

Abstract
Objectives
To discover biomarkers involved in the pathophysiology of ANCA-associated vasculitis (AAV) and determine if low-density granulocytes (LDGs) contribute to gene expression signatures in AAV.
Methods
The source of clinical data and linked biospecimens was a randomized controlled treatment trial in AAV. RNA-sequencing of whole blood from patients with AAV was performed during active disease at the baseline visit (BL) and during remission 6 months later (6M). Gene expression was compared between patients who met versus did not meet the primary trial outcome of clinical remission at 6M (responders vs. nonresponders). Measurement of neutrophil-related gene expression was confirmed in PBMCs to validate findings in whole blood. A negative selection strategy isolated LDGs from PBMC fractions.
Results
Differential expression between responders (n=77) and nonresponders (n=35) was detected in 2,346 transcripts at BL visit (p<0.05). Unsupervised hierarchical clustering demonstrated a cluster of granulocyte-related genes, including myeloperoxidase (MPO) and proteinase 3 (PR3). A granulocyte multi-gene composite score was significantly higher in nonresponders than responders (p<0.01) and during active disease compared to remission (p<0.01). This signature strongly overlapped an LDG signature identified previously in lupus (FDRGSEA<0.01). Transcription of PR3 measured in PBMCs was associated with active disease and treatment response (p<0.01). LDGs isolated from patients with AAV spontaneously formed neutrophil extracellular traps containing PR3 and MPO.
Conclusions
In AAV an increased expression of a granulocyte gene signature is associated with disease activity and decreased response to treatment. The source of this signature is likely LDGs, a potentially pathogenic cell type in AAV.
Keywords: vasculitis, antineutrophil cytoplasmic antibody (ANCA), proteinase 3, myeloperoxidase, biomarkers, low-density granulocyte (LDG), neutrophil extracellular traps (NETs), granulomatosis with polyangiitis (GPA, Wegener’s), microscopic polyangiitis (MPA)
Granulomatosis with polyangiitis (GPA, Wegener’s) and microscopic polyangiitis (MPA) are two forms of anti-neutrophil cytoplasmic antibody (ANCA) - associated vasculitis [1]. Advances in therapy have transformed ANCA-associated vasculitis (AAV) from a frequently fatal disease into a chronic illness; however, a proportion of patients with AAV do not achieve remission after an initial course of therapy and even fewer achieve sustained remission over long-term follow up [2]. The Rituximab in ANCA-Associated Vasculitis (RAVE) trial expanded standard of care treatment options for patients with AAV by demonstrating that rituximab was not inferior to cyclophosphamide as effective remission-induction therapy [3]. Despite the success of the trial, only 115 of 197 (58%) study patients in both treatment groups achieved the primary trial outcome defined as complete remission off glucocorticoids at the month 6 study visit, and only 71 patients (36%) remained in stable remission throughout 18 months of follow-up [4].
There are currently few clinical and serologic markers that predict clinical outcomes in AAV, and none of these markers have been shown to reliably guide treatment decisions [5–8]. While ANCA has an undisputed diagnostic role in AAV [9], the value of serial ANCA measurement to predict clinical outcomes is controversial [10]. Identification of novel therapeutic targets and biomarkers that predict clinical outcomes to guide patient-specific therapeutic decisions is a high priority in AAV.
Neutrophils have an important role in the pathogenesis of AAV [11]. Patients with AAV typically have antibodies directed against proteinase 3 (PR3) or myeloperoxidase (MPO), which are neutrophil granular proteins. In vitro studies have shown an activating effect of ANCA on cytokine-primed neutrophils [12], and the pathogenic potential of ANCA has been established in animal models [13, 14]. The understanding of the potential roles of neutrophils in AAV has been expanded with the discovery of neutrophil extracellular traps (NETs)[15]. NETs are a meshwork of chromatin fibers that contain granule-derived peptides and enzymes and are extruded by neutrophils following various sources of stimulation [16]. NETs play a role in host defense against pathogens; however, evidence also implicates NETs in the pathogenesis of many autoimmune diseases, including AAV [17, 18]. Upon binding to the surface membrane of neutrophils, ANCA can directly stimulate extrusion of NETs (NETosis) in vitro [19–21]. NETs can be seen at sites of active glomerulonephritis and within disease-associated thrombi in AAV [20, 22].
Low-density granulocytes (LDGs) are a distinct subset of neutrophils that co-localize with peripheral blood mononuclear cells (PBMCs) in density gradient preparations [23]. Circulating LDGs are not detected in healthy controls; however, LDGs are abundant in the blood of patients with systemic lupus erythematosus (SLE) where they have been characterized as pro-inflammatory, are capable of synthesizing type I interferons, and are cytotoxic to endothelial cells [24–26]. In contrast to normal-density neutrophils, which typically undergo NETosis in ex-vivo analyses only upon exposure to pro-inflammatory stimulants, LDGs undergo spontaneous NETosis without stimulation [24]. To date, LDGs have not been described in the blood of patients with AAV; however, two whole-genome gene expression profiling studies in AAV have identified granulocyte signatures in PBMC fractions isolated by density gradient preparations, implying that LDGs may be the source cell of these expression signatures [27, 28].
