

Abstract
High-voltage-activated (HVA) calcium channels play an important role in synaptic transmission. Activation of Mas-related G-protein-coupled receptor subtype C (MrgC; mouse MrgC11, rat homolog rMrgC) inhibits HVA calcium current (ICa) in small-diameter dorsal root ganglion (DRG) neurons, but the intracellular signaling cascade underlying MrgC agonist-induced inhibition of HVA ICa in native DRG neurons remains unclear. To address this question, we conducted patch-clamp recordings in MrgA3-eGFP-wild-type mice, in which most MrgA3-eGFP+ DRG neurons co-express MrgC11 and can be identified for recording. We found that the inhibition of HVA ICa by JHU58 (0.001–100 nM, a dipeptide, MrgC-selective agonist) was significantly reduced by pretreatment with a phospholipase C inhibitor (U73122, 1 μM), but not by its inactive analogue (U73343) or vehicle. Further, in rats that had undergone spinal nerve injury, pretreatment with intrathecal U73122 nearly abolished the inhibition of mechanical hypersensitivity by intrathecal JHU58. The inhibition of HVA ICa in MrgA3-eGFP+ neurons by JHU58 (100 nM) was partially reduced by pretreatment with a Gβγ blocker (gallein, 100 μM). However, applying a depolarizing prepulse and blocking the Gαi and Gαs pathways with pertussis toxin (0.5 μg/mL) and cholera toxin (0.5 μg/mL), respectively, had no effect. These findings suggest that activation of MrgC11 may inhibit HVA ICa in mouse DRG neurons through a voltage-independent mechanism that involves activation of the phospholipase C, but not Gαi or Gαs, pathway.
Keywords: MrgC, dorsal root ganglion, calcium channel, PLC, pain
INTRODUCTION
Recent studies suggest that Mas-related G protein-coupled receptors (Mrg) may play an important role in the function of nociceptive neurons and in the modulation of pain (Dong et al., 2001; Lembo et al., 2002; Grazzini et al., 2004). In particular, the subtype C of Mrg receptors (MrgC) may be a compelling new target for pain therapy by virtue of its restricted distribution in small-diameter primary sensory neurons, including both IB4-positive (non-peptidergic) and calcitonin gene-related peptide-positive (peptidergic) subpopulations in mice and rats (Han et al., 2013; He et al., 2014a; He et al., 2014b). Intrathecal administration of agonists to MrgC (mouse MrgC11, rat homolog rMrgC) produces analgesic effects in rodent models of inflammatory and neuropathic pain(Chen et al., 2006; Guan et al., 2010a; Jiang et al., 2013; He et al., 2014b). Recent studies have begun to reveal cellular mechanisms that may underlie MrgC agonist-induced analgesia in animal models of these pain types (Jiang et al., 2013; Wang et al., 2013; Li et al., 2014).
In our previous studies, we showed that JHU58, a novel, dipeptide, MrgC-selective agonist, dose-dependently inhibits N-type, but not L- or P/Q-type, high-voltage-activated (HVA) calcium channels in native mouse dorsal root ganglion (DRG) neurons (Li et al., 2014). Activation of HVA calcium channels plays a vital role in excitatory synaptic transmission and the presynaptic release of neurotransmitters (Abernethy and Schwartz, 1999; McGivern, 2006; Hoppa et al., 2012). In line with these findings, JHU58 attenuated both spontaneous and evoked excitatory postsynaptic currents in substantia gelatinosa dorsal horn neurons of spinal slices from wild-type mice that had undergone nerve injury, but not in those from similarly treated Mrg-clusterΔ−/− (Mrg KO) mice(Li et al., 2014; He et al., 2014b). The inhibition of HVA calcium current (ICa) and the alleviation of neuropathic pain-related behavior in animal models by JHU58 were both Mrg-dependent (Li et al., 2014; He et al., 2014b). These findings suggest that MrgC agonism at central terminals of primary sensory fibers may decrease peripheral excitatory inputs onto dorsal horn neurons.
HVA calcium channels can be modulated by different G-protein-dependent pathways (Dolphin and Scott, 1989; Hille et al., 1995; Herlitze et al., 1996). Mu-opioids are known to inhibit HVA calcium channels in DRG neurons primarily through the Gαi-dependent pathway(Dolphin and Scott, 1989); this inhibition leads to attenuation of neuronal excitability and reduction of excitatory neurotransmitter release(Su et al., 1998; Morikawa et al., 2006; Callaghan et al., 2008). MrgC, a G-protein-coupled receptor that is similar to human MrgX1 in expression and function, can act via both Gαq/11 and Gαi pathways(Dong et al., 2001; Han et al., 2002; Zylka et al., 2003). However, the major intracellular signaling cascade by which an MrgC agonist inhibits HVA ICa in native DRG neurons remains unclear. It has been difficult to identify MrgC-bearing DRG neurons for recording because only a subset of native DRG neurons express MrgC (Liu et al., 2009; Han et al., 2013). Intriguingly, MrgA3 is highly colocalized with MrgC11 in mouse DRG neurons (Liu et al., 2009; Han et al., 2013; Li et al., 2014). We recently developed MrgA3-eGFP-wild-type mice, in which most MrgA3 promoter-driven eGFP+ neurons co-express MrgC11(Han et al., 2013; Li et al., 2014). Because MrgA3-eGFP+ neurons can be seen by endogenous green fluorescence, we can prospectively identify these neurons that co-express MrgC11 and examine their functions without having to randomly sample a large number of neurons for electrophysiologic recording. This approach may facilitate mechanistic studies aimed at further dissecting the intracellular events involved in the actions of MrgC agonists.
To assess the possible involvement of different transduction pathways in the inhibition of HVA ICa by MrgC agonism, we used pharmacologic approaches to examine whether pretreatment with selective Gαi, Gαq/11, Gαs, and Gβγ pathway blockers reduces the ability of MrgC agonists to inhibit HVA ICa in MrgA3-eGFP+ wild-type DRG neurons. Our findings suggest that JHU58 inhibits HVA ICa through phospholipase C (PLC)-dependent mechanisms in this subset of mouse DRG neurons.
