell Biol. 1996;16:2744–2755. doi: 10.1128/mcb.16.6.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefansson B, Brautigan DL. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle. 2007;6:1386–1392. doi: 10.4161/cc.6.11.4276. [DOI] [PubMed] [Google Scholar]
- 7.Wu N, et al. MicroRNA-373, a new regulator of protein phosphatase 6, functions as an oncogene in hepatocellular carcinoma. FEBS J. 2011;278:2044–2054. doi: 10.1111/j.1742-4658.2011.08120.x. [DOI] [PubMed] [Google Scholar]
- 8.Garipler G, Mutlu N, Lack NA, Dunn CD. Deletion of conserved protein phosphatases reverses defects associated with mitochondrial DNA damage in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2014;111:1473–1478. doi: 10.1073/pnas.1312399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacinto E. What controls TOR? IUBMB Life. 2008;60:483–496. doi: 10.1002/iub.56. [DOI] [PubMed] [Google Scholar]
- 10.Rohde JR, et al. TOR controls transcriptional and translational programs via Sap-Sit4 protein phosphatase signaling effectors. Mol Cell Biol. 2004;24:8332–8341. doi: 10.1128/MCB.24.19.8332-8341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bastians H, Ponstingl H. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J Cell Sci. 1996;109:2865–2874. doi: 10.1242/jcs.109.12.2865. [DOI] [PubMed] [Google Scholar]
- 12.Morales-Johansson H, Puria R, Brautigan DL, Cardenas ME. Human protein phosphatase PP6 regulatory subunits provide Sit4-dependent and rapamycin-sensitive sap function in Saccharomyces cerevisiae. PLoS ONE. 2009;4:e6331. doi: 10.1371/journal.pone.0006331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wengrod J, et al. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci Signal. 2015;8:ra27. doi: 10.1126/scisignal.aaa0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas P, Moorhead G, Xu X, Lees-Miller S. Choreographing the DNA damage response: PP6 joins the dance. Cell Cycle. 2010;9:1221–1222. doi: 10.4161/cc.9.7.11321. [DOI] [PubMed] [Google Scholar]
- 15.Zhong W, et al. Hypertrophic growth in cardiac myocytes is mediated by Myc through a Cyclin D2-dependent pathway. EMBO J. 2006;25:3869–3879. doi: 10.1038/sj.emboj.7601252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douglas P, et al. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 2010;30:1368–1381. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191:1315–1332. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammond D, et al. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J Cell Sci. 2013;126:3429–3440. doi: 10.1242/jcs.128397. [DOI] [PubMed] [Google Scholar]
- 19.Bhandari D, et al. Sit4p/PP6 regulates ER-to-Golgi traffic by controlling the dephosphorylation of COPII coat subunits. Mol Biol Cell. 2013;24:2727–2738. doi: 10.1091/mbc.E13-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamoun M, Filali M, Murray MV, Awasthi S, Wadzinski BE. Protein phosphatase 2A family members (PP2A and PP6) associate with U1 snRNP and the spliceosome during pre-mRNA splicing. Biochem Biophys Res Commun. 2013;440:306–311. doi: 10.1016/j.bbrc.2013.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohama T, Wang L, Griner EM, Brautigan DL. Protein Ser/Thr phosphatase-6 is required for maintenance of E-cadherin at adherens junctions. BMC Cell Biol. 2013;14:42. doi: 10.1186/1471-2121-14-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kajihara R, et al. Protein phosphatase 6 controls BCR-induced apoptosis of WEHI-231 cells by regulating ubiquitination of Bcl-xL. J Immunol. 2014;192:5720–5729. doi: 10.4049/jimmunol.1302643. [DOI] [PubMed] [Google Scholar]
- 23.Couzens AL, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- 24.McConnell JL, Wadzinski BE. Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol. 2009;75:1249–1261. doi: 10.1124/mol.108.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anand P, Boylan JM, Ou Y, Gruppuso PA. Insulin signaling during perinatal liver development in the rat. Am J Physiol Endocrinol Metab. 2002;283:E844–E852. doi: 10.1152/ajpendo.00111.2002. [DOI] [PubMed] [Google Scholar]