The objectives of this study were to identify potential clinical biomarkers in AAV through whole-genome gene expression profiling and to determine if LDGs are present in AAV and contribute to gene expression signatures derived from blood.
METHODS
Patient selection and characterization
Patients were selected from the RAVE trial. Study design details of this trial have been reported [3]. For this study, a subset of patients in the RAVE trial was selected based upon clinical outcome data at month 6. Patients in either treatment assignment group who met the primary outcome in the RAVE trial of disease remission off glucocorticoids at month 6 were classified as responders. Patients who did not meet the primary outcome in the RAVE trial were termed nonresponders.
Sample collection, storage, and processing
Peripheral venipuncture was used to collect 3mL blood samples directly into Tempus blood RNA tubes™ at the baseline visit (prior to treatment with rituximab or cyclophosphamide) and at month 6. RNA was isolated in two separate batches using Applied Biosystems RNA chemistry. A complete blood cell count (CBC) with differential was measured concurrently with collection of each RNA sample. Serum samples were tested for ANCA by means of direct and capture enzyme-linked immunosorbent assays (ELISA). PBMCs were isolated by Ficoll/Hypaque density gradient preparations at each study center and stored in liquid nitrogen until further use.
Deep sequencing, alignment, and quantification of large RNA
Deep sequencing of large RNA (RNA-Seq) was performed by Expression Analysis Inc (Durham, NC) using an Illumina Genome Analyzer IIX as outlined in the Illuimina TruSeq™ RNA Sample Preparation Guide. Globin reduction was performed to reduce the proportion of large RNA in whole blood from globin genes which can interfere with the detection of less abundant gene transcripts [30]. cDNA libraries were built for single-read and paired-end sequencing using standard procedures. Starting with 500ng of total RNA, mRNA was purified by polyA selection, chemically fragmented, and reverse transcribed using oligo-DT primers. Following second strand synthesis, adapters were ligated to the 3′ end. Polymerase chain reaction (PCR) was performed to amplify and enrich the ligated material to create the final cDNA library.
Sequencing reads were aligned to the human genome using Bowtie2 software [31]. Local alignment was applied without overlap, treating overlapping mates as discordant. Discordant or unpaired alignment was not allowed. Bam files were imported into Partek Genomic Suites for RNA quantification analysis. Ensembl transcripts (release 64) were used for the annotation source. Transcript-level raw data without normalization was used for differential expression analyses.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis
RNA was isolated from PBMCs using DNA-Free RNA kit (Zymo Research, Irvine, CA). Total RNA (500 ng) was reverse transcribed using iScript RT single strand cDNA (Bio-Rad). qPCR was performed using TaqMan Gene Expression Master Mix (Applied Biosystem, Foster City, CA), human GAPDH primers (Hs99999905_m1) as internal control, and sequence-specific primers for MPO (Hs00924296_m1), PR3 (Hs01597752_m1), Cathelicidin (Hs00189038_m1), and Calprotectin S100A8 (Hs00374264_g1). Samples were run in duplicate using a CFX96 C1000 Touch Real Touch Thermal Cycler (Bio-Rad). Data was analyzed using Bio-Rad CFX Manager software.
Isolation of LDGs
LDGs were isolated from fresh peripheral blood samples obtained in an independent group of five patients with AAV sequentially evaluated at the National Institutes of Health (NIH). None of these patients were participants in the RAVE trial. All of the patients fulfilled the modified 1990 American College of Rheumatology (ACR) Classification Criteria for Wegener’s granulomatosis [29, 32] and were evaluated at random points in the course of disease. Healthy controls were recruited through an NIH healthy volunteer program. AVV-LDGs were isolated from the PBMC layer as previously described [24]. Briefly, “buffy coat” was incubated in sodium chloride solutions to eliminate red blood cells. PBMCs were incubated with a cocktail of biotinylated antibodies for 30 minutes on ice. Cells were then incubated with magnetic beads for 15 minutes on ice and passed through a MACS column (Miltenyi Biotec) per the manufacturer’s instructions. LDG purity was assessed by flow cytometry as described [24]. Control and AVV normal-density neutrophils were isolated from the red-blood cell layer by dextran sedimentation.
Immunofluorescence analysis
Cells were seeded on coverslips and stimulated with 40 nM of PMA. Neutrophils and LDGs were incubated with 5% CO2 at 37°C for 60 min. Cells were fixed with 4% paraformaldehyde in PBS overnight at 4°C. After washing, cells were blocked with 0.2% porcine skin gelatin (Sigma) in PBS for 30 min and incubated for 1 h at 37°C with anti-human elastase (Abcam), anti-MPO (DAKO) or anti-PR3 (Santa Cruz) antibodies diluted in blocking buffer. After 3 washes with PBS for 5 min at room temperature, cells were incubated for 30 min at 37°C with either Alexa-555-conjugated anti-rabbit IgG or Alexa-488-conjugated anti-mouse IgG secondary antibody. Nuclei were co-stained with 1:1000 fluorescence dye Hoechst 33342. Coverslips were washed with PBS 3 times for 5 min at room temperature and were mounted on a glass slide using Prolong-gold. Images were acquired on a Zeiss LSM780 confocal laser-scanning microscope and quantification performed as previously described [25].