EXPERIMENTAL PROCEDURES
Animals and surgery
All procedures were approved by the Johns Hopkins University Animal Care and Use Committees as consistent with the National Institutes of Health Guide for the Use of Experimental Animals. Animals received food and water ad libidum and were housed on a 12-h day–night cycle in isolator cages.
MrgA3-eGFP-wild-type mice
We purchased a mouse BAC (bacterial artificial chromosome) clone (RP23-311C15) containing the entire MrgA3 gene from the Children’s Hospital Oakland Research Institute. The BAC clone was modified by homologous recombination in bacteria to generate the MrgA3 GFP-Cre transgenic line as described in our previous studies(Han et al., 2013; Li et al., 2014).
L5 spinal nerve ligation (SNL) model of neuropathic pain
Male Sprague-Dawley rats (200–350 g, Harlan, Indianapolis, IN) were anesthetized with 2% isoflurane. The left L5 spinal nerve was ligated with a 6-0 silk suture and cut distally as described in our previous studies(Guan et al., 2010b; He et al., 2014b). The muscle layer was closed with 4-0 chromic gut suture and the skin closed with metal clips.
Intrathecal catheter implantation
A saline-filled piece of PE-10 tubing (5–6 cm) was inserted into the intrathecal space of rats through a small slit in the atlanto-occipital membrane. After completing the experiment, we confirmed intrathecal drug delivery by injecting lidocaine (400 μg/20 μL, Hospira, Lake Forest, IL), which resulted in a temporary motor paralysis of the lower limbs.
Paw withdrawal threshold (PWT) test
Drug effects were tested in nerve-injured rats at the maintenance phase of neuropathic pain (4–5 weeks post-SNL)(Guan et al., 2008; Guan et al., 2010b). All behavioral tests were conducted in the morning by an experimenter blind to drug treatment conditions. We determined hypersensitivity to punctuate mechanical stimulation with the up-down method by applying a series of von Frey filaments (0.38–13.1 g) to the test area on the plantar surface of the hind paw for 4–6 s each. We calculated the PWT according to the formula provided by Dixon(Dixon, 1980). Rats that underwent SNL but did not develop mechanical hypersensitivity (>50% reduction of PWT from pre-SNL baseline) by day 5 and rats that showed impaired motor function or deteriorating health after treatment were eliminated from the subsequent behavioral studies, and data were not analyzed.
Culture of dissociated DRG neurons
Acutely dissociated DRG neurons from mice (3–4 weeks old) were collected in cold DH10 (90% DMEM/F-12, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, Invitrogen, Grand Island, NY) and treated with enzyme solution (5 mg/mL dispase, 1 mg/mL collagenase Type I in HPBS without Ca2+ and Mg2+, Invitrogen) at 37°C for 30 min. After trituration and centrifugation, cells were resuspended in DH10, plated on glass coverslips coated with poly-D-lysine and laminin, and cultured in an incubator (95% O2 and 5% CO2) at 37°C for 24 h with nerve growth factor (25 ng/mL) and glial cell-derived neurotrophic factor (50 ng/mL).
Transfecting MrgC11 gene into DRG neurons from Mrg-clusterΔ−/− mice
The MrgC11 gene, which is deleted in Mrg-clusterΔ−/− mice, has been cloned into mammalian expression vectors. MrgC11 expression constructs were electroporated into the Mrg mutant DRG neurons by using Mouse Neuron Nucleofector Kit (Amaxa Biosystems) as described in our previous studies (Liu et al., 2009; Li et al., 2014). By fusing GFP to the C-termini of MrgC11 coding sequences, we are able to visualize the transfected cells and proper membrane localization of MrgC11 during electrophysiologic recording. Previous studies have shown that GFP does not disturb normal function of the receptors(Dong et al., 2001; Han et al., 2002).
Whole-cell voltage-clamp recordings from DRG neurons
The coverslip was transferred into a perfusion chamber with extracellular solution (in mM: 145 TEA-Cl, 5 CaCl2, 0.8 MgCl2, 10 HEPES, 5 glucose; pH adjusted to 7.39 with TEA-OH, and osmolarity adjusted to 320 mOsm with sucrose). GFP-expressing neurons with cell body diameters between 22 and 25 μm were recorded in the whole-cell voltage-clamp configuration. The intracellular pipette solution contained (in mM): 135 CsCl, 1 CaCl2, 2 MgCl2, 1.5 MgATP, 0.3 Na2GTP, 11 EGTA, 10 HEPES, with pH of 7.3 (adjusted by CsCl) and osmolarity of 310 mOsm (adjusted by sucrose). Electrodes were pulled (Model pp-830, Narishige International USA, Inc. Long Island, NY) from borosilicate glass (WPI, Inc., Sarasota, FL) and had a resistance of 2–4 MΩ. Whole-cell currents were measured with an Axon 700B amplifier and the pCLAMP 9.2 software package (Molecular Devices, Sunnyvale, CA). Current traces were sampled at 10 kHz and low-pass filtered at 2 kHz. Low-voltage-activated (LVA) Ica was evoked at −40 mV (20 ms) from a holding potential of −80 mV, and HVA Ica was evoked at 10 mV (20 ms) from a holding potential of −60 mV. All experiments were performed at room temperature (~25°C). The liquid junction potential, cell membrane capacitance, and series resistance were electronically compensated. An experimenter blind to genotype and/or drug treatment conditions performed all electrophysiologic recordings.
Drugs
Stock solutions were freshly prepared as instructed by the manufacturer. BAM8-22, JHU58 (a dipeptide MrgC-selective agonist), pertussis toxin (PTX, a Gαi pathway blocker), cholera toxin (CTX, a Gαs pathway blocker), vasoactive intestinal polypeptide (VIP), morphine, DAMGO, and tetrodotoxin (TTX) were all diluted in saline or extracellular solution. U73122 (a PLC inhibitor), U73343 (an inactive analogue of U73122), and m-3M3FBS (a PLC activator) were initially dissolved in 50% dimethylsulfoxide (DMSO) in 0.9% sterile saline. PTX was added into DH10 cell culture medium for 18–24 h before recording and was also included in the intracellular solution. CTX was applied in the same way as PTX. BAM8-22 and JHU58 were synthesized by Johns Hopkins University. Other drugs were purchase from Sigma-Aldrich (St. Louis, MO) or Tocris Bioscience (Bristol, UK).
