

Abstract
The reactivity between a thiolate-ligated five-coordinate complex [FeII(SMe2N4(tren))]+ (1) and dioxygen is examined in order to determine if O2 activation, resembling that of the metalloenzyme cytochrome P450, can be promoted even when O2 binds cis, as opposed to trans, to a thiolate. Previous work in our group showed that [FeII(SMe2N4-(tren))]+ (1) reacts readily with superoxide (O2−) in the presence of a proton source to afford H2O2 via an FeIII–OOH intermediate, thus providing a biomimetic model for the metalloenzyme superoxide reductase (SOR). Addition of O2 to 1 affords binuclear μ-oxo-bridged [FeIII(SMe2N4(tren))]2(μ2-O)(PF6)2•3MeCN (3). At low temperatures, in protic solvents, an intermediate is detected, the details of which will be the subject of a separate paper. Although the thiolate ligand does not appear to perturb the metrical parameters of the unsupported μ-oxo bridge (Fe–O=1.807(8) Å, and Fe–O–Fe= 155.3(5)° fall in the usual range), it decreases the magnetic coupling between the irons (J = −28 cm−1) and creates a rather basic oxo site. Protonation of this oxo using strong (HBF4, HCl) or weak (HOAc, NH4PF6, LutNHCl) acids results in bridge cleavage to cleanly afford the corresponding monomeric anion-ligated (OAc− (6), or Cl− (7)) or solvent-ligated (MeCN (4)) derivatives. Addition of OH− converts [FeIII(SMe2N4-(tren))(MeCN)]2+ (4) back to μ-oxo 3. Thus, μ-oxo bridge cleavage is reversible. The protonated μ-hydroxo-bridged intermediate is not observed. In an attempt to prevent μ-oxo dimer formation, and facilitate the observation of O2-bound intermediates, a bulkier tertiary amine ligand, tren-Et4= N-(2-amino-ethyl)-N-(2-diethylamino-ethyl)-N′,N′-diethyl-ethane-1,2-diamine, and the corresponding [FeII(SMe2N4(tren-Et4))]+ (5) complex was synthesized and structurally characterized. Steric repulsive interactions create unusually long FeII-N(3,4) amine bonds in 5 (mean distance = 2.219(1) Å). The [(tren-Et4)N4SMe2]1− ligand is unable to accommodate iron in the +3 oxidation state, and consequently, in contrast to most thiolate-ligated Fe(II) complexes, [FeII(SMe2N4(tren-Et4))]+ (5) does not readily react with O2. Oxidation of 5 is irreversible, and the potential (Epa = +410 mV (vs SCE)) is anodically shifted relative to 1 (E1/2 = −100 mV (vs SCE))
The bioinorganic chemistry of iron is rich and diverse,1–7 catalyzing reactions that range from O2-transport and activation, to hydrogen atom abstraction, alkane functionalization, and substrate hydrolysis. Non-heme iron enzymes are involved in a number of important biosynthetic pathways including DNA synthesis and repair,1,8 serotonin,9 leukotriene,1,10–12 plant hormone,13,14 and antibiotic synthesis.4,14 Cysteinate-ligated non-heme iron enzymes such as nitrile hydratase,6,15 superoxide reductase,16–23 and peptide deformylase24,25 represent a new class of these enzymes.6 Superoxide reductases (SORs)16–23 are non-heme iron enzymes that contain iron in an environment that is structurally very similar to that of the heme-enzyme cytochrome P450 (Scheme 1).26–29 In the catalytically active state, both have four equatorial sp2-nitrogens (histidine (SOR)16,23 or pyrrole (cyt P450) nitrogens), and an apical cysteinate trans to an open coordination site. Both enzymes react with oxygen-derived substrates to afford related intermediates. Cytochrome P450 reacts with dioxygen (plus an electron and proton; O2 + H+ + e− = HO2) to afford an FeIII–OOH intermediate, which undergoes O–O bond cleavage to afford a high-valent Fe–oxo upon the addition of protons.26–31 Superoxide reductase (SOR) reacts with superoxide (plus a proton = HO2) to afford an FeIII–OOH intermediate, which undergoes Fe–O bond cleavage to afford H2O2 (Scheme 2a).17–19,21 Why the O–O bond cleaves with cyt P450,26,27 whereas the Fe–O bond cleaves with SOR is a question worth addressing. Site specific delivery of protons to the proximal as opposed to distal oxygen would be expected to favor Fe–O bond cleavage.27 However, it is difficult to imagine how site-directed proton delivery would occur with SOR when the metal ion is sitting on the protein surface exposed to solvent. Spin-state differences might contribute to differences in the O–O versus Fe–O bond cleaving properties,32 as well as differences in the ligand environment. The P450 FeIII–OOH is S = 1/2,28 whereas the SOR FeIII-OOH is believed to be S = 5/2.17 Antibonding electrons present in the high-spin (S = 5/2) structure would weaken the Fe–O bond relative to the low-spin structure. A conjugated porphyrin ligand would help to stabilize the high-valent “FeV═O” intermediate that would result from O–O bond cleavage. A non-heme environment, although more flexible,33 is less capable of supporting a higher-valent “FeV═ O” oxidation catalyst34 (although it would be capable of supporting an FeIV═O).7,35,36 The cysteinate present both in the SOR and P450 active sites would also contribute to the stabilization of a high-valent Fe═O.37 Thiolates have been shown to be quite effective at stabilizing iron in higher oxidation states.6 The positioning of the P450 cysteinate trans to the dioxygen binding site is also believed to play an important role in controlling O–O bond cleavage.28,30,38–41 However, since the planar architecture of the porphyrin ligand constrains the cysteinate to a trans position relative to incoming dioxygen molecule, it is impossible to determine empirically whether the stereochemical relationship between the O2 and the cysteinate is critical to P450’s function.
Scheme 1.

Scheme 2.

Recently, we described a functional SOR model, [FeII-(SMe2N4(tren))]+ (1),42 that reacts with superoxide O2− in MeOH at –90 °C to generate the first example of an FeIII– OOH containing sulfur in the coordination sphere, [FeIII– (SMe2N4(tren))(OOH)]+ (2; Scheme 2b).43 Although the thiolate is coordinated cis to the open coordination site in 1, as opposed to trans, as it is with the SOR enzyme,17,19,21,22,44 this appears to have little effect on its superoxide reduction chemistry. Intermediate 2 is low-spin (S = 1/2), displays a νO–O at 784 cm−1 (that shifts to 753 cm−1 upon isotopic labeling with 18O2−), a S → FeIII charge-transfer band at 452-(2900) nm,45 and a coordinated diatomic oxygen ligand (with one short, and one long Fe–O distance at 1.86(3) Å, and 2.78(3) Å, respectively, as determined by EXAFS).43 Herein, we examine the reactivity between our thiolate-ligated five-coordinate complex [FeII(SMe2N4(tren))]+ (1) and dioxygen, in order to determine if O2 activation can be promoted when O2 binds cis to a thiolate. Also described are our attempts at trapping out intermediates in this reaction by incorporating bulky tertiary amines in place of primary amines. Reactions between 1 and dioxygen or superoxide are dramatically altered by a simple modification of our ligand system.