Statistical analysis
The Bioconductor package edgeR (release 2.14) was used for differential expression analyses of read counts [33]. Generalized linear models (GLMs) were applied to the non-normally distributed read counts data. Since RNA was isolated in two separate batches for these experiments, adjustments for batch effect differences were made using an additive model within edgeR. Different cut-offs to define statistical significance for differential gene expression analyses were explored. A multi-gene composite score was created by calculating z-scores on a per gene per sample basis. Mean granulocyte composite score was compared between responders and nonresponders and between active disease and remission using the Wilcoxon rank sum and Wilcoxon signed rank tests.
Linear regression models were used to determine the association between the granulocyte gene composite score (dependent variable) and the following independent variables derived from the baseline study visit: age, ANCA titer by direct or capture ELISA, Birmingham Vasculitis Activity Score modified for Wegener’s granulomatosis (BVAS/WG), mean dose of glucocorticoids over 14 days prior to baseline visit blood sample collection, absolute neutrophil count, absolute lymphocyte count, hemoglobin level, B cell count, platelet count, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). A p value of < 0.1 was used to define the threshold for incorporation of a variable in the multivariable linear regression models and a p value of < 0.05 was used to define statistical significance in the regression models. Logistic regression was used to determine the association between clinical outcome status (nonresponders vs responders) and the granulocyte gene composite score.
Enrichment of relevant gene set signatures was tested using Gene Set Enrichment Analysis (GSEA v2.1.0)[34]. GSEA is a computational method that can be used to determine if a specific set of genes shows concordant differences between two phenotypes (i.e. nonresponders vs responders). Gene sets with an estimated false discovery rate (FDR) of < 0.05 were considered significant, per GSEA guidelines. Data were analyzed through the use of QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity).
In the RAVE trial, PBMCs were collected concurrently with whole blood RNA at some study visits. A subset of 5 nonresponders and 5 responders was selected for gene expression analyses using PBMCs instead of whole blood as the sample source. Nonresponders and responders were matched on age, gender, ANCA specificity (PR3 vs MPO), disease subtype (GPA vs MPA), and treatment assignment (cyclophosphamide vs rituximab). Expression of the following neutrophil-related genes was studied in the PBMC fraction: PR3, MPO, CAMP, and calprotectin (S100A8). Differential expression of mRNA as measured by qPCR in the PBMC samples was compared using one-way analysis of variance (ANOVA).
Ethics and Informed Consent
All patients enrolled in the RAVE trial or evaluated at the NIH provided written informed consent for collection and future use of samples and data. Participating ethics boards approved the research.
RESULTS
Subject characteristics
The baseline clinical characteristics of the study population are provided in Table 1. There were 112 patients included in the study (responders = 77; nonresponders = 35). Specific reasons for inclusion into the nonresponder group included: major disease flare before study month 6 (n=9), treatment crossover for flare (n=7), BVAS/WG > 0 at month 6 (n=18), and requirement at month 6 for prednisone 15 mg daily (n=1). There were few statistically significant differences between responders and nonresponders. The majority of patients (88%) had received high doses of glucocorticoids in the two-week period prior to baseline sample collection, but there were no differences between responders and nonresponders in the proportion of patients in each group who had received glucocorticoids at study entry, nor in the total amount of glucocorticoids that patients had received for the episode of disease activity resulting in enrollment in the trial. The percentage of patients with new (versus relapsing) disease at the time of study enrollment and the hemoglobin levels at baseline were significantly higher in the responder group.
Table 1.