Data analysis
The methods for statistical comparisons in each study are given in the figure legends. The number of animals used in each study was based on our experience with similar studies and power analysis calculations. We randomized animals to the different treatment groups and blinded the experimenter to drug treatment to reduce selection and observation bias. After the experiments were completed, no data point was excluded. Representative data are from experiments that were replicated biologically at least three times with similar results. STATISTICA 6.0 software (StatSoft, Inc., Tulsa, OK) was used to conduct all statistical analyses. The Tukey honestly significant difference (HSD) post-hoc test was used to compare specific data points. Bonferroni correction was applied for multiple comparisons. Two-tailed tests were performed, and data are expressed as mean ± SEM; P<0.05 was considered significant in all tests.
RESULTS
JHU58 inhibition of HVA Ica in MrgA3-eGFP+ DRG neurons does not require Gαi pathway
Previous studies have shown that MrgC11 is the mouse receptor for JHU58 (Li et al., 2014; He et al., 2014b). Consistent with our previous findings (Li et al., 2014), bath application of JHU58 induced a quick and dose-dependent decrease of HVA ICa in MrgA3-eGFP+ DRG neurons that mostly co-express MrgC11 in wild-type mice (Fig. 1A–D). JHU58-induced inhibition diminished after washout with extracellular solution, suggesting that the drug action is reversible (Fig. 1B,C). MrgA3-eGFP+ DRG neurons exhibited small or virtually no LVA ICa (Fig. 1A). To block the Gαi pathway, we pre-incubated DRG neurons overnight with PTX (0.5 μg/mL) as in previous studies(Dolphin and Scott, 1989; Chen and Ikeda, 2004). PTX was also included in the intracellular solution during patch clamp recording of these neurons. Compared to the respective vehicle control (n=5), PTX treatment significantly reduced the inhibition of HVA ICa by morphine (100 nM, n=11; Fig. 1B–D). PTX treatment also reduced the inhibition of HVA ICa by DAMGO (100 nM), a highly selective mu-opioid receptor agonist, in MrgA3-eGFP+ neurons. DAMGO decreased HVA ICa by 25.9 ± 1.9% (n=5) in vehicle-pretreated neurons but by only 15.7 ± 2.6% (n=4) in PTX-treated neurons. These findings indicate that PTX effectively blocked Gαi pathways in DRG neurons. However, the same PTX treatment did not significantly reduce the ability of JHU58 (0.001–100 nM, n=6–12/dose, one dose/neuron) to inhibit HVA ICa in MrgA3-eGFP+ neurons, as compared to the control (n=8–12/group; Fig. 1D).
Fig. 1. Pertussis toxin (PTX) does not block JHU58-induced inhibition of high-voltage-activated (HVA) calcium currents (ICa) in MrgA3-eGFP+ wild-type DRG neurons.

(A) Upper: Image of an MrgA3-eGFP+ DRG neuron (arrow) visible in cell culture by co-expressed green fluorescent protein. Lower: Traces of small low-voltage-activated (LVA) ICa (−80 to −40 mV) and large HVA ICa (−60 to 10 mV, 20 ms) evoked by depolarization before and after bath application of JHU58 (100 nM). (B) MrgA3-eGFP+ DRG neurons were incubated overnight with PTX (left) or vehicle (control, right). The traces show HVA ICa evoked by depolarization after bath application of JHU58 (100 nM) and morphine (100 nM), as well as HVA ICa before drug treatment (baseline). (C) Time course of changes in HVA ICa amplitude after JHU58 and morphine in DRG neurons pretreated with PTX (left) or vehicle (right). (D) Incubation of DRG neurons overnight with PTX (0.5 μg/ml) significantly decreased the inhibition of HVA ICa by morphine (100 nM) but did not affect the dose-dependent inhibition of HVA ICa by JHU58 (0.001–100 nM, n=8–12/dose), as compared to that in the control group. The number of neurons in each group is indicated in the figure. Two-way ANOVA.
BAM8-22 inhibition of HVA Ica in MrgC11-GFP+ DRG neurons does not require Gαi pathway
BAM8-22 is a large peptide agonist to both MrgC11 and human homolog MrgX1 and inhibits HVA ICa in rat DRG neurons that heterologously express MrgX1. Because this effect of BAM8-22 was reported to be sensitive to PTX (Chen and Ikeda, 2004), we next examined whether inhibition of HVA Ica by BAM8-22–induced activation of MrgC11 also involves the Gαi pathway. The Mrg-mutant DRG neurons were transfected with MrgC11 and can be visualized by co-expressed GFP for recording (Fig. 2A). As in our previous study (Li et al., 2014), bath application of BAM8-22 (3 μM) significantly inhibited HVA Ica in MrgC11-transfected cells, but not in non-transfected cells. The peak inhibitory effect usually occurred at 60 s after drug application (Fig. 2B). Importantly, the reductions in HVA ICa induced by BAM8-22 (3 μM) were similar in MrgC11-transfected neurons pretreated with PTX (0.5 μg/mL, n=12, ICa inhibition: 58.1 ± 3.9% of pre-drug) and those pretreated with vehicle (control, n=9, ICa inhibition: 55.8 ± 7.8% of pre-drug; Fig. 2C), suggesting that PTX did not block the BAM8-22–induced inhibition of HVA ICa.
Fig. 2. Pertussis toxin (PTX) does not reduce BAM8-22–induced inhibition of high-voltage-activated (HVA) calcium currents (ICa) in MrgC11-transfected DRG neurons.
