

Abstract
An immune tolerant tumor microenvironment promotes immune evasion of lung cancer. Agents that antagonize immune tolerance will thus aid the fight against this devastating disease. Members of the tumor necrosis factor receptor (TNFR) family modulate the magnitude, duration and phenotype of immune responsiveness to antigens. Among these, GITR expressed on immune cells functions as a key regulator in inflammatory and immune responses. Here, we evaluate the GITR agonistic antibody (DTA-1) as a mono-therapy and in combination with therapeutic vaccination in murine lung cancer models. We found that DTA-1 treatment of tumor-bearing mice increased: (i) the frequency and activation of intratumoral natural killer (NK) cells and T lymphocytes, (ii) the antigen presenting cell (APC) activity in the tumor, and (iii) systemic T-cell specific tumor cell cytolysis. DTA-1 treatment enhanced tumor cell apoptosis as quantified by cleaved caspase-3 staining in the tumors. DTA-1 treatment increased expression of IFNγ, TNFα and IL-12 but reduced IL-10 levels in tumors. Furthermore, increased anti-angiogenic chemokines corresponding with decreased pro-angiogenic chemokine levels correlated with reduced expression of the endothelial cell marker Meca 32 in the tumors of DTA-1 treated mice. In accordance, there was reduced tumor growth (8-fold by weight) in the DTA-1 treatment group. NK cell depletion markedly inhibited the antitumor response elicited by DTA-1. DTA-1 combined with therapeutic vaccination caused tumor rejection in 38% of mice and a 20-fold reduction in tumor burden in the remaining mice relative to control. Mice that rejected tumors following therapy developed immunological memory against subsequent re-challenge. Our data demonstrates GITR agonist antibody activated NK cell and T lymphocyte activity, and enhanced therapeutic vaccination responses against lung cancer.
Keywords: APC, lung cancer, NK, T cell activation, vaccination responses
Abbreviations: Ab, antibody; APC, antigen presenting cell; BMA, bone marrow adherent; CTL, cytotoxic T lymphocyte; DC, dendritic cell; DTA-1, anti-GITR antibody; ERK, extracellular signal-regulated kinase; GITR, glucocorticoid‐induced TNFR‐related gene;IFNγ, interferon γ; JNK, janus kinase; MAPK, mitogen-activated protein kinase; MDSC, myeloid derived suppressor cell; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NK, natural killer; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1;TNFα, Interferon Alpha; TNFR, tumor necrosis factor receptor; TCR, T cell receptor; Treg, regulatory T cell; P38, p38 mitogen-activated protein kinase(s)
Introduction
Lung cancer causes more deaths than the next 3 most common cancers (colon, breast and prostate) combined. Worldwide more than 1.1 million deaths are attributed to lung cancer annually.1 Despite existing therapeutic efforts, the long-term survival rates for lung cancer patients remains low, thus new strategies are desperately needed to fight this lethal disease. Cancer immunotherapy, the harnessing of the immune system to combat cancer, offers an attractive therapeutic option for long-term immunological protection against this malignancy. Agents that activate the immune system against lung carcinoma will be an important addition to the existing armaments to combat this disease.
Tumor growth and invasion leads to inflammatory responses but the immune system generally develops tolerance to cancer. Genetic alterations in oncogenes and tumor suppressor genes or epigenetic changes in the tumor modulate tumor growth and invasion that orchestrate the persistence of inflammatory leukocytic infiltrates in the tumor. The tumor programs the infiltrates to sustain dysregulated inflammation that is hypo-responsive to the tumor. Both host antigen presenting cell (APC) and T-cell mediated antitumor activities are downregulated in cancer patients 2,3 and murine lung cancer models.4 These cellular infiltrates modulate tumor development and progression. In multiple human cancers, tumor-infiltrating effector T cells are associated with improved prognoses and intratumoral infiltration by relatively high numbers of activated T lymphocytes 5,6 and APCs 7 herald a better prognosis in lung cancer patients.