Baseline Subject Characteristics
| Nonresponders N=35 |
Responders N=77 |
P-value | |
|---|---|---|---|
|
| |||
| Age (years ± standard deviation) | 51 ± 14.8 | 52 ± 17.9 | 0.78 |
|
| |||
| Gender (%) | |||
| Female | 46 | 56 | 0.43 |
| Male | 54 | 44 | |
|
| |||
| Race (White %) | 91 | 96 | 0.37 |
|
| |||
| Treatment arm (%) | |||
| Cyclophosphamide/azathioprine | 66 | 47 | 0.10 |
| Rituximab | 34 | 53 | |
|
| |||
| ANCA subtype (%) | |||
| MPO | 20 | 35 | 0.17 |
| PR3 | 80 | 65 | |
|
| |||
| Disease type (%) | |||
| MPA | 14 | 26 | 0.26 |
| GPA | 86 | 74 | |
|
| |||
| New diagnosis (%) | 31 | 57 | 0.02 |
|
| |||
| Organ system involvement (%) | |||
| - Constitutional | 60 | 53 | 0.64 |
| - Cutaneous | 17 | 18 | >.99 |
| - Mucous membranes, eyes | 23 | 19 | 0.87 |
| - Ear, nose, throat | 57 | 56 | >.99 |
| - Cardiovascular | 3 | 1 | 0.53 |
| - Gastrointestinal | 3 | 3 | >.99 |
| - Pulmonary | 63 | 57 | 0.72 |
| - Renal | 54 | 65 | 0.39 |
| - Nervous | 17 | 22 | 0.73 |
|
| |||
| Received glucocorticoids prior to baseline visit (%) | 89 | 87 | >.99 |
| Mean cumulative dose of glucocorticoids from 14 days before baseline sample collection | |||
| - Prednisone (mg) | 290 ± 228.9 | 332 ± 322.5 | 0.47 |
| - Methylprednisolone (g) | 0.7 ± 1.11 | 1.0 ± 1.32 | 0.28 |
|
| |||
| Total white blood cell count | 12 ± 5.4 | 12 ± 4.5 | 0.69 |
|
| |||
| Absolute neutrophil count | 9 ± 5.4 | 10 ± 4.7 | 0.84 |
|
| |||
| Hemoglobin | 12 ± 1.5 | 11 ± 1.8 | 0.01 |
|
| |||
| Platelet | 356 ± 129.9 | 379 ± 133.3 | 0.42 |
|
| |||
| Lymphocyte count | 1.3 ± 1.0 | 1.3 ± 1.0 | 0.96 |
|
| |||
| BVAS/WG score | 7.3 ± 2.8 | 8.3 ± 3.5 | 0.19 |
|
| |||
| ANCA titer | 221 ± 145.8 | 189 ± 122.0 | 0.26 |
|
| |||
| Erythrocyte sedimentation rate (mm/hr) | 38 | 44 | 0.34 |
|
| |||
| C-reactive protein (mg/dL) | 2.2 | 7.4 | 0.12 |
Key: ANCA = antineutrophil cytoplasmic antibody, MPO = myeloperoxidase, PR3 = proteinase 3, BVAS/WG = Birmingham Vasculitis Activity Score for Wegener’s granulomatosis.
Identification of granulocyte gene signature
After filtering transcripts expressed in <50% of both responders and nonresponders, there were 44,532 total aligned reads. Differential expression between responders and nonresponders was seen in 2,346 transcripts at the baseline visit at a threshold p<0.05. Pathway analysis of differentially expressed genes revealed upregulation of pathways related to bacterial defense, myeloid differentiation, and neutrophil activation in nonresponders (Supplemental Figure 1 and Supplementary Table 1).
Unsupervised hierarchical clustering of differentially expressed genes demonstrated a distinct cluster of 179 genes that predominantly included granulocyte-related genes and included MPO and PR3, the major auto-antigens in AAV (see Supplemental Table 2 for the complete list of genes). A multi-gene composite score was made using expression data from 11 granulocyte genes purposefully selected within the cluster of differentially-expressed granulocyte genes to represent a spectrum of neutrophil granular proteins. A list of the 11 genes and the differential expression values between nonresponders and responders is provided in Table 2.
Table 2.
Differences in expression between nonresponders and responders for the 11 granulocyte multi-gene composite score components
| Gene | Fold Change | Likelihood Ratio | P-value |
|---|---|---|---|
| Myeloperoxidase (MPO) | 2.42 | 14.10 | <0.001 |
| Proteinase 3 (PR3) | 2.65 | 10.33 | 0.001 |
| Elastase (ELANE) | 2.43 | 11.62 | <0.001 |
| Bactericidal permeability inducing protein (BPI) | 2.20 | 12.61 | <0.001 |
| Azurocidin 1 (AZU1) | 2.64 | 12.78 | <0.001 |
| Cathepsin G (CTSG) | 2.46 | 12.31 | <0.001 |
| Lactoferrin (LTF) | 1.81 | 6.93 | 0.008 |
| Defensin A3 (DEFA3) | 2.15 | 9.21 | 0.002 |
| Defensin A4 (DEFA4) | 2.56 | 13.48 | <0.001 |
| Lipocalin-2 (LCN2) | 1.93 | 8.73 | 0.003 |
| Cathelicidin (CAMP) | 1.66 | 6.76 | 0.009 |
The mean granulocyte multi-gene composite score was higher in nonresponders [0.11; range −0.43 to 7.59] than responders [−0.27; range −0.46 to 3.70] (p=0.02). Nine of 26 nonresponders (26%) and 4 of 73 responders (5%) had granulocyte composite scores of > 1.2 (p<0.01 by Fisher’s exact test). Clinical and demographic characteristics of patients with a baseline granulocyte composite scores >1.2 did not differ significantly from patients with lower granulocyte composite scores (data not shown).
Whole blood RNA from the month 6 study visit was available only in the responder group. At this time point, all of these patients were in clinical remission off glucocorticoids. The median month 6 granulocyte multi-gene composite score for responders was higher at the baseline visit during active disease [−0.17; range −0.46 to 3.70] compared to the month 6 visit during clinical remission [−0.39; range −0.47 to 0.53] (p<0.01).
Association of granulocyte multi-gene composite score with clinical outcomes
Linear regression models were used to determine the association between the granulocyte gene composite score (dependent variable) and several clinical variables. Age, BVAS/WG, absolute neutrophil count (ANC), and platelet count were significantly associated with the granulocyte gene composite score in univariable analyses, and only ANC remained significantly associated in a multivariable regression model (Table 3).