Experimental Section
General Methods
All reactions were performed under an atmosphere of dinitrogen in a glovebox or by using standard Schlenk techniques, or by using a custom-made solution cell equipped with a threaded glass connector sized to fit a dip probe. Reagents purchased from commercial vendors were of the highest purity available and used without further purification. Tetrahydrofuran (THF), Et2O, and MeCN were rigorously degassed and purified using solvent purification columns housed in a custom stainless steel cabinet, dispensed via a stainless steel Schlenk-line (Glass-Contour). Methanol (MeOH) was distilled from magnesium methoxide. 1H NMR spectra were recorded on a Bruker AF 300 and Bruker DPX 200 FT-NMR spectrometers and are referenced to an external standard of TMS (paramagnetic compounds) or to residual protio-solvent (diamagnetic compounds). Chemical shifts are reported in ppm, and coupling constants (J) are in Hz. IR spectra were recorded on a Perkin-Elmer 1700 FT-IR spectrometer as KBr pellets. Cyclic voltammograms were recorded in MeCN (100 mM Bun4N(PF6) solutions) on a PAR 273 potentiostat utilizing a glassy carbon working electrode, a platinum auxiliary electrode, and an SCE reference electrode. Magnetic moments (solution state) were obtained using the Evans method as modified for superconducting solenoids.46,47 Temperatures were obtained using Van Geet’s method.48 Solid-state magnetic measurements were obtained with polycrystalline samples in gel caps using a Quantum Design MPMS S5 SQUID magnetometer. Ambient temperature electronic absorption spectra were recorded on a Hewlett-Packard model 8450 spectrometer, interfaced to an IBM PC. Low-temperature electronic absorption spectra were recorded using a Varian Cary 50 spectro-photometer equipped with a fiber optic cable connected to a “dip” ATR probe (C-technologies), with a custom-built two-neck solution sample holder equipped with a threaded glass connector (sized to fit the dip probe). Thiolate-ligated [FeII(SMe2N4(tren))](PF6) (1) and [FeIII(SMe2N4(tren))(MeCN)]2+ (4) were synthesized as previously described.42,43
Preparation of [FeIII(SMe2N4(tren))]2(μ-O)(PF6)2•MeCN (3)
A solution of 2.00 mmol of [FeII(SMe2N4(tren))](PF6) (1, 864 mg) in 30 mL of methanol was stirred in air for 30 min, and then stirred under N2 for an additional 4 h. The volatiles were then removed, and the resulting brick red powder was dissolved in 10 mL of MeCN, filtered through a piece of glass wool, and layered with 50 mL of Et2O. The two layers diffused together over several days affording brick red crystals of 3 (863 mg, 98.1%). Electronic absorption λmax, nm (ε, cm−1 M−1): 243 (sh); 289(9350); 331(8825); 483(5290). IR (KBr pellet): vC═N 1649 cm−1; vC═N 1594 cm−1. Magnetic moment (solid state, 300 K): 2.5 μB/Fe. Anal. for Fe2C21H46N8S2OP2F12 Calcd: C, 29.09; H, 5.55; N, 12.33. Found: C, 29.68; H, 5.25; N, 11.72.
Alternative Preparation of [FeIII(SMe2N4(tren))]2(μ-O)(BPh4)2-(3-BPh4)
Tetrabutylammonium hydroxide (14 µL, 0.097 mmol) was added to a MeCN solution of [FeIII(SMe2N4(tren))(MeCN)]-(BPh4)2 (4, 50 mg, 0.051 mmol) at ambient temperature, and then stirred under N2 for 4 h. The volatiles were then removed, and the resulting brick red powder was dissolved in MeOH, filtered over Celite, and layered with 25 mL of Et2O. A red solid was isolated affording 3 (17 mg, 53%).
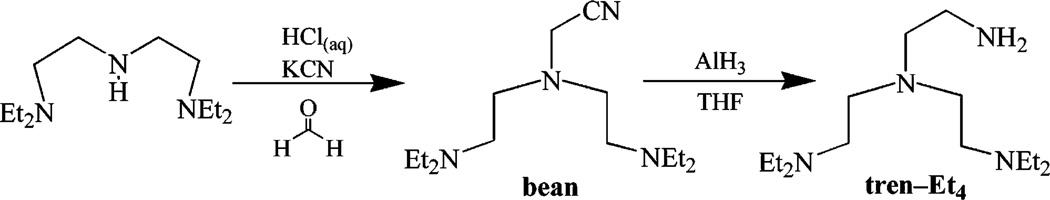
Preparation of [Bis-(2-diethylamino-ethyl)-amino]-acetonitrile (Bean)
An aqueous solution (20 mL) of N,N,N′,N’-tetraethyldi-ethylenetriamine (10.77 g, 50 mmol) was cooled to 0 °C, and HCl (32% aq solution, 5.70 g, 50 mmol) was added in one portion. The solution was then cooled to −10 °C, and an aqueous solution (30 mL) of KCN (3.26 g, 50 mmol) was added slowly. To this was added an aqueous formaldehyde solution (37%, 4.06 g, 50 mmol) at such a rate that the solution temperature did not exceed 0 °C. This was then brought to room temperature and stirred overnight. Methylene chloride (100 mL) was then added, and the organic layer was extracted and washed with water two times (10 mL each time), which was followed by brine twice (10 mL each time). The organic layer was then dried with MgSO4 and the solvent removed by rotary evaporation yielding a pale yellow oil (10.25 g, 80.9%). 300 MHz 1H NMR (CDCl3): 3.74 (s, 2H, CH2CN), 2.61-2.41 (m, 16 H, CH2), 0.96 (t, 12 H, J = 7 Hz, CH3). 50 MHz 13C NMR (CDCl3): 115.9 (C≡N), 52.7 (CH2), 51.4(CH2), 47.5 (CH2), 43.1 (CH2), 11.9 (CH3). HR-MS (FAB) Calcd: 255.2549. Found: 255.2548.
Preparation of N-(2-Amino-ethyl)-N-(2-diethylamino-ethyl)-N′,N′-diethyl-ethane-1,2-diamine (Tren-Et4)
Lithium aluminum hydride (4.55 g, 120 mmol) was suspended in THF (100 mL) and cooled to −15 °C. To this suspension, was added H2SO4 (96% aq, 6.13 g, 60 mmol) dropwise over the period of 30 min. The reaction mixture was then brought to 0 °C and stirred for an additional hour. To this, bean (8.56 g, 33.8 mmol) was added dropwise via syringe at a rate such that the solution did not rise above 20 °C. The reaction was then brought to room temperature and stirred overnight. The remaining AlH3 was then quenched with a large excess of H2O and filtered. The residue was washed with a large volume of methylene chloride and the organic layer extracted. This layer was then dried with MgSO4 and the solvent removed by rotary evaporation yielding tren-Et4 as a musky smelling yellow oil that was spectroscopically pure (7.04 g, 81.0%). 300 MHz 1H NMR (CDCl3): 2.76-2.48 (m, 20H, CH2), 2.34 (bs, 2H, NH2), 1.07 (t, 12H, J = 6 Hz, CH3). 50 MHz 13C NMR: 57.2 (CH2), 52.5 (CH2), 50.9 (CH2), 46.8 (CH2), 39.3 (CH2), 11.1 (CH3). HR-MS (FAB) Calcd: 259.2862. Found: 259.2862.
Preparation of [FeII(SMe2N4(tren-Et4))](PF6) (5)
Sodium hydroxide (80 mg, 2.00 mmol) was dissolved in 10 mL of MeOH followed by the addition of 3-methyl-3-mercapto-2-butanone (236 mg, 2.00 mmol).10 Tren-Et4 (514 mg, 2.00 mmol) was then added to the thiolate solution and the reaction mixture cooled to −20 °C. A cold (−20 °C) solution of FeCl2 (252 mg, 2.00 mmol in 5 mL of MeOH) was then added to the ligand solution dropwise, producing a yellow solution, which progressed to a dark brown solution over 1 h. After 24 h, NaPF6 (334 mg, 2.00 mmol) was added, and the reaction stirred another hour. The reaction mixture was then filtered, and all volatiles were removed by vacuum at room temperature affording a brown tar. This was dissolved in MeCN, filtered through a bed of Celite, and concentrated to ~5 mL. The solution was then layered with Et2O (20 mL), and the two layers were allowed to diffuse together over several days. The pale green filtrate was then separated from the resulting white and black solids. Green crystals of 5 were obtained in low yields by slow evaporation of solvent over several days (161 mg, 13%). 200 MHz 1H NMR (MeCN-d3): 43.9 (bs), 7.46 (s), 5.46 (s), 3.67 (s), 3.47 (s), 1.99 (s), 1.73(s), −4.68 (bs), −16.46 (bs). Electronic absorption λmax nm (ε cm −1 M −1): 223 (7900), 314 (745), 330 (790), 369 (370). IR (KBr pellet): vC═N 1633 cm−1. Magnetic moment (MeCN solution, 302 K): 4.96 μB. Anal. for FeC19H41N4SPF6 Calcd: C, 40.87; H, 7.40; N, 10.03. Found: C, 41.07; H, 7.48; N, 10.22.