- 26.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 27.Lamming DW, et al. Hepatic signaling by the mechanistic target of rapamycin complex 2 (mTORC2) FASEB J. 2013;28:300–315. doi: 10.1096/fj.13-237743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23:3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 29.Demirkan G, Yu K, Boylan JM, Salomon A, Gruppuso PA. Phosphoproteomic Profiling of In Vivo Signaling in Liver by the Mammalian Target of Rapamycin Complex 1 (mTORC1) PLoS ONE. 2011;6:e21729. doi: 10.1371/journal.pone.0021729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Yu K, et al. Integrated platform for manual and high-throughput statistical validation of tandem mass spectra. Proteomics. 2009;9:3115–3125. doi: 10.1002/pmic.200800899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 34.Helou YA, Nguyen V, Beik SP, Salomon AR. ERK positive feedback regulates a widespread network of tyrosine phosphorylation sites across canonical T cell signaling and actin cytoskeletal proteins in Jurkat T cells. PLoS ONE. 2013;8:e69641. doi: 10.1371/journal.pone.0069641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen V, et al. A new approach for quantitative phosphoproteomic dissection of signaling pathways applied to T cell receptor activation. Mol Cell Proteomics. 2009;8:2418–2431. doi: 10.1074/mcp.M800307-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 38.Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209:497–499. doi: 10.1126/science.6248960. [DOI] [PubMed] [Google Scholar]
- 39.Jimenez RH, et al. Regulation of gene expression in hepatic cells by the mammalian Target of Rapamycin (mTOR) PLoS ONE. 2010;5:e9084. doi: 10.1371/journal.pone.0009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gruppuso PA, Boylan JM, Sanders JA. The physiology and pathophysiology of rapamycin resistance: Implications for cancer. Cell Cycle. 2011;10:1050–1058. doi: 10.4161/cc.10.7.15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 43.Shen CH, et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell. 2013;52:161–172. doi: 10.1016/j.molcel.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salton M, Lerenthal Y, Wang SY, Chen DJ, Shiloh Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle. 2010;9:1568–1576. doi: 10.4161/cc.9.8.11298. [DOI] [PubMed] [Google Scholar]
- 45.Sakita-Suto S, et al. Aurora-B regulates RNA methyltransferase NSUN2. Mol Biol Cell. 2007;18:1107–1117. doi: 10.1091/mbc.E06-11-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cai D, et al. Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell. 2014;157:1146–1159. doi: 10.1016/j.cell.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mumby MC, Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol Rev. 1993;73:673–699. doi: 10.1152/physrev.1993.73.4.673. [DOI] [PubMed] [Google Scholar]
- 49.Arndt KT, Styles CA, Fink GR. A suppressor of a HIS4 transcriptional defect encodes a protein with homology to the catalytic subunit of protein phosphatases. Cell. 1989;56:527–537. doi: 10.1016/0092-8674(89)90576-x. [DOI] [PubMed] [Google Scholar]
- 50.Sutton A, Immanuel D, Arndt KT. The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol Cell Biol. 1991;11:2133–2148. doi: 10.1128/mcb.11.4.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov SV, et al. Pro-tumorigenic effects of miR-31 loss in mesothelioma. J Biol Chem. 2010;285:22809–22817. doi: 10.1074/jbc.M110.100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen Y, et al. Serine/threonine protein phosphatase 6 modulates the radiation sensitivity of glioblastoma. Cell Death Dis. 2011;2:e241. doi: 10.1038/cddis.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J Biol Chem. 2012;287:9230–9239. doi: 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jimenez RH, et al. Rapamycin response in tumorigenic and non-tumorigenic hepatic cell lines. PLoS ONE. 2009;4:e7373. doi: 10.1371/journal.pone.0007373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanders JA, et al. The inhibitory effect of rapamycin on the oval cell response and development of preneoplastic foci in the rat. Exp Mol Pathol. 2012;93:40–49. doi: 10.1016/j.yexmp.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gentric G, Desdouets C. Polyploidization in liver tissue. Am J Pathol. 2014;184:322–331. doi: 10.1016/j.ajpath.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 57.Duncan AW, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. doi: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong J, et al. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell Cycle. 2011;10:1411–1419. doi: 10.4161/cc.10.9.15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Douglas P, et al. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep. 2014;34 doi: 10.1042/BSR20140051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broglie P, Matsumoto K, Akira S, Brautigan DL, Ninomiya-Tsuji J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem. 2010;285:2333–2339. doi: 10.1074/jbc.M109.090522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kajino T, et al. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem. 2006;281:39891–39896. doi: 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bauer M, Press AT, Trauner M. The liver in sepsis: patterns of response and injury. Curr Opin Crit Care. 2013;19:123–127. doi: 10.1097/MCC.0b013e32835eba6d. [DOI] [PubMed] [Google Scholar]