Table 3.
Linear Regression Models showing the associations between the Granulocyte Multi-Gene Composite Score and Clinical/Laboratory Data
| Univariable Model | Multivariable Model | |||||
|---|---|---|---|---|---|---|
| Predictor Variable | Parameter Estimate | Standard Error | P-value | Parameter Estimate | Standard Error | P-value |
| Absolute neutrophil count | 0.04 | 0.01 | <0.01 | 0.03 | <0.01 | 0.01 |
| Platelet count | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.14 |
| BVAS/WG Score | 0.04 | 0.02 | 0.01 | 0.03 | 0.02 | 0.05 |
| Age | 0.01 | <0.01 | 0.07 | 0.01 | <0.01 | 0.09 |
| Hemoglobin | −0.05 | 0.03 | 0.12 | These variables were not included in the multivariable models due to lack of significant association (p>0.1) in the univariable models. | ||
| Absolute lymphocyte count | 0.11 | 0.08 | 0.15 | |||
| Erythrocyte sedimentation rate | <0.01 | 0.10 | 0.36 | |||
| Cumulative glucocorticoid use | <0.01 | <0.01 | 0.46 | |||
| B cell count | <0.01 | <0.01 | 0.49 | |||
| ANCA titer | <0.01 | <0.01 | 0.76 | |||
| C-reactive protein | <0.01 | 0.06 | 0.80 | |||
Logistic regression was used to determine the association between clinical outcome status (nonresponders vs responders) and the granulocyte gene composite score. In univariable models, clinical outcome status was only significantly associated with the granulocyte gene composite score and hemoglobin level. A 1-unit increase in the granulocyte gene composite score at baseline was significantly associated with a 1.77 times increased odds for not meeting the primary outcome in the RAVE trial (OR=1.77; 95%CI=1.15 – 2.73; p=0.01); this association remained significant after adjustment for age, BVAS/WG, absolute neutrophil count, hemoglobin level, and glucocorticoid use (OR=2.13; 95%CI 1.16 – 3.90, p=0.01). Logistic regression analysis using granulocyte composite score as a dichotomous outcome variable (> 1.2 versus ≤1.2) showed no significant associations with any of the clinical, demographic, or laboratory variables including ANCA subtype and absolute neutrophil count (data not shown). These analyses demonstrate that the multi-gene granulocyte composite score was significantly associated with treatment response status in the RAVE trial even after adjustments for potential confounding variables.
Enrichment of low-density granulocyte signature
Given the presence of a granulocyte gene signature detected in whole blood in patients with AAV in the RAVE trial, GSEA was used to identify potential cell populations as sources of the signature. Differential expression of granulocyte genes measured in the PMBC fraction from patients with AAV compared to healthy controls has been reported in two independent whole-genome microarray studies [27, 28]. The list of differentially expressed granulocyte gene sets from both of these prior studies was significantly enriched in the nonresponder group in RAVE (enrichment scores for both studies = 0.86, FDRGSEA < 0.001). A gene signature derived by comparing differential whole-genome gene expression from isolated LDGs versus autologous normal-density neutrophils in patients with SLE has been reported [25]. Forty-one of the 281 genes that defined an LDG signature in the lupus cohort overlapped with differential gene expression in the RAVE nonresponders, including 9 of the 11 granulocyte composite score genes (enrichment score = 0.79, GSEAFDR <0.001). There was substantial overlap in differentially expressed genes between the present study, the two published studies of PBMCs in AAV, and the study of LDGs in SLE (FIGURE 1). Nine genes were present in all 4 datasets: BPI, CAMP, CEACAM6, DEFA4, HP, MS4A3, PGLYRP1, RETN, and TCN1. See Supplementary Figure 2 for further information regarding enrichment analyses.
Figure 1. Gene Set Enrichment Analyses.
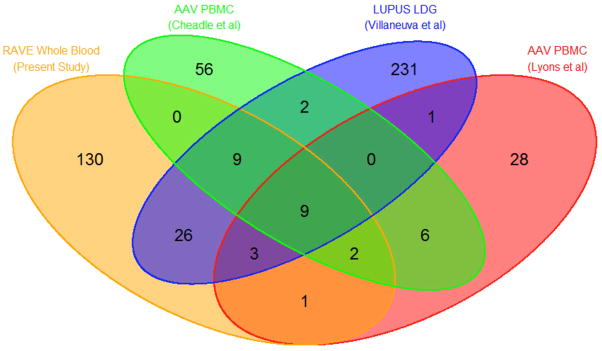
Venn diagram illustrates overlap in the number of differentially expressed genes identified in four independent datasets. The RAVE dataset compared whole blood gene expression between treatment responders and nonresponders in ANCA-associated vasculitis (AAV). The Cheadle and Lyons studies compared expression in PBMCs between patients with AAV and healthy controls (33,34). The Villanueva et al study compared expression between LDGs and autologous normal-density neutrophils in patients with SLE (25). Overlapping ovals indicates the number of common genes shared between datasets.