The immune system plays a crucial role in tumor immune surveillance. The clinical application of immune-based therapeutic strategies is to elicit immune activation for tumor rejection. Immune modulatory monoclonal antibodies that target immune evasion pathways in cancer have shown efficacy in clinical trials.8-10 Cancer cells often express a protein called programmed cell death ligand 1 (PD-L1) that facilitates their ability to evade the immune system. An immunotherapeutic strategy targeting this immune evasion pathway and stimulating the immune activation of T cells has demonstrated promise in early phase clinical trials against lung cancer. This therapy works by releasing the brakes on T-cell immune activation by blockade of the co-inhibitory checkpoint programmed cell death 1 (PDCD1, better known as PD-1) and its ligand (PD-L1, also kown as CD274/B7-H1) to disable the mechanisms of tumor immune escape and improve antitumor immune activity.9
Another prominent pathway of immune modulation is through the activation of glucocorticoid-induced tumor necrosis factor (TNF) receptor related gene or ‘GITR’, a target listed by the National Cancer Institute as the 12th most promising immunotherapy for cancer. GITR also known as TNF receptor superfamily member 18 (TNFRSF18) is expressed on innate and adaptive components of the immune system including CD4+ T cells, CD8+ T cells, natural killer (NK) cells, B cells, macrophages and dendritic cells (DCs) 11,12 and functions as a key regulator of inflammatory and immune responses. On T cells, GITR acts as a co-stimulatory molecule 11,13 enhancing T-cell proliferation and cytokine production in response to T-cell receptor (TCR) stimulation. Moreover, GITR cross-linking inhibits T-cell receptor-induced apoptosis 11 and sustains T-cell survival and responsiveness 14-16 by triggering 3 distinct mitogen activated protein kinase (MAPK) pathways (ERKs, JNKs and p38) and activating nuclear factor κB (NF-κB).
In this study, we evaluated the GITR agonist antibody (GITR Ab, termed DTA-1) as monotherapy or in combination with therapeutic vaccination in the murine Lewis Lung Cancer model. Both innate and T cell immune responses are downregulated in lung cancer. We posited that agonistic activation of GITR on immune cells would stimulate the antitumor activities of these effectors in murine lung cancer models. The rationale for evaluating GITR agonist in combination with the vaccine approach is that immune modulatory monoclonal antibodies (mAbs) administered as monotherapy achieve antitumor benefit but only in a subset of patients. For example, in early studies, an anti-PD-1 drug known as nivolumab (BMS-936558) was efficacious in causing tumor shrinkage in 1 out of 5 people with non-small cell lung cancer (NSCLC), while a drug targeting PD-L1 (known as BMS-936559) shrank tumors in 1 out of 10 people.9 Thus, a combination with other therapies is essential to improve the management and clinical outcome of patients who do not respond or those whose disease eventually progresses.17 Our results demonstrate that GITR agonist Ab mediated tumor reduction in lung cancer is NK cell and T cell dependent. Furthermore, GITR Ab augmented therapeutic vaccination responses against lung cancer. Our data provide support for the development of agents that target GITR as a therapeutic avenue against lung cancer.
Results
Agonistic Anti-GITR antibody therapy inhibits 3LL tumor growth
First, we evaluated the biological effect of administering agonistic anti-GITR (DTA-1) antibody on 3LL tumor growth in C57BL/6 host mice. DTA-1 inhibited tumor volume (Fig. 1A) and weight (8-fold, day 21; Fig. 1B) in comparison to controls . Immunocytochemistry of the showed increased tumor-infiltrating T cells and enhanced staining for cleaved caspase 3, a marker indicative of apoptotic tumor cells, in the DTA-1 treatment group relative to the control (Fig. 1C).Furthermore, mice in the control treatment group had visible lung metastases from cancer cells that presumably migrated from the s.c. implantation site whereas the lungs from the DTA-1 treated group did not show any visible carcinoma growth, suggesting that DTA-1 inhibited the migration of cancer cells in vivo (Fig. 1D). Tumors from the DTA-1 treated group had enhanced expression of caspase 8 (7.5 fold) in comparison to control (Fig. 1E) and increased APC activity in the tumor (16-fold; Fig. 1F). The levels of apoptotic tumor cells were also found to be increased following DTA-1 treatment (in comparison to controls) as indicated by elevated annexin V and propidium iodide (PI) positive apoptotic tumor cells at both early (8-fold) and late (5-fold) time points in vivo (Fig. 1G i–iv).
Figure 1.