μ-Oxo Bridge Cleaving Reactions
Acids (2 equiv, in 0.125 equiv aliquots) were injected via syringe into a cold (−20 °C) MeCN solution containing [FeIII(SMe2N4(tren))]2μ-O(PF6)2•MeCN (3) in a custom-made two-neck vial equipped with a septum cap, and threaded dip-probe feed-through adaptor. Reactions were monitored by electronic absorption spectroscopy, and spectra were recorded after the addition of each 0.125 equiv aliquot.
X-ray Crystallographic Structure Determination
A dark red crystal of 3 was cut to 0.41 × 0.24 × 0.05 mm3, submerged in mineral oil, placed on a glass capillary, and mounted over a stream of cold nitrogen gas (−130(2) °C). The crystal-to-detector distance was set to 30 mm, and exposure time was 50 s deg−1 for all data sets with a scan width of 1°. The data collection was 72.6% complete to 26.37° in 𝓜. A total of 77312 partial and complete reflections were collected covering the indices h = −11 to 11, k = −21 to 20, l = −45 to 45. Of these, 12010 reflections were symmetry independent, and the Rint = 0.1519 indicated that the data was fair. Indexing and unit cell refinements indicated a monoclinic P lattice in the space group P21/c. Pale green crystals of 5 were submerged in mineral oil. A suitable single crystal (0.43 × 0.29 × 0.22 mm3) was selected, placed on a glass capillary, and mounted over a stream of cold nitrogen gas (−130(2) °C). The crystal-to-detector distance was set to 30 mm, and exposure time was 10 s deg−1 for all data sets with a scan width of 2°. The data collection was 96.3% complete to 28.28° in 𝓜. A total of 20333 partial and complete reflections were collected covering the indices, h = −11 to 10, k = −13 to 14, and l = −18 to 18. Of the reflections, 5300 were symmetry-independent, and the Rint = 0.045 indicated the data quality was excellent. Indexing and unit cell refinements indicated a monoclinic P lattice in the space group Pc.
The data for both 3 and 5 were integrated and scaled using Denzo-hkl-SCALEPACK, and an absorption correction was performed using SORTAV. Solution by direct methods produced a complete heavy atom phasing model. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares methods, while all hydrogen atoms were then placed using a riding model. Crystal data for 3 and 5 are presented in Table 1. Selected bond distances and angles are contained in Table 2.
Table 1.
Crystal Data for [FeIII(SMe2N4(tren))]2(μ-O)(PF6)2•MeCN (3) and [FeII(SMe2N4(tren-Et4))](PF6) (5)
| 3 | 5 | |
|---|---|---|
| formula | C24H53F12Fe2-N9OP2S2 | C19H41F6FeN4PS |
| MW (g/mol) | 949.51 | 558.44 |
| Temp (K) | 130(2) | 130(2) |
| unit cella | monoclinic | monoclinic |
| space group | P21/c | Pc |
| a (Å) | 12.6470(9) | 8.4540(2) |
| b (Å) | 17.004(2) | 10.8940(3) |
| c (Å) | 36.359(5) | 14.4500(4) |
| α (deg) | 90 | 90 |
| β (deg) | 90 | 106.1270(19) |
| γ (deg) | 90 | 90 |
| V (Å3) | 7816.5(16) | 1278.44(6) |
| Z | 8 | 2 |
| σcalc (mg/m3) | 1.614 | 1.415 |
| Rb | 0.1108 | 0.0406 |
| Rw | 0.2369 | 0.1074 |
| GOF | 0.949 | 1.091 |
In all cases, Mo Kα(λ = 0.71070 Å) radiation was used.
R = Σ||Fo| − |Fc||/Σ|Fo|; Rw=[Σw(|Fo| − |Fc|)2/ΣwFo2]1/2, where w−1 = [σ2count + (0.05F2)2]/4F2.
Table 2.
Selected Geometrical Parameters for the Cations of [FeII(SMe2N4(tren))](PF6) (1), [FeIII(SMe2N4(tren))]2(μ-O)(PF6)2•MeCN (3), [FeIII(SMe2N4(tren))(MeCN)](BPh4)2•MeCN (4), and [FeII(SMe2N4(tren-Et4))](PF6)(5)a
| 1 | 3 | 4 | 5 | |
|---|---|---|---|---|
| Fe(1)–S(1) | 2.3281(9) | 2.317(4) | 2.146(1) | 2.3171(9) |
| Fe(1)–N(1) | 2.091(3) | 2.186(10) | 1.912(2) | 2.104(3) |
| Fe(1)–N(2) | 2.268(3) | 2.239(10) | 2.026(2) | 2.227(3) |
| Fe(1)–N(3) | 2.131(3) | 2.208(10) | 2.002(2) | 2.219(3) |
| Fe(1)–N(4) | 2.117(3) | 2.173(10) | 2.018(2) | 2.220(3) |
| Fe(1)–O(1) | N/A | 1.807(8) | N/A | N/A |
| S(1)–Fe(1)–N(2) | 163.02(7) | 154.5(4) | 171.96(6) | 161.53(7) |
| S(1)–Fe(1)–N(1) | 84.02(8) | 80.4(3) | 86.59(6) | 83.41(7) |
| N(1)–Fe(1)–X | N/A | 178.6(4) | 177.31(8) | N/A |
| Fe(1)–O(1)–Fe(2) | N/A | 155.3(5) | N/A | N/A |
Bond lengths and bond angles are given in Å and deg, respectively.
Results and Discussion
Reaction of [FeII(SMe2N4(tren))](PF6) with Dioxygen
Previously,42,43 we showed that superoxide (O2−) reacts with the five-coordinate ferrous complex [FeII(SMe2N4(tren))](PF6) (1) (Scheme 2b) via an inner-sphere mechanism to afford a transient hydroperoxide intermediate 2. A ferrous superoxide-ligated intermediate is not detected in this reaction. Hydrogen peroxide (detected by mass spectrometry, 1H NMR, and IR) is released from intermediate 2 upon protonation49 to afford the six-coordinate, solvent-ligated ferric complexes [FeIII-(SMe2N4(tren))(solv)]2+ (solv = MeCN (4) or MeOH)42,43 Addition of dioxygen to complex 1, on the other hand, affords a different product at ambient temperature. Complete formation of this product requires 0.25 equiv of O2 per iron (as measured by O2 consumption using a known-volume bulb). This stoichiometry is consistent with the formation of a μ-oxo dimer, as was confirmed by X-ray crystallography.