Validation of granulocyte gene signature in PBMCs
To determine if the granulocyte gene signature identified in whole blood originated from LDGs, neutrophil-related gene expression was measured in the PBMC fraction in a subset of responders and nonresponders. Significant differential transcription of PR3 was observed between responders and nonresponders. Expression of PR3 was 17-fold upregulated in 4 of 5 nonresponders compared to responders (p<0.01). There was non-statistically significant increased expression for S100A8 in nonresponders (p=0.18). No significant differences were observed for MPO or CAMP (FIGURE 2A).
Figure 2. Differential Expression of Neutrophil Related mRNA in the PBMC Fraction.

Expression of proteinase 3 (PR3) and calprotectin (S100A8) but not myeloperoxidase (MPO) or cathelicidin (CAMP) are differentially upregulated in PBMCs in treatment nonresponders compared to responders in the RAVE trial (Figure 2A). PBMCs expression of PR3 and S100A8 is differentially upregulated within the same patients with AAV during active disease compared to remission (Figure 2B).
PBMCs were available for paired sample analyses in 4 responders from both baseline (active disease) and month 6 (remission) study visits. Transcription of PR3 was significantly increased in all 4 patients during active disease versus remission (p<0.01). Differences in S100A8 expression were observed in 3 of 4 patients. There were no differences for MPO or CAMP (FIGURE 2B). These experiments validate the findings in whole blood and provide additional evidence that LDGs are the source of the whole blood granulocyte signature.
Direct isolation of low-density granulocytes in independent cohort
To confirm whether LDGs are present in patients with AAV and to account for the neutrophil signature observed in PBMCs, normal-density neutrophils and LDGs were isolated in 60 ml of blood from 5 patients with AAV. Three of these patients were evaluated at 2 separate study visits 6-month apart. The clinical characteristics of these patients are provided (Supplementary Table 3). LDGs were identified and quantified in all 5 patients with AAV at every study visit (Supplementary Table 3). Spontaneous NETosis was observed in LDGs for all 5 patients at every visit (FIGURE 3A). Unstimulated normal-density neutrophils and LDGs from patients with AAV underwent significantly more NET formation compared to unstimulated normal-density neutrophils from healthy controls (FIGURE 3B). NETs derived from LDGs in patients with AAV, irrespective of ANCA subtype, externalized both PR3 and MPO (FIGURE 3C).
Figure 3. Demonstration of LDGs in Patients with AAV.

Immunofluorescence microphotographs of normal-density neutrophils from a healthy control, normal-density neutrophils and LDGs from a patient with AAV (Figure 3A). The top row depicts unstimulated neutrophils and the bottom row depicts neutrophils stimulated with PMA. NET formation (long strands) in the absence of PMA stimulation is observed in in both neutrophils and LDGs in AAV but not in control neutrophils, and is enhanced by PMA stimulation. Blue = DAPI stain (DNA); red = anti-myeloperoxidase stain. Unstimulated normal-density neutrophils (Vas-Neu) and LDGs (Vas-LDG) from patients with AAV undergo significantly more ex vivo NET formation than neutrophils from healthy controls (Ctrl-Neu) (p<0.01) and LDGs from patients with AAV undergo significantly more NET formation than autologous normal-density neutrophils (p<0.05) (Figure 3B). LDGs isolated from a patient with AAV demonstrate spontaneous NET formation and both PR3 and MPO co-localize within NETs (Figure 3C).
DISCUSSION
Transcriptomic analysis of whole blood from patients with AAV revealed a granulocyte gene expression signature associated with disease activity and treatment response. In the RAVE trial, patients with high levels of granulocyte gene expression during the baseline study visit were less likely to meet the trial’s primary outcome of complete remission at the month 6 study visit. In contrast, clinical features of disease, standard laboratory assessments including ANCA, and disease-specific activity indices did not predict treatment response in these patients. Differential gene expression of granulocyte-related genes in the PBMC fraction validates the granulocyte signature observed in whole blood and localizes the signature to a recently-described subset of neutrophils known as LDGs. In particular, expression of PR3 in the PBMC fraction was 17-fold higher in patients who did not meet the primary outcome in RAVE compared to treatment responders. LDGs were directly isolated from peripheral blood in all patients with AAV in an independent cohort of patients, marking the first time these cells have been reported in AAV. LDGs from patients with AAV readily underwent NETosis in the absence of added stimulation and produced NETs containing both PR3 and MPO, the major antigenic targets of ANCA.