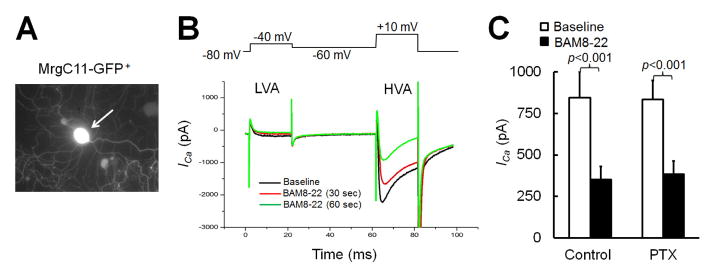
(A) An MrgC11-transfected DRG neuron from an Mrg mutant mouse is visible by co-expressed GFP in cell culture. (B) MrgC11 restored responsiveness of Mrg mutant DRG neurons to BAM8-22, as measured by the ability of BAM8-22 to attenuate HVA ICa. Traces show small low-voltage-activated (LVA) ICa (−80 to −40 mV) and large HVA ICa (−60 to 10 mV, 20 ms) evoked by depolarization before (baseline) and after bath application of BAM8-22 (3 μM). (C) BAM8-22 (3 μM) induced similar peak inhibition of HVA ICa in MrgC11-transfected DRG neurons pretreated with PTX (0.5 μg/ml, n=12) and vehicle (control, n=9). Paired t-test.
The inhibition of HVA ICa in MrgA3-eGFP+ DRG neurons by JHU58 involves PLC-dependent mechanisms
MrgC11 also couples with the Gαq/11 pathway(Han et al., 2002; Chen and Ikeda, 2004), which works by activating PLC. We next examined whether the JHU58-induced inhibition of HVA ICa in MrgA3-eGFP+ DRG neurons involves PLC-dependent mechanisms. In MrgA3-eGFP+ neurons, inhibition of HVA ICa by different doses of JHU58 (0.001–100 nM, n=4–9/dose, one dose/neuron) was significantly reduced by acute pretreatment with a PLC inhibitor (U73122, 1 μM, 2 min, bath application; Fig. 3A,B), but not by pretreatment with the vehicle of U73122. Importantly, pretreatment with U73343 (1 μM), an inactive analogue of U73122, did not affect JHU58 inhibition of HVA ICa (Fig. 3D). Bath application of PLC activator m-3M3FBS (50 μM) inhibited HVA ICa in the absence of JHU58 (Fig. 3D). Extracellular solution (vehicle of JHU58) did not affect HVA ICa.
Fig. 3. JHU58-induced inhibition of high-voltage-activated (HVA) calcium currents (ICa) in MrgA3-eGFP+ wild-type DRG neurons involves phospholipase C (PLC)-dependent mechanisms.

(A) MrgA3-eGFP+ DRG neurons were pretreated with U73122 (1 μM) or vehicle and then treated with JHU58 (100 nM) by bath application. The traces show HVA ICa at baseline and after depolarization (−60 to 10 mV, 20 ms). (B) Time course of changes in HVA ICa amplitude after JHU58 (100 nM) in MrgA3-eGFP+ DRG neurons pretreated with and without U73122 (1 μM, bath application, 2 min). (C) Blocking the PLC pathway by acute pretreatment with U73122 significantly reduced JHU58-induced inhibition of HVA ICa in MrgA3-eGFP+ wild-type DRG neurons, as compared to that in vehicle-pretreated neurons. Drug treatment groups were compared to vehicle control with two-way ANOVA. (D) Upper: Sample traces of HVA ICa before (baseline) and after bath application of JHU58 (100 nM), pretreatment with U73343 (an inactive analogue of U73122, 1 μM) followed by bath application of JHU58, and bath application of m-3M3FBS (a PLC activator, 50 μM). Cells were held at −60 mV and were depolarized to +10 mV for 20 ms to evoke HVA ICa. Lower: Quantification of HVA ICa inhibition by JHU58, U73343-JHU58, and m-3M3FBS. In each case, current inhibition was normalized to the pre-drug baseline. The number of neurons in each group is indicated in the figure.
The inhibition of HVA ICa in MrgA3-eGFP+ DRG neurons by JHU58 is not voltage-dependent
HVA ICa inhibition by G-protein-coupled receptor activation can involve agonist-induced release of Gβγ subunits, in addition to release of Gα subunit. Therefore, we examined whether inhibition of HVA ICa by JHU58 (100 nM) in MrgA3-eGFP+ neurons can be reduced by acute pretreatment with a Gβγ blocker (gallein, 100 μM, 2 min, bath application, n=8; Fig. 4A,B). Unexpectedly, gallein alone gradually decreased HVA ICa before JHU58 was applied (Fig. 4C). Under these conditions, gallein attenuated the JHU58-induced decrease in HVA ICa amplitude (as a percent of pre-drug baseline; Fig. 4C).
Fig. 4. Effects of gallein and prepulse stimulation on JHU58-induced inhibition of high-voltage-activated (HVA) calcium currents (ICa) in MrgA3-eGFP+ wild-type DRG neurons.

(A) MrgA3-eGFP+ wild-type DRG neurons were treated with JHU58 (100 nM, bath application) before and after acute treatment with a Gβγ blocker (gallein, 100 μM, 2 min, bath application). The traces show HVA ICa at baseline and after depolarization (−60 to 10 mV, 20 ms). (B) Time course of changes in HVA ICa amplitude induced by JHU58 applied before and after acute treatment with gallein. Note that HVA ICa amplitude gradually decreased after gallein treatment. (C) Compared to that before gallein, JHU58-induced inhibition of HVA ICa (n=8) was reduced after gallein. Paired t-test. (D) A sample recording trace of the paired-pulse protocol used to examine the voltage dependence of JHU58-induced inhibition of HVA ICa. A prepulse (+100 mV, 50 ms) was inserted between two depolarization test pulses as previously described (−60 mV to +10 mV). (E) Quantification of HVA ICa inhibition by JHU58 before and after the prepulse. HVA ICa inhibition by JHU58 was normalized to baseline values.