X-ray quality crystals of the final product formed in the reaction between 1 and dioxygen [FeIII(SMe2N4(tren))]2(μ2-O)](PF6)2•3MeCN (3, Figure 1) were produced via slow diffusion of Et2O into a MeCN solution. This complex crystallized in the P21/c space group, with two structurally distinct binuclear iron complexes per asymmetric unit. Metrical parameters for the two dimers differ only slightly (see Supporting Information). Binuclear [FeIII(SMe2N4(tren))]2- (μ2-O)2+ is dicationic and contains an unsupported μ-oxo bridge that connects two pseudo-octahedrally coordinating FeIII ions. As shown in the ORTEP diagram of Figure 1, the two halves of the dimer are twisted (~90°) relative to one another, presumably in order to relieve steric repulsive interactions between the gem dimethyls adjacent to the thiolate sulfurs.50 The bridging oxo-ligand is trans to the imine-nitrogens and cis to the thiolate-sulfurs. Average Fe–N (2.172(3) Å) and Fe–S (2.304(1) Å) bond lengths in 3 are longer than low-spin complexes containing FeIII in this ligand environment (Table 2). This is, in part, because the irons are either high-spin (S = 5/2) or intermediate-spin (S = 3/2) (vide infra),6–8 and in part due to the trans effect of the oxo and thiolate ligands. The bridging oxo ligand is at an average distance of 1.807(4) Å from the iron centers, with an average Fe–O-Fe bond angle of 152.51(8)°. These metrical parameters are typical of μ-oxo dimers50–54 and reflect extensive π-bonding within the Fe–O-Fe core.9 Diferric μ-oxo bridges are quite flexible, support Fe–O–Fe angles ranging from 114° to 180°, and typically possess unusually short Fe–O distances.51 Unsupported μ-oxo-bridged structures tend to have Fe–O–Fe angles closer to linear: the smallest Fe– O–Fe angle in an unsupported bridge structure is 139°.51 Thiolate-ligated μ-oxo dimers are extremely rare.52,55 This is because O2 oxidation reactions typically afford disulfides, metallosulfoxides (M – S(═O)R), or metallosulfones (M – SO2R), when thiolates are in the coordination sphere.56 The only other two examples of thiolate-ligated binuclear μ-oxo-bridged iron complexes are [(bme-daco)FeIII]2(μ-O) reported by Darensbourg52 and [(bmmp-TASN)Fe]2(μ-O) reported by Grapperhaus.55 In all three of these examples, the thiolate sulfur does not appear to perturb the strong Fe–O(oxo) bonds: Fe–O bond lengths in these complexes fall in the usual range. Bond lengths and angles for μ-oxo-bridged 3 are compared with those of its reduced Fe(II) precursor 1, as well as oxidized [FeIII(SMe2N4(tren))(MeCN)]2+ (4), in Table 2.
Figure 1.

ORTEP of the μ-oxo dimer [FeIII(SMe2N4(tren))]2μ-O2+ (3) and atom labeling scheme. All hydrogen atoms have been removed for clarity.
Mechanism of μ-Oxo 3 Formation via Reaction of 1 with O2
The generally accepted mechanism for O2-induced μ-oxo dimer formation (in aprotic solvents) comes from the well-developed iron porphyrin literature57,58 and is believed to involve initial coordination of O2 to FeII to afford an FeIII–O2−(superoxide) intermediate. Reaction of this FeIII-O2-intermediate with 1 equiv of FeII starting material would afford a peroxide-bridged Fe2III(μ-O22−) ferric dimer, that could cleave to afford 2 equiv of an Fe(IV)═O (oxo). Reaction of the highly reactive Fe(IV)═O with 1 equiv of FeII starting material would afford the thermodynamically stable μ-oxo-bridged binuclear product. In protic solvents, the mechanism is likely to involve protonation of peroxide and/ or superoxide intermediates, rather than peroxide-dimer formation. The inner-sphere mechanism for O2− reduction by our SOR model complex42,43 1 suggests that O2 reduction could occur via the initial coordination of O2 to the metal. The site to which O2 (or O2−) binds (cis with respect to the thiolate sulfur) has been established by structurally characterizing more stable analogues, including an Fe–NO,59 and an Fe–OAc derivative.60 If the reaction between 1 and O2 is monitored spectrophotometrically at low temperatures, in aprotic solvents such as MeCN (at −20 °C) and THF (at −78 °C), no intermediates are observed. Instead, 1 rapidly (within seconds) converts directly to 3, even at these lower temperatures. In protic solvents such as MeOH, on the other hand, a transient intermediate is observed (by electronic absorption spectroscopy (Figure S-3), and resonance Raman)45 when O2 is added to 1 at −78 °C. This supports the possibility that μ-oxo dimer formation from 1 involves an inner-sphere mechanism. Further characterization of this intermediate, as well as the kinetics of its formation, is ongoing.
Electronic, Magnetic, and Redox Properties μ-Oxo-Bridged 3
Solutions of μ-oxo-bridged 3 are brick-red in MeOH, MeCN, THF, pyridine, and DMF. In MeCN, the electronic absorption spectrum of 3 (Figure S-1) is dominated by an intense charge-transfer band in the visible region, at 483(5290) nm, which is presumably S → FeIII in nature. This band shifts to 500(5500) nm in MeOH (Figure S-2). Binuclear 3 is irreversibly reduced at a potential of −1.08 V versus SCE (in MeCN), indicating that Fe3+ ion is highly stabilized by this ligand environment. The irreversibility of this wave suggests that reduction induces cleavage of the μ-oxo bridge. The magnetic properties of 3 indicate that the coupling between the Fe3+ ions is weak. The ambient temperature MeCN solution μeff) 2.5 μB per iron. A negative x-intercept in the 1/χ versus temperature (SQUID) plot (Figure S-7) indicates that the two metals are antiferromagnetically coupled in the solid state. Linear fits to the 1/χ versus T data over the temperature range 55–350 K give a μeff = 2.5 μB per iron (Figure S-8), and a coupling constant of J = −28 cm−1. This J-value falls below the usual range for μ-oxo dimers containing an unsupported bridge.51 However, the reported range (−65 cm−1 > J > −195 cm−1) includes only N-,O-ligated Fe(III) μ-oxo systems,51 suggesting that thiolate-sulfurs weaken this coupling. The increased lability of the μ-oxo bridge (vide infra) in thiolate-ligated 3 would be consistent with weaker coupling.
Reactivity of μ-Oxo-Bridged 3
Although μ-oxo dimers tend to be quite stable (representing the “thermodynamic pit” of FeIII chemistry) and unreactive,51 there are a few rare reported examples of reactivity.53,61–63 Reversible μ-oxo bridge cleavage is seen upon the addition of a proton source (H2Im+Cl−) to [(TPP)Fe]2(μ-O) in aprotic solvents to afford 2 equiv of [(TPP)Fe(HIm)2] (+H2O).61 Reed has shown that equilibria involving μ-oxo [(TPP)Fe]2(μ-O) to [(TPP)Fe–(H2O)]+ interconversion will favor the bridge-cleaved aquo complex in rigorously dried aprotic solvents when acidic anions are present.62 Protonation of [(TPP)Fe]2(μ-O) using [H(mesitylene)][B(C6F5)4] affords the protonated monohy-droxy-bridged porphyrin [(TPP)Fe]2(μ-OH)+.62 The unsupported μ-oxo bridge of [(N4Py)2Fe2(μ-O)]+4 (J = −104 cm−1) is quite labile, and bridge cleavage occurs readily upon the addition of H2O or MeOH.53 This is despite the presence of rather short Fe–O bonds (1.803(1) Å) and is attributed to the relief of steric repulsive interactions between the two halves of the [(N4Py)2Fe2(μ-O)]+4 dimer. It is likely that the large cationic charge of [(N4Py)2Fe2(μ-O)]+4 also contributes to its facile bridge cleavage.64 Octacationic 5,10,15,20-tetrakis(2,4,6-trimethyl-3,5-bis{ a-N,N,N-trimethylammoniummethyl}phenyl)porphinatoiron(III) is resistant to μ-oxo dimer formation, even in water. This is both due to the ligand’s steric bulk and extensive charge build-up.64 Oxidation of μ-oxo dimers with H2O2 can lead to high-valent, doubly bridged Fe2IV(μ-O)2 diamond cores, via the insertion of an oxygen atom into the singly bridged Fe2III(μ-O) core.63
Reversible μ-Oxo Bridge Cleavage and Formation
The μ-oxo bridge of 3 can be reversibly cleaved with acids (HX) to afford either the corresponding anion-bound (Fe–X), or solvent-bound (Fe–MeCN) products, depending on the acids (NH4+, HOAc, HCl, LutNH+, HBF4) used (Scheme 3; Figure 2 and Figure 3). Addition of hydroxide converts these products back to μ-oxo-bridged 3 (Figure 4). For example, addition of 2 equiv of HOAc(aq) to dimeric 3 at −20 °C in MeCN cleanly affords (as demonstrated by the sharp isosbestic point at 520 nm) monomeric acetate-ligated [FeIII-(SMe2N4(tren))(OAc)]+ (6).60 This is illustrated in the electronic absorption spectrum of Figure 2. The final product in this reaction was identified by spectral comparison with authentic samples of 6 (prepared as described elsewhere).60 The reaction is complete upon the addition of 2 equiv of HOAc/dimer. When HCl(aq) is used in place of acetic acid, clean conversion to [FeIII(SMe2N4(tren))Cl]+ (7) is observed as demonstrated by the sharp isosbestic point at 520 nm (Figure S-4 of Supporting Information), and comparison with authentic samples of 7 (prepared as described elsewhere).59 Again, the reaction is complete upon the addition of 2 equiv of HCl at ambient temperature. These reactions occur despite the presence of water in HOAc(aq) and HCl(aq), suggesting that the μ-oxo bridge of 3 is more basic than [(TPP)FeIII]2-(μ-O).62 This is supported by the fact that weaker acids, such as NH4+ (Figure 3) and LutNH+ (Figure S-5), will also cleave the μ-oxo bridge of 3. Given that the oxo is coordinated to two Lewis acidic Fe3+ ions, this behavior is surprising. The thiolates most likely contribute to this increased basicity.37 This is supported by a recent EXAFS study which shows that the cysteinate of chloroperoxidase contributes to an increased basicity of the Fe(IV)=O oxo intermediate, resulting in its protonation at physiological pH.37 Chloride-ligated 7 is the final product59 observed in the LutNHCl protonation reaction (Figure S-5). When anhydrous acids containing noncoordinating anions, such as HBF4 (Figure S-6), are used to protonate 3 in MeOH or MeCN, the solvent-ligated complex [FeIII(SMe2N4(tren))(MeCN)]+ (4) or [FeIII(SMe2N4-(tren))(MeOH)]+ forms.42 As shown in Figure 3, solvent-ligated 4 also forms when NH4PF6 is used to protonate the μ-oxo bridge. In noncoordinating solvents, such as THF, on the other hand, protonation of 3 using acids containing noncoordinating anions results in loss of the charge-transfer band, bleaching of the solution, and formation of intractable solids, even at low temperatures.