The source of a granulocyte signature identified in whole blood could be normal-density neutrophils, LDGs, or both. Comparisons with other transcriptomic studies strongly suggest that LDGs and not normal-density neutrophils are the source of the granulocyte signature identified in this study. Two prior studies examined whole-genome gene expression in sorted normal-density neutrophils versus PBMCs. In a study by Lyons et al, granulocyte-related gene expression was detected in the PBMC fraction of blood from patients with lupus and AAV compared to healthy controls and correlated with the number of quantity of granulocytes identified in PBMCs by flow cytometry [28]. In a study by Cheadle et al, transcription of granulocyte genes in the PBMC fraction, including PR3 and MPO, differentiated patients with AAV from healthy controls [27]. In both studies, granulocyte expression signatures identified in PBMCs, not in normal-density neutrophils, strongly overlapped the granulocyte gene signature identified in whole blood in our study. These data suggest that LDGs, which by definition are neutrophils that co-localize in the PBMC fraction, are the source of granulocyte gene expression signatures in AAV. A gene expression signature derived directly from isolated LDGs in patients with SLE [25] also strongly overlaps the granulocyte signatures identified in AAV in each of these studies, providing additional evidence that LDGs are the specific cellular source of these signatures. Although enrichment of degranulated neutrophils could contribute to gene expression signatures identified in this study, electron microscopy studies demonstrate that LDGs are not simply degranulated neutrophils [23]. Spontaneous NET formation observed in LDGs isolated from patients with AAV requires the presence of neutrophil granular proteins, which are abundantly transcribed in LDGs.
Increased transcription of PR3, and to a lesser extent calprotectin, in PBMCs was associated with disease activity and treatment response. Similar associations were not observed for MPO or CAMP, despite the fact that these genes were components of the granulocyte expression signature identified in whole blood. Other studies have found differences between PR3 and MPO as potential biomarkers in AAV. Independent of ANCA specificity, transcription of PR3 but not MPO and presence of neutrophil surface membrane PR3 but not surface membrane MPO differentiates AAV from healthy blood donors and disease controls [35, 36]. Additionally, increased transcription of PR3 in the PBMC fraction has been previously shown to correlate with disease activity in AAV [27]. Whether PR3 expression could be a useful surrogate marker for LDGs needs to be assessed in prospective studies.
A major strength of this study is the use of samples collected within a large clinical trial in AAV linked to systematically-recorded clinical and laboratory data. Regression models were used to assess the associations between the granulocyte signature and clinical/laboratory data. The granulocyte signature was weakly associated with baseline BVAS/WG score and platelet count, suggesting that the signature captures elements of baseline disease activity. Historically, baseline disease activity scores in AAV do not predict treatment response [5]. In keeping with this observation, granulocyte gene expression, but not the baseline BVAS/WG score, was significantly associated with treatment response.
Glucocorticoids are a potential confounder of granulocyte gene expression. Glucocorticoids promote demargination of circulating neutrophils and enhance release of immature neutrophils from bone marrow [37]. The majority of patients in this study were receiving high doses of glucocorticoids at the time of the baseline sample collection. However, cumulative glucocorticoid use in the 2 weeks prior to the baseline visit was not directly associated with the granulocyte multi-gene composite score, and no differences in glucocorticoid use were observed between treatment responders and nonresponders, lessening concerns for potential confounding by glucocorticoids of the association between the granulocyte expression signature and treatment response.
Use of whole blood as a tissue source for gene expression is potentially a study limitation. Blood is comprised of many cell populations, and it can be challenging to differentiate changes in transcript abundance in blood caused by regulation of gene transcriptional activity from changes secondary to relative abundance of cell populations expressing transcripts at constant levels. However, in this study absolute neutrophil and lymphocyte counts at baseline did not differ between treatment responders and nonresponders. Although hemoglobin levels were significantly higher in nonresponders, the association between treatment response and the granulocyte signature score remained significant in multivariable models that adjusted for differences in hemoglobin levels. Heterogeneity of cell populations in whole blood can also reduce the power of transcriptomic studies to identify significant differential gene expression across phenotypes. In this study, a lenient p value of <0.05 was used to define significant differential expression, increasing the potential for false discovery. However, validation of differential granulocyte gene expression in a different tissue source (PBMCs), consistency in findings across different transcriptomic datasets in AAV, and functional studies of LDGs lessen the concern for false discovery. The associations between granulocyte gene expression derived from whole blood and clinical outcomes, while statistically significant, were only observed in a minority of treatment nonresponders (26%). The high doses of glucocorticoids at baseline may have reduced some of the significant differences in gene expression between the responder and non-responder groups. More specific measures of LDGs, rather than indirect measures in whole blood, may be required to accurately predict clinical outcomes for individual patients with AAV. LDGs were not isolated directly in fresh samples collected within the RAVE trial, and these cells could not be isolated from frozen PBMC because they do not survive freeze-thaw cycles.
In conclusion, granulocyte-related gene expression was associated with disease activity and relatively poor response to treatment in AAV. LDGs are the likely source of granulocyte gene expression in this study and previous transcriptomic studies that have identified similar gene expression signatures in AAV. LDGs may play an important role in disease pathogenesis in AAV by promoting direct toxicity to endothelial cells and, potentially, by direct involvement in the causal pathway for the generation of ANCAs through autoantigen externalization due to enhanced NET formation. Future efforts to further characterize the function of LDGs and to explore their potential as biomarkers in AAV are warranted.
Supplementary Material
Footnotes
Financial supports of conflicts disclosure:
This research was performed as a project of the Immune Tolerance Network (NIH contract N01-AI-15416; protocol number ITN-21AI). This research was also supported through the Intramural Research Program at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and was supported by the National Center for Advancing Translational Sciences, NIH, through BU-CTSI Grant Number KL2 TR000158. Dr. Grayson received support from a Rheumatology Scientist Development Award from the Research and Education Foundation of the American College of Rheumatology.