To determine if the HVA ICa inhibition by JHU58 is voltage-dependent, we examined whether application of a strongly depolarizing prepulse reverses the inhibition by JHU58. In a paired-pulse protocol, a prepulse (+100 mV, 50 ms) was applied between two depolarization test pulses as previously described (−60 mV to +10 mV, Fig. 4D).The depolarizing prepulse may convert HVA calcium channels inhibited through the membrane-delimited pathway from the “willing” to the “reluctant” mode. However, the prepulse did not decrease the JHU58-induced HVA ICa inhibition (Fig. 4E).
JHU58 inhibition of HVA Ica in MrgA3-eGFP+ DRG neurons does not involve Gαs pathway
To block the Gαs pathway, we pre-incubated neurons overnight with CTX (0.5 μg/mL)(Zhu and Ikeda, 1994). CTX was also included in the intracellular solution during patch clamp recording. Compared to the control (vehicle of CTX, n=4), CTX significantly reduced the inhibition of HVA ICa by VIP (5 μM, n=9, Fig. 5A–C), suggesting that the toxin treatment was effective. However, CTX did not reduce JHU58 (0.001–100 nM)-induced inhibition of HVA ICa (n=6–12/dose, one dose/neuron, Fig. 5C).
Fig. 5. Cholera toxin (CTX) does not block JHU58-induced inhibition of high-voltage-activated (HVA) calcium currents (ICa) in MrgA3-eGFP+ wild-type DRG neurons.

(A) MrgA3-eGFP+ DRG neurons were pretreated with CTX (0.5 μg/ml) or vehicle (control). The traces show HVA ICa evoked by depolarization after bath application of JHU58 (100 nM) and vasoactive intestinal polypeptide (VIP, 5 μM). The trace of HVA ICa before drug treatment (baseline) is also shown. (B) Time course of changes in HVA ICa amplitude after application of JHU58 (100 nM) and VIP (5 μM) in neurons pretreated with CTX (0.5 μg/mL, left) or vehicle (right) overnight. (C) Incubation of neurons overnight with CTX significantly decreased the inhibition of HVA ICa by VIP (5 μM) but did not affect the dose-dependent inhibition of HVA ICa by JHU58 (0.001–100 nM, n=6–12/dose), as compared to that of the control group. The number of neurons in each group is indicated in the figure. Two-way ANOVA.
U73122 reduces pain inhibition by intrathecal JHU58
We next examined whether pain inhibition produced by intrathecal JHU58 injection is also dependent on activation of the PLC pathway in vivo. At the peak to maintenance phase of neuropathic pain (day 14–21 post-SNL), rats pretreated with vehicle (n=6) exhibited significantly increased ipsilateral PWT at 30 min after JHU58 (0.1 mM, 15 μL) injection. However, pretreatment of rats with an intrathecal injection of low-dose U73122 (1 μg/10 μL, n=6) blocked this JHU58-induced pain inhibition (Fig. 6A,B). U73122 at this dose did not significantly affect PWT at 30 min post-injection, as compared to that of vehicle-treated rats.
Fig. 6. U73122 reduces JHU58-induced inhibition of neuropathic mechanical hypersensitivity.
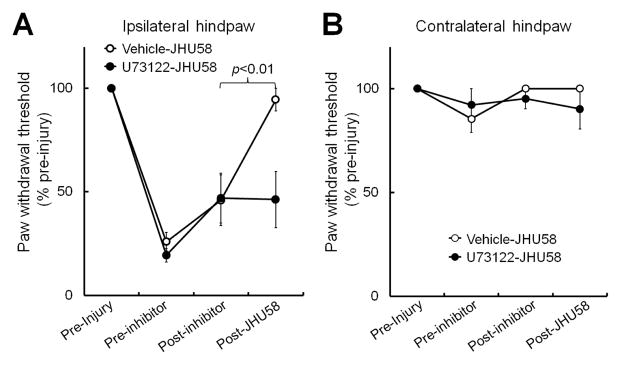
At 14–21 days after spinal nerve ligation, rats were administered a low dose of U73122 (1 μg/10μL, n=6) by intrathecal injection. (A) U73122 did not significantly change the ipsilateral paw withdrawal threshold (PWT) at 30 min after injection, as compared to that after vehicle treatment (n=6). However, U73122 pretreatment blocked pain inhibition by the subsequent intrathecal injection of JHU58 (0.1 mM, 15 μL). The ipsilateral PWT at 30 min after JHU58 injection was significantly increased in animals that received vehicle pretreatment, but not in those pretreated with U73122. (B) Contralateral PWT was not significantly altered after drug treatment. One-way repeated measures ANOVA.
DISCUSSION
Our previous studies suggested that MrgC agonists inhibit HVA Ica in DRG neurons and attenuate synaptic transmission in superficial dorsal horn of mice (Li et al., 2014; He et al., 2014b). These actions may contribute to the pain inhibitory effects of an MrgC agonist when it is delivered intrathecally (Chen et al., 2006; Guan et al., 2010a; He et al., 2014b). Yet, how MrgC activation leads to inhibition of downstream HVA calcium channels remains an open question. Here, we used pharmacologic approaches to show for the first time that MrgC agonists inhibit HVA ICa predominantly through Gαq/11-dependent mechanisms, but not through Gαi or Gαs pathways, in mouse DRG neurons. Additionally, we found that HVA ICa inhibition is not voltage-dependent.