Administration of anti-GITR agonistic antibody reduction of 3LL tumor burden. 3LL (2 × 105) cells were inoculated s.c. on the right supra scapular area of C57BL/6. Mice bearing established tumors were administered anti-glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR) antibody (DTA-1) or isotype control antibody via i.p. route every other day for 2 weeks. (A, B) In comparison to controls DTA-1 administration led to inhibition in tumor volume (A) and tumor weight (B). (C) Immunocytochemistry of the tumor sections showed increased T cells and cleaved caspase 3 in the tumor areas. (D) Histological examination of lung tissue metastases. Tumors from the DTA-1 treated group had enhanced expression of caspase 8 (E) and antigen presenting cell (APC) activity was measured by the ability of APC to process and present chicken ovalbumin and activate MHC Class I OVA peptide specific reporter CD8-T cell line B3Z to secrete IL-2 (pg/ml). (F) in comparison to control. Apoptosis quantified by the % of annexin V and propidium iodide (PI) double positive cells (Annexin V/PI+ve) stained gated on the CD45-ve cells as determined by immunostaining and cytofluorimetric analysis showed increased apoptotic tumor cells in the DTA-1 treatment group in comparison to control (Gi-iv). Values are shown as the mean ± SEM (n = 8 mice/group); statistical analysis was performed by Student's test; *P < 0.05).
Anti-GITR agonistic antibody treatment augments NK and T-cell effectors activation in tumor-bearing mice
We next sought to evaluate the impact of DTA-1 treatment on the frequency and activation status of innate and immune effectors in tumor-bearing mice. We found that DTA-1 treatment in the tumor relative to the control increased: (i) the frequency of activated NK cells expressing IFNγ (2-fold), granzyme (2-fold) and perforin (5-fold) (Fig. 2A i–viii); (ii) the percentage of CD4+CD107a+ (3.6-fold) and CD8+CD107a+ (4.5-fold) cells (Fig. 2B i-vi); and (iii) modulated the expression of CD8+ cytokines and effector molecules, such as IFNγ (3-fold), perforin (1.5-fold) and granzyme (2-fold) as well as reduced IL-10 (6.1-fold; Fig. 2C i–xi). In comparison to controls, DTA-1 increased the frequency of CD8+ (2.4-fold), NK (2-fold), and CD4+ (1.5.-fold) immune cells without altering the frequency of F480 macrophages or CD11c+ DCs in the tumor (Fig. 2D). DTA-1 did not alter the frequency of CD4+CD25+Foxp3+ Treg (data not shown) but reduced the frequency of CD11b+Gr1+ expressing myeloid-derived suppressor cells (2-fold) in the tumor (Fig. 2E i–iv). The cytokine levels of IFNγ, IL-10, TNFα and IL-12 were subsequently determined in the tumors and spleens following treatment. DTA-1 increased IFNγ (4-fold), TNFα (4.6-fold) and IL-12 (4.8-fold) but reduced IL-10 (39-fold) cytokines at the protein level in the tumors (Fig. 2F). A similar cytokine pattern was observed systemically in spleens in DTA-1 treated mice in comparison to controls (Fig. 2G). Furthermore, DTA-1 treatment enhanced the specific cytolytic activity of systemic splenic T cells against parental tumor cells, killing [E:T at 10:1, (3-fold) and at 5:1, (2.4-fold) (Fig. 2H). In comparison, there was no difference in T-cell cytolysis against the non-related B16 tumors between the DTA-1 treated and control groups (data not shown). To determine the significance of increased activated NK cells, CD4+ T or CD8+ T lymphocytes in the tumor following DTA-1 treatment, individual antibody depletion of these effectors was subsequently performed by immune cell-depleting antibody injection. Depletion of NK, CD4+ and CD8+ T lymphocytes inhibited the anti-tumor activity of DTA-1 treatment with NK and CD8+ T-cell depletion being more effective than CD4+ T-cell depletion (Fig. 2I).
Figure 2.