Scheme 3.

Figure 2.

Electronic absorption spectrum showing that the μ-oxo bridge of dimeric 3 is cleaved upon the addition of 2 equiv of HOAc in MeCN solvent to cleanly afford monomeric [FeIII(SMe2N4(tren))(OAc)]+ (6).
Figure 3.

Electronic absorption spectrum showing that the μ-oxo bridge of dimeric 3 is cleaved upon the addition of 2 equiv of NH4PF6 in MeCN solvent to cleanly afford monomeric solvent-ligated [FeIII(SMe2N4(tren))-(MeCN)]+2 (4).
Figure 4.

Electronic absorption spectrum showing that reaction of 2 equiv of Bu4NOH with monomeric [FeIII(SMe2N4(tren))(MeCN)]+2 (4) in MeCN affords the μ-oxo dimer [FeIII(SMe2N4(tren))]2μ-O2+ (3) at ambient temperature.
Bridge cleavage in these reactions most likely involves initial protonation of the bridging oxo to afford a transient hydroxo-bridged intermediate. Hydroxo-bridged intermediates are not observed, however, even at low temperatures with only 1 equiv of acid. Although hydroxo-bridged binuclear iron (FeIII2) complexes are known, these are usually only stable when additional bridge supporting ligands (e.g., RCO2−, PO43−, etc.) are present.65 Unsupported hydroxo diferric bridge structures FeIII–(μ-OH)–FeIII are unstable, because significant weakening of the Fe–O bonds occurs upon protonation, with a concomitant decrease in magnetic coupling.65 Also, the extremely acidic proton on the bridging hydroxy of an FeIII–(μ-OH)–FeIII unit usually favors protonation of water to afford H3O+ and the deprotonated μ-oxo FeIII–(μ-O)–FeIII core. It is only in the absence of more basic proton acceptor sites that μ-hydroxo-bridged structures are stable. Thus, it is not surprising that the μ-oxo bridge of 3 cleaves in MeCN upon the addition of protons. That hydroxide intermediates would be unobservable is consistent with Norton’s observation that protonation of bridging-oxos can be surprisingly slow.66 If protonation of 3’s oxo is rate-limiting, then one would not expect to see an intermediate hydroxo. Also, given that the oxo of 3 is buried within the dimeric core it is not surprising that protonation of the oxo would be slow. It is surprising, however, that the more exposed thiolate ligands of 3 are not protonated with stronger acids.
Oxo-bridged binuclear 3 can be regenerated upon the addition of OH− to solvent-ligated [FeIII(SMe2N4(tren))-(MeCN)]2+ (4; Scheme 3). This reaction is clean, as shown by the isosbestic points in the electronic absorption spectrum shown in Figure 4. Intermediates, such as hydroxide-bound monomeric [FeIII(SMe2N4(tren))(OH)]+, are not observed, even under dilute conditions at low temperatures.
Oxo-bridged 3 is robust toward further oxidative damage by dioxygen or peroxides. Solutions of 3 are stable for several weeks in air, and stable (for several minutes) in the presence of a slight excess (1.1 equiv) of hydrogen peroxide. This is in stark contrast to other known FeIII–thiolates,67–72 which readily react with O2 or peroxide to afford metal-bound sulfoxides or sulfones, or insoluble rustlike products. Addition of greater than 1.4 equiv of H2O2 to 3 affords intractable solids even at lower temperatures; no higher valent Fe(IV) species are observed.63 A similar result is obtained when pentane solutions of HOOBut are added to 3, even at low temperatures (−90 °C).
Synthesis of a Sterically Encumbered Ligand Tren-Et4
In an attempt to prevent μ-oxo dimer formation and facilitate the observation of dioxygen-bound intermediates, a bulkier version of the tris(2-aminoethyl) amine (tren) ligand was synthesized. Steric bulk has been shown to prevent μ-oxo dimer formation with N-,O-ligated Fe(III) systems.73–77 The most straightforward way to incorporate bulk was to place alkyl substituents on the nitrogens. In order to maintain a single primary amine site for condensation with the thioke-tone arm, and avoid isomeric mixtures, an asymmetrical bis-N-substituted tris-amine, N-(2-amino-ethyl)-N-(2-diethyl-aminoethyl)-N′,N′-diethyl-ethane-1,2-diamine (tren-Et4), was synthesized (as outlined in Scheme 4).
Scheme 4.