As has been shown in other neuronal types (Bean, 1989; Hille, 1994), we showed that HVA calcium channels in mouse MrgA3-eGFP+ DRG neurons may be modulated by various G-protein-dependent mechanisms (Gαi: morphine and DAMGO; Gαq/11: JHU58; Gαs: VIP; Gβγ: gallein). HVA calcium channels, especially the N-type, are important to producing the heightened neurotransmitter release from central terminals of DRG neurons (Braunwald, 1982; Delmas et al., 2000; McGivern, 2006). Our previous study suggested that JHU58, an MrgC agonist, selectively and dose-dependently inhibits N-type, but not L- or P/Q-type, HVA calcium channels in DRG neurons (Li et al., 2014). Mu-opioid receptor agonists, such as morphine, may inhibit spinal nociceptive transmission at least in part by inhibiting N-type channels. Morphine acts predominantly through the Gαi pathway to exert this and other intracellular effects (Surratt and Adams, 2005; Pan et al., 2008; Kanbara et al., 2014). In our study, PTX, which inhibits the Gαi pathway, reduced both morphine- and DAMGO-induced inhibition of HVA ICa in MrgA3-eGFP+ DRG neurons. A previous study showed that activation of exogenously expressed human MrgX1 receptors inhibited HVA ICa in rat DRG neurons and that this effect was also dependent on the Gαi pathway (Chen and Ikeda, 2004). MrgC is a functional homolog of human MrgX1(Dong et al., 2001; Lembo et al., 2002), and HVA calcium channels are important downstream molecular targets of the MrgC receptor. Yet, our findings suggest that the Gαi pathway may not contribute to JHU58-induced inhibition of HVA ICa in MrgA3-eGFP+ DRG neurons that co-express MrgC11, as the drug effect was not reduced by PTX. The reason for the discrepancies in Gα-protein coupling between our finding and that of the previous study is unclear, but it is possible that MrgC11 and human MrgX1 modulate HVA calcium channels through different Gα pathways, owing to the species difference. We also do not know whether overexpression of exogenous MrgX1 in rodent DRG neurons by recombinant adenoviruses might alter activation of Gα proteins or the coupling of Gα proteins to downstream targets (e.g., calcium channels). Finally, although MrgA3-eGFP+ neurons have facilitated our ability to record from MrgC11-bearing DRG neurons, which were previously difficult to identify, it should be noted that MrgC11 is also present in DRG neurons that do not co-express MrgA3 (Liu et al., 2009). The Gα protein that contributes to inhibition of HVA calcium channels after MrgC11 activation in these neurons remains to be determined (Han et al., 2013).
Our previous study showed that MrgC11, but not MrgA3 or other Mrg subtypes, is the mouse receptor for JHU58 and is essential to JHU58–induced inhibition of HVA ICa. JHU58 inhibited HVA ICa in eGFP+ DRG neurons of MrgA3-eGFP-wild-type mice, but not in those from MrgA3-eGFP-Mrg mutant mice, which express only the reporter gene product eGFP (Li et al., 2014). Further, PTX did not block the inhibition of HVA ICa by BAM8-22, another potent agonist of MrgC and MrgX1, in Mrg mutant DRG neurons transfected with MrgC11. However, the same PTX treatment significantly reduced the morphine- and DAMGO-induced inhibition of HVA ICa in MrgA3-eGFP+ neurons, suggesting that PTX effectively blocked the Gαi pathway. In contrast to PTX, a nonselective PLC inhibitor, U73122, did block the inhibition of HVA ICa by JHU58. We further characterized the role of the PLC pathway in JHU58-induced inhibition of HVA ICa by testing U73343 and m-3M3FBS. U73343, an inactive analogue of U73122, did not block the inhibitory effects of JHU58 on HVA ICa, whereas m-3M3FBS, a PLC activator that stimulates Ca2+ release and inositol phosphate formation in the cell, reduced HVA ICa. Together, these findings suggest that activation of PLC by MrgC agonism may lead to inhibition of HVA calcium. Importantly, intrathecal injection of low-dose U73122, which by itself did not significantly affect animal pain behavior, blocked JHU58-induced inhibition of mechanical hypersensitivity in rats after nerve injury. A previous study showed that intrathecal injection of low-dose U73122 also attenuated the analgesia induced by intrathecal injection of a delta-opioid receptor agonist (Narita et al., 2000). Our findings suggest that the pain inhibition by intrathecal JHU58 is dependent on activation of the PLC pathway. Nevertheless, we cannot rule out the possibility that, in our experimental setting, the effect of U73122 in vivo may also involve an unknown postsynaptic mechanism, such as modulation of dorsal horn neurons (Narita et al., 2000; Shi et al., 2008). Details regarding involvement of the PLC-IP3 pathway in MrgC agonist-produced cellular effects still need to be determined. Future studies may examine whether inhibiting other downstream effectors of the Gαq/11 pathway, such as by depleting the intracellular calcium store with thapsigargin and blocking IP3 receptor with heparin, also reduce JHU58-induced inhibition of HVA ICa, as U73122 does. However, in our experimental setting, ICa fluctuates substantially in neurons treated with thapsigargin or heparin, thereby preventing us from examining their effects on JHU58-induced inhibition of HVA ICa. Nevertheless, the molecular mechanisms that underlie Gαq/11-mediated modulation of HVA calcium channels in DRG neurons warrant future study.
We also examined the role of Gαs and Gβγ in JHU58-induced inhibition of HVA ICa. Inhibiting Gαs with CTX did not block JHU58-induced inhibition of HVA ICa. Although acute treatment with the Gβγ blocker gallein partially reduced the inhibition of HVA ICa by JHU58 from pre-JHU58 level, gallein itself gradually decreased HVA ICa amplitude without showing a recovery before JHU58 testing. Under these conditions, it is difficult to interpret the role of Gβγ in MrgC agonism-induced inhibition of HVA ICa, which may be complex. Thus, additional studies are required. Finally, we tested whether the inhibition of HVA ICa after MrgC activation might also occur through voltage-dependent pathways. However, this was apparently not the case, as the effect of JHU58 was not reduced by a strong depolarizing prepulse.
Modulation of HVA calcium channels via G-protein-coupled receptor represents an important mechanism for regulating neurotransmitter release and synaptic plasticity. The reduction of HVA ICa, after activation of MrgC receptors may be an important cellular mechanism that leads to pain inhibition. Our previous studies showed a decrease in the frequency but not the amplitude of mEPSC in substantia gelatinosa neurons in the presence of MrgC agonists (He et al., 2014b). Further, we observed an increase in the paired-pulse ratio and a decrease in the amplitude of evoked-EPSCs after MrgC agonist treatment (Li et al., 2014). These findings indicate that excitatory presynaptic transmission is depressed in the substantia gelatinosa, perhaps by a reduction in the probability of glutamate release from the primary afferent terminals. Nevertheless, mechanisms for MrgC agonist-induced pain inhibition in vivo remain largely unclear, especially under pathologic pain conditions.