References
- 1.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis and rheumatism. 2013;65(1):1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman GS, Leavitt RY, Fleisher TA, Minor JR, Fauci AS. Treatment of Wegener’s granulomatosis with intermittent high-dose intravenous cyclophosphamide. The American journal of medicine. 1990;89(4):403–10. doi: 10.1016/0002-9343(90)90367-m. [DOI] [PubMed] [Google Scholar]
- 3.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. The New England journal of medicine. 2010;363(3):221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Specks U, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. The New England journal of medicine. 2013;369(5):417–27. doi: 10.1056/NEJMoa1213277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogan SL, Falk RJ, Chin H, Cai J, Jennette CE, Jennette JC, et al. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Annals of internal medicine. 2005;143(9):621–31. doi: 10.7326/0003-4819-143-9-200511010-00005. [DOI] [PubMed] [Google Scholar]
- 6.McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DR, Willcocks LC, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nature medicine. 2010;16(5):586–91. doi: 10.1038/nm.2130. 1p following 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li ZY, Chang DY, Zhao MH, Chen M. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated vasculitis: a study of 439 cases in a single Chinese center. Arthritis & rheumatology. 2014;66(7):1920–6. doi: 10.1002/art.38621. [DOI] [PubMed] [Google Scholar]
- 8.Pagnoux C, Hogan SL, Chin H, Jennette JC, Falk RJ, Guillevin L, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: comparison of two independent cohorts. Arthritis and rheumatism. 2008;58(9):2908–18. doi: 10.1002/art.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney international. 1998;53(3):743–53. doi: 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 10.Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis--a meta-analysis. Rheumatology. 2012;51(1):100–9. doi: 10.1093/rheumatology/ker280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falk RJ, Jennette JC. ANCA disease: where is this field heading? Journal of the American Society of Nephrology: JASN. 2010;21(5):745–52. doi: 10.1681/ASN.2009121238. [DOI] [PubMed] [Google Scholar]
- 12.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(11):4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. The American journal of pathology. 2005;167(1):39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. The Journal of clinical investigation. 2002;110(7):955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 16.Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell death and differentiation. 2011;18(4):581–8. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. Journal of immunology. 2012;189(6):2689–95. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? The Journal of cell biology. 2012;198(5):773–83. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. Journal of the American Society of Nephrology: JASN. 2014;25(5):990–7. doi: 10.1681/ASN.2013060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine. 2009;15(6):623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley JM, Monach PA, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(51):20736–41. doi: 10.1073/pnas.1109227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Frontiers in immunology. 2012;3:333. doi: 10.3389/fimmu.2012.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Seminars in immunopathology. 2013;35(4):455–63. doi: 10.1007/s00281-013-0375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. Journal of immunology. 2010;184(6):3284–97. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. Journal of immunology. 2011;187(1):538–52. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Annals of the rheumatic diseases. 2014 doi: 10.1136/annrheumdis-2013-204837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheadle C, Berger AE, Andrade F, James R, Johnson K, Watkins T, et al. Transcription of proteinase 3 and related myelopoiesis genes in peripheral blood mononuclear cells of patients with active Wegener’s granulomatosis. Arthritis and rheumatism. 2010;62(6):1744–54. doi: 10.1002/art.27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyons PA, McKinney EF, Rayner TF, Hatton A, Woffendin HB, Koukoulaki M, et al. Novel expression signatures identified by transcriptional analysis of separated leucocyte subsets in systemic lupus erythematosus and vasculitis. Annals of the rheumatic diseases. 2010;69(6):1208–13. doi: 10.1136/ard.2009.108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Group WR. Design of the Wegener’s Granulomatosis Etanercept Trial (WGET) Controlled clinical trials. 2002;23(4):450–68. doi: 10.1016/s0197-2456(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 30.Vartanian K, Slottke R, Johnstone T, Casale A, Planck SR, Choi D, et al. Gene expression profiling of whole blood: comparison of target preparation methods for accurate and reproducible microarray analysis. BMC genomics. 2009;10:2. doi: 10.1186/1471-2164-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis and rheumatism. 1990;33(8):1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 33.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohlsson S, Hellmark T, Pieters K, Sturfelt G, Wieslander J, Segelmark M. Increased monocyte transcription of the proteinase 3 gene in small vessel vasculitis. Clinical and experimental immunology. 2005;141(1):174–82. doi: 10.1111/j.1365-2249.2005.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller Kobold AC, Kallenberg CG, Tervaert JW. Leucocyte membrane expression of proteinase 3 correlates with disease activity in patients with Wegener’s granulomatosis. British journal of rheumatology. 1998;37(8):901–7. doi: 10.1093/rheumatology/37.8.901. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa M, Terashima T, D’Yachkova Y, Bondy GP, Hogg JC, van Eeden SF. Glucocorticoid-induced granulocytosis: contribution of marrow release and demargination of intravascular granulocytes. Circulation. 1998;98(21):2307–13. doi: 10.1161/01.cir.98.21.2307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.