Synthesis and Properties of [FeII(SMe2N4(tren-Et4))](PF6) (5)
Tertiary amine/thiolate ligated [FeII(SMe2N4(tren-Et4))]-(PF6) (5) was prepared in a manner analogous to 1 via the metal-templated Schiff-base condensation between 3-methyl-3-mercapto-2-butanone and tren-Et4.42,43 Solutions of 5 are chartreuse in MeCN, and characterized by an intense LMCT band at 223(7900) nm, and weak LMCT bands at 314(745) nm and 330(790) nm. The effective moment of 5 is 4.92 μB in MeCN solution indicating that it is high-spin (S = 2), and, most likely, mononuclear at ambient temperature. This is supported by its 1H NMR spectrum, which displays paramagnetically shifted peaks in the region 50 to −20 ppm (Figure S-9). A monomeric structure was ultimately proven by X-ray crystallography. As shown in the ORTEP diagram of Figure 5, tertiary amine-ligated 5 contains FeII in a distorted trigonal bipyramidal coordination environment (τ = 0.67)78 similar to that of primary amine-ligated 1 (τ = 0.68). Selected geometric parameters of 5 are compared to those of 1 in Table 2. The Fe–S bond length (2.317(1) Å) in 5 is typical for an FeII–thiolate bond and is comparable to that of 1 (2.328(1) Å; Table 2). The imine Fe–N(1) bond length (Table 2) is also within the normal range. Most notable about the structure of 5 is that the iron amine bonds (Fe– N(2), Fe–N(3), and Fe–N(4); Table 2) are unusually long. The mean Fe–N(2,3,4) bond length is 2.22(3) Å, whereas the usual range for Fe–N bonds in high-spin FeII amine complexes is 2.00–2.18 Å.79–82 The trans influence of the thiolate ligand is most likely responsible for the elongation of the apical Fe–N(2) bond: this distance is approximately the same in structures 1 and 5 (Table 2) indicating that the bulky ethyl substituents have very little to do with the observed elongation. The terminal iron–amine bonds Fe– N(3,4) (Figure 5; Table 2), on the other hand, are noticeably (0.1 Å) longer in tertiary amine ligated 5 (mean distance = 2.219(1) Å) versus primary amine ligated 1 (mean distance = 2.11(2) Å), demonstrating that steric bulk, imposed by the ethyl groups, weakens the Fe–N(3,4) bonds. Similarly elongated Fe(II)–N bonds are observed in the sterically encumbered complexes (Mes2ArCO2)2FeII(TMEDA)2 (2.19-(2) Å),83 [FeII(N(CH2C(O)NtBu)3)]1− (2.098(3) Å),84 and [FeII(6-Me-TPA)(MeCN)2]2+ (2.15(1) Å).85 If this were an electronic effect, then one would expect the tertiary amine Fe–N bonds to be shorter (as opposed to longer) than the primary amine Fe–N bonds, since tertiary amines are better σ-donors. That sterics are responsible for the observed elongation of Fe–N bonds is supported by recent DFT calculations comparing tertiary versus secondary amine cyclam complexes.86 One would expect steric repulsive interactions to be even more problematic in the higher-valent Fe(III) derivative of 5 (vide infra), since the optimum Fe(III)–N bond length would require that the tertiary amines move even closer to one another. Since reactions between 5 and O2 or O2− involve oxidation of the metal center, the instability of the higher oxidation state would be expected to affect reactivity with these substrates.
Figure 5.

ORTEP of the ferrous complex [FeII(SMe2N4(tren-Et4))]+ (5) and atom labeling scheme. All hydrogen atoms have been removed for clarity.
Reactivity of [FeII(SMe2N4(tren-Et4))](PF6) (5) with Dioxygen and Superoxide
In contrast to most Fe(II) thiolate complexes,55,69,71,72,80,87–91 thiolate-ligated [FeII(SMe2N4(tren-Et4))](PF6) (5) does not react with dioxygen. Solutions of 5 are stable in air for hours. Complex 1, on the other hand, reacts rapidly (within seconds) with dioxygen at ambient temperature to cleanly afford the stable μ-oxo dimer 3. Also in contrast to primary amine ligated 1, tertiary amine-ligated 5 does not react readily with superoxide. When KO2 (solubilized as its 18-crown-6 salt) is injected into an MeCN solution of 5, there is no immediate reaction. Instead, 5 gradually (over 30 min at −20°C) converts to an intractable deep-brown oily solid. Complex 1, on the other hand, reacts rapidly (at diffusion controlled rates) with O2− to afford an FeIII–OOH intermediate.43 Initially, it was thought that the absence of N–H protons in 5 was responsible for its sluggish reactivity with O2−, and the mechanism was thought to involve H-atom abstraction. However, this mechanism was ruled out by the fact that no reaction occurs between primary amine-ligated 1 and superoxide in rigorously dried THF, in the absence of an external proton donor.49 Alternatively, the Et-groups alter reactivity by modifying the steric and electronic properties of the metal center. More specifically, one would expect the frontier orbitals involved in binding and activating superoxide, and the stability of the Fe(III)-peroxide intermediate, to be perturbed by the Et-substituents of 5.
In order to assess the stability of the oxidized Fe(III) derivative of 5, both the chemical oxidation of 5 (using NO-(PF6)), and a direct synthetic route to [FeIII(SMe2N4(tren-Et4))]2+ (via the insertion of FeIII into the [(tren-Et4)N4SMe2]1− ligand), were attempted. Both of these methods resulted only in the formation of intractable solids. Unfavorable steric repulsive interactions between the bulky ethyl groups are most likely responsible for this instability. This is supported by the crystal structure of reduced 5 which revealed (vide supra) that steric repulsive forces do not allow the nitrogens to attain their idealized FeII–N bond length, resulting in unusually long FeII–N(Et)2 bonds. These repulsive forces would increase prohibitively as the nitrogens moved closer to the metal ion in an attempt to adopt the optimized FeIII–N bond length. Thus, it would appear that the [(tren-Et4)-N4SMe2]1− ligand is unable to accommodate iron in the Fe3+ oxidation state. Similar behavior is seen with [FeII(6-Me-TPA)(MeCN)2]2+ (8),85 and [FeII(N(CH2C(O)NtBu)3)]1–84 where steric repulsive forces disfavor shorter bonds and higher oxidation states, resulting in cathodically shifted redox potentials. Introduction of a methyl group on one of the pyridine rings of the TPA ligand of 8 causes the Fe(II)–N bonds to elongate by 0.16 Å (from 1.99(1) to 2.15(1) Å) relative to the unsubstituted ligand. The redox potential of 5 (Epa; irrev) is also cathodically shifted (from −100 mV with primary amine-ligated 1 to Epa = +410 mV with tertiary amine-ligated 5 (Figure 6)). This indicates that the Fe3+ oxidation state is ~12 kcal/mol less stable in the tertiary amine ligand [(tren-Et4)N4SMe2]1− relative to the primary amine ligand [(tren)N4SMe2]1−. Furthermore, the cyclic voltammogram wave associated with 5 (Figure 6) is irreversible indicating that the FeIII derivative, once generated, rapidly (on the order of seconds) decomposes. This instability of FeIII in the [(tren-Et4)N4SMe2]1− ligand is unusual in that thiolates typically stabilize iron in higher oxidation states.6 This is demonstrated by the rather anodic redox potentials (−400 to −1000 mV vs SCE) of FeIII–thiolate complexes.68,90,92–94 The instability of FeIII in the [(tren-Et4)-N4SMe2]1− ligand thus explains the lack of reactivity between 5 and O2 or O2−.
Figure 6.

Cyclic voltammogram of [FeII(SMe2N4(tren-Et4))]+ (5) in MeCN, with Bu4N(PF6) supporting electrolyte (0.100 M), over the range +700 to −300 mV vs SCE.
Summary and Conclusions
This work describes the reaction between five-coordinate, thiolate-ligated [FeII(SMe2N4(tren))]+ (1) and dioxygen to afford a rare example of a μ-oxo dimer containing a thiolate in the coordination sphere, [FeIII(SMe2N4(tren))]2(μ2-O)2+ (3). An intermediate is detected in this reaction at low temperatures, suggesting that this reaction involves the initial coordination of O2 to the open coordination site at iron. In a related reaction, superoxide (O2−) has been shown to oxidatively add to this site at low temperatures to afford a spectroscopically detectable FeIII–OOH intermediate. Although the thiolate does not appear to noticeably perturb the metrical parameters of μ-oxo-bridged 3, it does decrease the magnetic coupling between the two iron centers and creates a rather basic oxo site. Protonation of this oxo using strong (HBF4, HCl) or weak (HOAc, NH4PF6, LutNHCl) acids results in μ-oxo bridge cleavage to afford the corresponding solvent- or anion-bound monomeric derivatives. That the μ-hydroxo-bridged intermediate is not detected in these protonation reactions is not suprising, given that protonated μ-hydroxo-bridged species tend to be unstable in the absence of additional bridging ligands. Mechanisms of μ-oxo dimer formation have been shown to involve peroxide and high-valent oxo intermediates. An attempt to stabilize these intermediates in the reaction affording 3 by increasing steric bulk instead creates a ligand ([(tren-Et4)N4SMe2]1−) that alters the electronic properties of the iron to the point where O2 oxidation chemistry is no longer favored.