- 64.Hayashi K, et al. Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene. 2014;0:1–9. doi: 10.1038/onc.2014.398. [DOI] [PubMed] [Google Scholar]
- 65.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 66.Stebbing J, et al. The regulatory roles of phosphatases in cancer. Oncogene. 2014;33:939–953. doi: 10.1038/onc.2013.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1A contains the full data set derived from microarray analysis. Triplicate RNA samples were prepared from control transduced cells and the two PP6KD cell lines, and analyzed as described in Materials and Methods. Table S1B contains the microarray data filtered to remove any probes that are not annotated. Probes for which all individual results were below the detection threshold of 50 were also removed. The mean control intensity was calculated, and results were ordered based on average intensity. Table S1C contains microarray data for PP6 catalytic subunits and regulatory components. The insert to the right shows the position of PP6 components in the hierarchy of control intensities.
Table S2A shows microarray results filtered for genes that were significant at FDR<0.05 for a fold-difference from the control group of >1.25. Ratios are shown for clones 18.5 and 19.5 as indicated in the first column. Table S2B contains the IPA input generated from the genes shown in Supplemental Table 1A. The expression ratios (PP6KD:Control) represent the mean of the ratios for clones 18.5 and 19.5. Tables S2C through S2F contain IPA results for individual gene ontology categories. Highly significant results are shown in dark blue; borderline significant results are shown in light blue. Level of significance was determined as the lower end of the range of P-values for 5 sets of control genes, which are shown in Table S1G. Control P-values were generated using randomly selected genes for which the fold-difference between PP6KD clones and control cells was between 0.95 and 1.05. Five sets of 93 randomly selected genes were used to generate these data. Table S2H shows the genes that contributed to the identification of selected, significant IPA categories. Red cells represent genes that were upregulated in the PP6KD cells; green represents downregulated genes.
Tables S3A through S3I show GSEA results for the major collections within the Molecular Signatures Database (MSigDB). Colored cells indicate genes that are common to both the 18.5 and 19.5 cell lines. Tables S3J and S3K show the gene enrichment data for KEGG “complement and coagulation cascade” and “DNA replication” categories, respectively.
Each tab represents the data for a biological replicate generated from the mock-transduced control cells, the 18.5 cell line, or the 19.5 cell line.
Table S5A contains the information used for data interpretation for all quantified phosphopeptides. Table S5B shows the correlation of results in the two PP6KD cell lines. Table S5C provides the distribution of ratios (PP6KD-to-control) for all peptides detected and quantified in both cell lines. These data were used to determine the inflection point, the calculation of which is shown in Table S5D. Table S5E contains data for the phosphopeptides for which the difference in abundance between the PP6KD cell lines and the control cell line was significant (FDR<0.05), beyond the inflection point, and concordant with regard to direction of change in the two PP6KD cell lines. Table S5F contains data for control phosphopeptides, which were identified based on a non-significant FDR and fold-change less than the inflection point. Table S5G contains data, including gene ontology categories for phosphopeptides deemed significant (FDR<0.05, direction of change concordant in both cell lines). Table S5H shows the results of the gene ontology analysis. Numbers under “Cont” and “KD” represent the number of phosphopeptides in each category for the control or PP6KD cell lines. The “Cont%” and “KD%” columns give the percentage of the total number of phosphopeptides in Tables S5F and S5G that are represented in the previous two columns in Table S5H, thus indicating the proportion of phosphopeptides representing each gene ontology category. These data were analyzed using a Fisher’s exact test to calculate the P value for each category.