Nerve injury may induce complex changes in the signaling pathway that elicits MrgC agonism-induced pain inhibition. For example, after an L5 SNL, MrgC expression is upregulated in neurons at the uninjured L4 DRG that transmits afferent sensory inputs from the periphery (He et al., 2014a). Additionally, N-type calcium channel expression is increased in chronic pain conditions (Luo et al., 2002) and may contribute to sustained neuronal firing and enhanced neurotransmitter release into spinal cord (Braunwald, 1982; Delmas et al., 2000; McGivern, 2006). It is possible that MrgC signals through two opposing pathways (Gαi and Gαq/11) and hence causes different cellular effects (Han et al., 2002). It remains to be determined if the Gαi pathway contributes to MrgC agonism-induced pain inhibition through other mechanisms, such as by inhibition of adenylyl cyclase. Future studies are needed to examine whether Gαi and Gαq/11 coupling with MrgC changes after tissue inflammation or nerve injury.
CONCLUSIONS
This study suggests that HVA calcium channels may be inhibited through a Gαq/11-dependent mechanism by the activation of MrgC11 in native mouse DRG neurons. The MrgC gene shares substantial homogeneity with the human MrgX1 gene(Dong et al., 2001; Lembo et al., 2002; Zylka et al., 2003). Nevertheless, because of species differences, the function of human homolog MrgX1 may not be fully inferred from studying rodent MrgC. It is challenging to record from DRG neurons of nonhuman primates, which do express MrgX1. Therefore, developing transgenic mice that express MrgX1 under the endogenous MrgC11 promoter may be an alternative strategy that can provide critical insights about the functions of MrgX1 in native DRG neurons.
Highlights.
The inhibition of HVA ICa in DRG neurons by an MrgC agonist (JHU58) was reduced by a phospholipase C inhibitor, U73122.
Blocking the Gαi and Gαs pathways did not reduce the JHU58-induced inhibition of HVA ICa in DRG neurons.
A depolarizing prepulse did not reduce the inhibition of HVA ICa by JHU58.
Intrathecal U73122 reduced the inhibition of mechanical hypersensitivity by JHU58 in nerve-injured rats.
Acknowledgments
The authors thank Claire F. Levine, MS (scientific editor, Department of Anesthesiology/CCM, Johns Hopkins University), for editing the manuscript and Yixun Geng for mouse genotyping and maintenance. This study was mainly supported by NIH grants NS70814 (Y.G.) and NS54791 (X.D.). This study was subsidized by grant NS26363 (S.N.R.) and grants from the Johns Hopkins Blaustein Pain Research Fund (Y.G.) and the National Natural Science Foundation of China: 81428008 (Y.W.). X.D. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
Author contributions
Z.L., S-Q. H., and P-Y.T. performed most of the experiments and were involved in writing a draft manuscript. Q.X., V.T., Y.F., B.S., and T.Z. also conducted some of the electrophysiology, molecular, and behavioral experiments. Z.T., S.N.R., and Y.W. were involved in experimental design and data analysis. Y.G. and X.D. designed and directed the project and wrote the final manuscript.
The work was conducted in the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University, School of Medicine, Baltimore, MD, USA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abernethy DR, Schwartz JB. Calcium-antagonist drugs. N Engl J Med. 1999;341:1447–1457. doi: 10.1056/NEJM199911043411907. [DOI] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Braunwald E. Mechanism of action of calcium-channel-blocking agents. N Engl J Med. 1982;307:1618–1627. doi: 10.1056/NEJM198212233072605. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ. Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J Neurosci. 2008;28:10943–10951. doi: 10.1523/JNEUROSCI.3594-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ikeda SR. Modulation of ion channels and synaptic transmission by a human sensory neuron-specific G-protein-coupled receptor, SNSR4/mrgX1, heterologously expressed in cultured rat neurons. J Neurosci. 2004;24:5044–5053. doi: 10.1523/JNEUROSCI.0990-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Cai Q, Hong Y. Intrathecal sensory neuron-specific receptor agonists bovine adrenal medulla 8–22 and (Tyr6)-gamma2-MSH-6-12 inhibit formalin-evoked nociception and neuronal Fos-like immunoreactivity in the spinal cord of the rat. Neuroscience. 2006;141:965–975. doi: 10.1016/j.neuroscience.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Buckley NJ, Brown DA. Calcium channel gating and modulation by transmitters depend on cellular compartmentalization. Nat Neurosci. 2000;3:670–678. doi: 10.1038/76621. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Scott RH. Modulation of Ca2+-channel currents in sensory neurons by pertussis toxin-sensitive G-proteins. Ann N Y Acad Sci. 1989;560:387–390. doi: 10.1111/j.1749-6632.1989.tb24117.x. [DOI] [PubMed] [Google Scholar]
- Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Grazzini E, Puma C, Roy MO, Yu XH, O’Donnell D, Schmidt R, Dautrey S, Ducharme J, Perkins M, Panetta R, Laird JM, Ahmad S, Lembo PM. Sensory neuron-specific receptor activation elicits central and peripheral nociceptive effects in rats. Proc Natl Acad Sci U S A. 2004;101:7175–7180. doi: 10.1073/pnas.0307185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Johanek LM, Hartke TV, Shim B, Tao YX, Ringkamp M, Meyer RA, Raja SN. Peripherally acting mu-opioid receptor agonist attenuates neuropathic pain in rats after L5 spinal nerve injury. Pain. 2008;138:318–329. doi: 10.1016/j.pain.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Liu Q, Tang Z, Raja SN, Anderson DJ, Dong X. Mas-related G-protein-coupled receptors inhibit pathological pain in mice. Proc Natl Acad Sci U S A. 2010a;107:15933–15938. doi: 10.1073/pnas.1011221107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Wacnik PW, Yang F, Carteret AF, Chung CY, Meyer RA, Raja SN. Spinal cord stimulation-induced analgesia: electrical stimulation of dorsal column and dorsal roots attenuates dorsal horn neuronal excitability in neuropathic rats. Anesthesiology. 2010b;113:1392–1405. doi: 10.1097/ALN.0b013e3181fcd95c. [DOI] [PubMed] [Google Scholar]
- Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, Kim Y, Nie H, Qu L, Patel KN, Li Z, McNeil B, He S, Guan Y, Xiao B, LaMotte RH, Dong X. A subpopulation of nociceptors specifically linked to itch. Nat Neurosci. 2013;16:174–182. doi: 10.1038/nn.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Dong X, Hwang JI, Zylka MJ, Anderson DJ, Simon MI. Orphan G protein-coupled receptors MrgA1 and MrgC11 are distinctively activated by RF-amide-related peptides through the Galpha q/11 pathway. Proc Natl Acad Sci U S A. 2002;99:14740–14745. doi: 10.1073/pnas.192565799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SQ, Han L, Li Z, Xu Q, Tiwari V, Yang F, Guan X, Wang Y, Raja SN, Dong X, Guan Y. Temporal changes in MrgC expression after spinal nerve injury. Neuroscience. 2014a;261:43–51. doi: 10.1016/j.neuroscience.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SQ, Li Z, Chu YX, Han L, Xu Q, Li M, Yang F, Liu Q, Tang Z, Wang Y, Hin N, Tsukamoto T, Slusher B, Tiwari V, Shechter R, Wei F, Raja SN, Dong X, Guan Y. MrgC agonism at central terminals of primary sensory neurons inhibits neuropathic pain. Pain. 2014b;155:534–544. doi: 10.1016/j.pain.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hille B, Beech DJ, Bernheim L, Mathie A, Shapiro MS, Wollmuth LP. Multiple G-protein-coupled pathways inhibit N-type Ca channels of neurons. Life Sci. 1995;56:989–992. doi: 10.1016/0024-3205(95)00038-8. [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Wang D, Zhou X, Huo Y, Chen T, Hu F, Quirion R, Hong Y. Effect of Mas-related gene (Mrg) receptors on hyperalgesia in rats with CFA-induced inflammation via direct and indirect mechanisms. Br J Pharmacol. 2013 doi: 10.1111/bph.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbara T, Nakamura A, Takasu K, Ogawa K, Shibasaki M, Mori T, Suzuki T, Hasegawa M, Sakaguchi G, Kanemasa T. The Contribution of Gi/o Protein to Opioid Antinociception in an Oxaliplatin-Induced Neuropathy Rat Model. J Pharmacol Sci. 2014;126:264–273. doi: 10.1254/jphs.14133fp. [DOI] [PubMed] [Google Scholar]
- Lembo PM, Grazzini E, Groblewski T, O’Donnell D, Roy MO, Zhang J, Hoffert C, Cao J, Schmidt R, Pelletier M, Labarre M, Gosselin M, Fortin Y, Banville D, Shen SH, Strom P, Payza K, Dray A, Walker P, Ahmad S. Proenkephalin A gene products activate a new family of sensory neuron--specific GPCRs. Nat Neurosci. 2002;5:201–209. doi: 10.1038/nn815. [DOI] [PubMed] [Google Scholar]
- Li Z, He SQ, Xu Q, Yang F, Tiwari V, Liu Q, Tang Z, Han L, Chu YX, Wang Y, Hin N, Tsukamoto T, Slusher B, Guan X, Wei F, Raja SN, Dong X, Guan Y. Activation of MrgC receptor inhibits N-type calcium channels in small-diameter primary sensory neurons in mice. Pain. 2014 doi: 10.1016/j.pain.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, Undem BJ, Kollarik M, Chen ZF, Anderson DJ, Dong X. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- McGivern JG. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov Today. 2006;11:245–253. doi: 10.1016/S1359-6446(05)03662-7. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Matsuzawa Y, Makita K, Katayama Y. Antimigraine drug, zolmitriptan, inhibits high-voltage activated calcium currents in a population of acutely dissociated rat trigeminal sensory neurons. Mol Pain. 2006;2:10. doi: 10.1186/1744-8069-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Ohsawa M, Mizoguchi H, Aoki T, Suzuki T, Tseng LF. Role of the phosphatidylinositol-specific phospholipase C pathway in delta-opioid receptor-mediated antinociception in the mouse spinal cord. Neuroscience. 2000;99:327–331. doi: 10.1016/s0306-4522(00)00202-5. [DOI] [PubMed] [Google Scholar]
- Pan HL, Wu ZZ, Zhou HY, Chen SR, Zhang HM, Li DP. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol Ther. 2008;117:141–161. doi: 10.1016/j.pharmthera.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi TJ, Liu SX, Hammarberg H, Watanabe M, Xu ZQ, Hokfelt T. Phospholipase C{beta}3 in mouse and human dorsal root ganglia and spinal cord is a possible target for treatment of neuropathic pain. Proc Natl Acad Sci U S A. 2008;105:20004–20008. doi: 10.1073/pnas.0810899105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Wachtel RE, Gebhart GF. Inhibition of calcium currents in rat colon sensory neurons by K- but not mu- or delta-opioids. J Neurophysiol. 1998;80:3112–3119. doi: 10.1152/jn.1998.80.6.3112. [DOI] [PubMed] [Google Scholar]
- Surratt CK, Adams WR. G protein-coupled receptor structural motifs: relevance to the opioid receptors. Curr Top Med Chem. 2005;5:315–324. doi: 10.2174/1568026053544533. [DOI] [PubMed] [Google Scholar]
- Wang D, Chen T, Yang J, Couture R, Hong Y. Activation of Mas-related gene (Mrg) C receptors enhances morphine-induced analgesia through modulation of coupling of mu-opioid receptor to Gi-protein in rat spinal dorsal horn. Neuroscience. 2013 doi: 10.1016/j.neuroscience.2013.08.069. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin-sensitive pathway. Neuron. 1994;13:657–669. doi: 10.1016/0896-6273(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Zylka MJ, Dong X, Southwell AL, Anderson DJ. Atypical expansion in mice of the sensory neuron-specific Mrg G protein-coupled receptor family. Proc Natl Acad Sci U S A. 2003;100:10043–10048. doi: 10.1073/pnas.1732949100. [DOI] [PMC free article] [PubMed] [Google Scholar]