Supplementary Material
Acknowledgment
J.S. wishes to thank the EPA for a graduate fellowship (91594801-0). NIH support (Grant GM 45881) is gratefully acknowledged.
Footnotes
Supporting Information Available: Crystallographic data for 3 and 5, electronic absorption spectrum of 3 (in MeCN and MeOH), magnetic data (1/χ vs T and μeff vs T), electronic absorption spectrum showing the intermediate that forms in the low T reaction between 1 and O2, electronic absorption spectrum showing that the μ-oxo bridge of dimeric [(FeIII(SMe2N4(tren))2(μ-O)]2+ (3) is cleaved upon the addition of 2 equiv of HCl(aq), HBF4, or LutNHCl, 1H NMR spectrum of 5, and electronic absorption spectrum of 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Waller BJ, Lipscomb JD. Chem. Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 3.Que L, Jr, Ho RYN. Chem. Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 4.Feig AL, Lippard SJ. Chem. Rev. 1994;94:759–805. [Google Scholar]
- 5.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S, Lehnert N, Neese F, Skulan AJ, Yang Y, Ahou J. Chem. Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs JA. Chem. Rev. 2004;104:825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovacs JA. Science. 2003;299:1024–1025. doi: 10.1126/science.1081792. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini M, Liehr IS, Fisher AL, Laub PB, Cooperman BS, Mierke DF. Biochemistry. 2000;39:12210–12215. doi: 10.1021/bi001323a. [DOI] [PubMed] [Google Scholar]
- 9.Kappock TJ, Caradonna JP. Chem. Rev. 1996;96:2659–2756. doi: 10.1021/cr9402034. [DOI] [PubMed] [Google Scholar]
- 10.Prigge ST, Boyington JC, Faig M, Doctor KS, Gaffney BJ, Amzel LM. Biochimie. 1997;79:629–636. doi: 10.1016/s0300-9084(97)83495-5. [DOI] [PubMed] [Google Scholar]
- 11.Nelson MJ, Seitz SP. Curr. Opin. Struct. Biol. 1994;4:878–884. doi: 10.1016/0959-440x(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 12.Goldsmith CR, Jonas RT, Stack TDP. J. Am. Chem. Soc. 2002;124:83–96. doi: 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]
- 13.Rocklin AM, Tierney DL, Kofman V, Brunhuber NMW, Hoffman BM, Christoffersen RE, Reich NO, Lipscomb JD, Que L., Jr Proc. Natl. Acad. Sci. U.S.A. 1999;96:7905–7909. doi: 10.1073/pnas.96.14.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hausinger RP. Crit. Rev. Biochem. Mol. Biol. 2004;39:1–47. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 15.Mascharak PK. Coord. Chem. Rev. 2002;225:201–214. [Google Scholar]
- 16.Yeh AP, Hu Y, Jenney FE, Jr, Adams MWW, Rees DC. Biochemistry. 2000;39:2499–2508. doi: 10.1021/bi992428k. [DOI] [PubMed] [Google Scholar]
- 17.Mathe C, Mattioli TA, Horner O, Lombard M, Latour J-M, Fontecave M, Niviere V. J. Am. Chem. Soc. 2002;124:4966–4967. doi: 10.1021/ja025707v. [DOI] [PubMed] [Google Scholar]
- 18.Nivie`re V, Asso M, Weill CO, Lombard M, Guigliarelli B, Favaudon V, Houeé-Levin C. Biochemistry. 2004;43:808–818. doi: 10.1021/bi035698i. [DOI] [PubMed] [Google Scholar]
- 19.Coulter ED, Emerson JP, Kurtz DM, Jr, Cabelli DE. J. Am. Chem. Soc. 2000;122:11555–11556. [Google Scholar]
- 20.Jenney FE, Jr, Verhagen MFJM, Cui X, Adams MWW. Science. 1999;286:306–309. doi: 10.1126/science.286.5438.306. [DOI] [PubMed] [Google Scholar]
- 21.Lombard M, Houee-Levin C, Touati D, Fontecave M, Niviere V. Biochemistry. 2001;40:5032–5040. doi: 10.1021/bi0023908. [DOI] [PubMed] [Google Scholar]
- 22.Niviere V, Lombard M, Fontecave M, Houee-Levin C. FEBS Lett. 2001;497:171–173. doi: 10.1016/s0014-5793(01)02468-1. [DOI] [PubMed] [Google Scholar]
- 23.Coelho AV, Matias P, Fulop V, Thompson A, Gonzalez A, Carrondo MA. J. Biol. Inorg. Chem. 1997;2:680–689. [Google Scholar]
- 24.Becker A, Schlichting I, Kabsch W, Groche D, Schultz S, Wagner AFV. Nat. Struct. Biol. 1998;5:1053–1058. doi: 10.1038/4162. [DOI] [PubMed] [Google Scholar]
- 25.Rajagopalan PTR, Yu XC, Pei D. J. Am. Chem. Soc. 1997;119:12418–12419. [Google Scholar]
- 26.Loew GH, Harris DL. Chem. Rev. 2000;100:407–419. doi: 10.1021/cr980389x. [DOI] [PubMed] [Google Scholar]
- 27.Harris DL, Loew GH. J. Am. Chem. Soc. 1998;120:8941–8948. [Google Scholar]
- 28.Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 29.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 30.Auclair K, Moënne-Loccoz P, Ortiz de Montellano PR. J. Am. Chem. Soc. 2001;123:4877–4885. doi: 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]
- 31.Ogliaro F, deVisser SP, Cohen S, Sharma PK, Shaik S. J. Am. Chem. Soc. 2002;124:2806–2817. doi: 10.1021/ja0171963. [DOI] [PubMed] [Google Scholar]
- 32.Clay M, Cosper CA, Jenney FE, Jr, Adams MWW, Johnson MK. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3796–3801. doi: 10.1073/pnas.0636858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Que L., Jr Nat. Struct. Biol. 2000;7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 34.Neese F, Zaleski JM, Zaleski KL, Solomon EI. J. Am. Chem. Soc. 2000;122:11703–11724. [Google Scholar]
- 35.Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Munck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 36.Decker A, Rohde J-U, Que L, Jr, Solomon EI. J. Am. Chem. Soc. 2004;126:5378–5379. doi: 10.1021/ja0498033. [DOI] [PubMed] [Google Scholar]
- 37.Green MT, Dawson JH, Gray HB. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 38.Urano Y, Higuchi T, Hirobe M, Nagano T. J. Am. Chem. Soc. 1997;119:12008–12009. [Google Scholar]
- 39.Higuchi T, Shimada K, Maruyama N, Hirobe M. J. Am. Chem. Soc. 1993;115:7551–7552. [Google Scholar]
- 40.Rapael AL, Gray HB. J. Am. Chem. Soc. 1991;113:1038–1040. [Google Scholar]
- 41.Matsui T, Nagano S, Ishimori K, Watanabe Y, Morishima I. Biochemistry. 1996;35:13118–13124. doi: 10.1021/bi960459z. [DOI] [PubMed] [Google Scholar]
- 42.Shearer J, Nehring J, Kaminsky W, Kovacs JA. Inorg. Chem. 2001;40:5483–5484. doi: 10.1021/ic010221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shearer J, Scarrow RC, Kovacs JA. J. Am. Chem. Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 44.Silaghi-Dumitrescu R, Silaghi-Dumitrescu I, Coulter ED, Kurtz DM., Jr Inorg. Chem. 2003;42:446–456. doi: 10.1021/ic025684l. [DOI] [PubMed] [Google Scholar]
- 45.Dey A, Chow M, Theisen RM, Kovacs JA, Solomon EI. Manuscript in preparation. [Google Scholar]
- 46.Evans DA. J. Chem. Soc. 1959:2005. [Google Scholar]
- 47.Live DH, Chan SI. Anal. Chem. 1970;42:791. [Google Scholar]
- 48.Van Geet AL. Anal. Chem. 1968;40:2227–2229. [Google Scholar]
- 49.Theisen RM, Kovacs JA. Inorg. Chem. submitted. [Google Scholar]
- 50.Mukherjee RN, Stack TDP, Holm RH. J. Am. Chem. Soc. 1988;110:1850–1861. [Google Scholar]
- 51.Kurtz DM., Jr Chem. Rev. 1990;90:585–606. [Google Scholar]
- 52.Musie G, Lai C-H, Reibenspies JH, Sumner LW, Darensbourg MY. Inorg. Chem. 1998;37:4086–4093. doi: 10.1021/ic980475f. [DOI] [PubMed] [Google Scholar]
- 53.Roelfes G, Lubben M, Chen K, Ho RYN, Mettsma A, Genseberger S, Hermant RM, Hage R, Mandal SK, Young VG, Jr, Zang Y, Kooijman H, Spek AL, Que L, Jr, Feringa BL. Inorg. Chem. 1999;38:1929–1936. doi: 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]
- 54.Norman RE, Holz RC, Menage S, O’Connor CJ, Zhang JH, Que L., Jr Inorg. Chem. 1990;29:4629–4637. [Google Scholar]
- 55.Grapperhaus CA, Li M, Patra AK, Poturovic S, Kozlowski PM, Zgierski MZ, Mashuta MS. Inorg. Chem. 2003;42:4382–4388. doi: 10.1021/ic026239t. [DOI] [PubMed] [Google Scholar]
- 56.Grapperhaus CA, Darensbourg MY. Acc. Chem. Res. 1998:31, 1451–459. [Google Scholar]
- 57.Chin D-H, La Mar GN, Balch AL. J. Am. Chem. Soc. 1980;102:4344–4350. [Google Scholar]
- 58.Balch AL, Chan Y-W, Cheng R-J, La Mar GN, Latos-Grazynski L, Renner MW. J. Am. Chem. Soc. 1984;106:7779–7785. [Google Scholar]
- 59.Villar G, Fitch S, Horner O, Latour J-M, Kovacs JA. Manuscript in preparation. [Google Scholar]
- 60.Shearer J, Fitch SB, Kaminsky W, Benedict J, Scarrow RC, Kovacs JA. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3671–3676. doi: 10.1073/pnas.0637029100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ostfeld D, Colfax JA. Inorg. Chem. 1978;17:1796–1799. [Google Scholar]
- 62.Evans DR, Reed CA. J. Am. Chem. Soc. 2000;122:4660–4667. [Google Scholar]
- 63.Que L, Jr, Tolman WB. Angew. Chem., Int. Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 64.Almarsson O, Adalsteinsson H, Bruice TC. J. Am. Chem. Soc. 1995;117:4524–4532. [Google Scholar]
- 65.Armstrong WH, Lippard SJ. J. Am. Chem. Soc. 1984;106:4632–4633. [Google Scholar]
- 66.Carroll MM, Norton JR. J. Am. Chem. Soc. 1992;114:8744–8745. [Google Scholar]
- 67.Tyler LA, Noveron JC, Olmstead MM, Mascharak PK. Inorg. Chem. 1999;38:616–617. doi: 10.1021/ic990794m. [DOI] [PubMed] [Google Scholar]
- 68.Jackson HL, Shoner SC, Rittenberg D, Cowen JA, Lovell S, Barnhart D, Kovacs JA. Inorg. Chem. 2001;40:1646–1653. doi: 10.1021/ic001271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Govindaswamy N, Quarless DA, Jr, Koch SA. J. Am. Chem. Soc. 1995;117:8468–69. [Google Scholar]
- 70.Millar M, Lee JF, Koch SA, Fikar R. Inorg. Chem. 1982;21:4105–6. [Google Scholar]
- 71.Herskovitz T, Depamphilis BV, Gillum WO, Holm RH. Inorg. Chem. 1975;14:1426–30. [Google Scholar]
- 72.Lane RW, Ibers JA, Frankel RB, Papaefthymiou GC, Holm RH. J. Am. Chem. Soc. 1977;99:84–98. doi: 10.1021/ja00443a017. [DOI] [PubMed] [Google Scholar]
- 73.Collman JP, Gagne RR, Halbert TR, Marchon JC, Reed CA. J. Am. Chem. Soc. 1973;95:7868–7870. doi: 10.1021/ja00804a054. [DOI] [PubMed] [Google Scholar]
- 74.MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 75.Ogo S, Wada S, Watanabe Y, Iwase M, Wada A, Harata M, Jitsukawa K, Masuda H, Einaga H. Angew. Chem., Int. Ed. Engl. 1998;37:2102–2104. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2102::AID-ANIE2102>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 76.Yeh C-Y, Chang CJ, Nocera DG. J. Am. Chem. Soc. 2001;123:1513–1514. doi: 10.1021/ja003245k. [DOI] [PubMed] [Google Scholar]
- 77.Shirin Z, Young VG, Jr, Borovik AS. Chem. Commun. 1997:1967–1968. [Google Scholar]
- 78.Addison AW, Rao TN, Reedijk J. J. Chem. Soc., Dalton Trans. 1984:1349. [Google Scholar]
- 79.Boeyens JCA, Forbes AGS, Hancock RD, Wieghardt K. Inorg. Chem. 1985;24:2926–2931. [Google Scholar]
- 80.Shoner SC, Nienstedt A, Ellison JJ, Kung I, Barnhart D, Kovacs JA. Inorg. Chem. 1998;37:5721–5725. [Google Scholar]
- 81.Matouzenko GS, Bousseksou A, Borshch SA, Perrin M, Zein S, Salmon L, Molnar G, Lecocq S. Inorg. Chem. 2004:227–236. doi: 10.1021/ic034450e. [DOI] [PubMed] [Google Scholar]
- 82.Di Vaira M, Ghilardi CA, Sacconi L. Inorg. Chem. 1976;15:1955. [Google Scholar]
- 83.Hagadorn JR, Que L, Jr, Tolman WB. Inorg. Chem. 2000;39:6086–6090. doi: 10.1021/ic000531o. [DOI] [PubMed] [Google Scholar]
- 84.Ray M, Hammes B, Yap GPA, Rheingold AL, Borovik AS. Inorg. Chem. 1998;37:1527–1532. [Google Scholar]
- 85.Zang Y, Kim J, Dong Y, Wilkinson EC, Appelman EH, Que L., Jr J. Am. Chem. Soc. 1997:4197–4205. [Google Scholar]
- 86.Clark T, Hennemann M, Eldik Rv, Meyerstein D. Inorg. Chem. 2002;41:2927–2935. doi: 10.1021/ic0113193. [DOI] [PubMed] [Google Scholar]
- 87.Hagen KS, Holm RH. J. Am. Chem. Soc. 1982;104:5496–7. [Google Scholar]
- 88.Millar M, Lee JF, Fikar R. Inorg. Chim. Acta. 1996;243:333. [Google Scholar]
- 89.Shoner S, Barnhart D, Kovacs JA. Inorg. Chem. 1995;34:4517–18. [Google Scholar]
- 90.Noveron JC, Olmstead MM, Mascharak PK. J. Am. Chem. Soc. 2001;123:3247–3259. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 91.Artaud I, Chatel S, Chauvin AS, Bonnet D, Kopf MA, Leduc P. Coord. Chem. Rev. 1999;190-192:577–586. [Google Scholar]
- 92.Mascharak PK, Harrop TC. Acc. Chem. Res. 2004;37:253–260. doi: 10.1021/ar0301532. [DOI] [PubMed] [Google Scholar]
- 93.Beissel T, Buerger KS, Voigt G, Wieghardt K, Butzlaff C, Trautwein AX. Inorg. Chem. 1993;32:124–6. [Google Scholar]
- 94.Noveron JC, Olmstead MM, Mascharak PK. Inorg. Chem. 1998;37:1138–1139. doi: 10.1021/ic971388a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.