(See previous page). Anti-GITR agonistic antibody treatment increased NK and T-cell activities in tumor-bearing mice. Mice bearing 7 day established 3LL tumors were treated with anti-glucocorticoid-induced tumor necrosis factor (TNF) receptor anti-GITR antibody or isotype control antibody for the duration of the experiment. DTA-1 increased the frequency of activated: NK cells expressing IFNγ, perforin and granzyme (Ai-viii), CD4+CD107a+ and CD8+ CD107a+ (Bi-vi), CD8+ cells expressing IFNγ, perforin and granzyme but reduced IL-10 expression (Ci-xi) in comparison to control. (D) DTA-1 administration increased the: frequency of NK, CD3+, CD4+, CD8+ cells but did not alter the frequency of F480+ macrophages or CD11c+ dendritic cells (DCs) in tumor-bearing mice. DTA-1 decreased the frequency of myeloid-derived suppressor cells (MDSCs) in the tumor in comparison to control (Ei-iv). (F-G) ELISA demonstrated increased IFNγ, IL-12, and TNFα but reduced IL-10 in the tumors and systemically in spleens following DTA-1 administration in comparison to controls. (H) Systemic T cell specific cytolysis against parental tumor cells. Purified splenic T cell effectors were co-cultured with CFSE-labeled tumor cells at effector-to-target (E:T) cell ratios of 10:1–5:1 for 4 h prior to analysis by flow cytometry. The cytolysis of non- related B16 tumors between the DTA-1 treated groups and controls was assayed as control (data not shown). (I) Individual depletion of NK, CD8+ and CD4+ T cells inhibited the anti-tumor activity of DTA-1. Seven-day tumor bearing mice were individually treated with respective depleting antibody (200 μg/dose via i.p. injection) or isotype control every 48 h for 2 weeks. Values are shown as the mean (n= 8 mice per group) ± SEM; statistical analysis was performed by Student's t-test; *P < 0.05 DTA-1 vs control.
Anti-GITR antibody treatment modulates angiogenic chemokines
The angiogenic signature in the tumors was next determined following therapy. The tumors of DTA-1 treated mice had decreased pro-angiogenic [Cxcl2 (6-fold), Cxcl5 (3.6-fold), Angpt2 (12.5-fold), Angpt1 (4.5-fold) and Vegf α(2.8–fold)] but increased anti-angiogenic [(Cxcl9 (2-fold) and Cxcl10 (3-fold)] transcript levels. (Fig. 3A i–ii) Consistent with this signature was reduced transcript encoding the endothelial marker MECA32 (2-fold) in the tumor compared to control (Fig. 3B i–iv).
Figure 3.

Anti-GITR agonistic antibody treatment alters the balance of pro and anti-angiogenic chemokines in the tumor. Real-time PCR for transcripts encoding angiogenesis-related chemokines. DTA-1 treatment of tumor-bearing mice decreased pro-angiogenic (VEGF-a, CXCL2, CXCL5, Ang1 and Ang2) (Ai) but increased anti-angiogenic chemokine (CXCL9 and CXCL10) mRNA levels (Aii) in the tumor in comparison to control. There was a decrease in the endothelial cell marker expression MECA 32 in the tumors of DTA-1 treated mice (B i–iv). Values are shown as the mean ± standard error of the mean (n= 8 mice per group) ± SEM; statistical analysis was performed by Student's t-test; *P < 0.05 (DTA-1 vs control).
Agonistic anti-GITR antibody monotherapy attenuates orthotopic tumor burden
The therapeutic antitumor efficacy of DTA-1 was subsequently determined in an established (7-day prior) orthotopic 3LL lung cancer model. DTA-1 treatment inhibited tumor burden (3-fold) compared to controls, as determined by histological examination and EpCam staining (Fig. 4A and B). In the control group, there was 10% decrease in the average body weight at the end of the experimental duration but no significant weight change was observed in the DTA-1 treatment group. Similar to our observations in the s.c model, we found increased activated NK and T cell effector frequency in the lung following treatment (data not shown).
Figure 4.
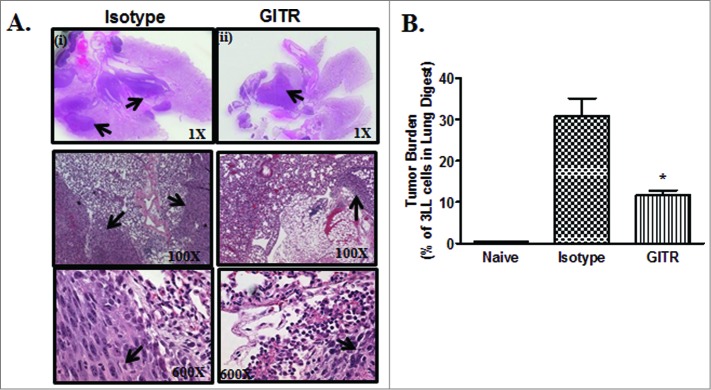
Anti-GITR agonistic antibody treatment inhibited 3LL orthotopic lung tumor growth. To examine the effect of DTA-1 treatment in an orthotopic setting, 104 3LL cells in 25 μL sterile NS diluent were injected by the transthoracic route in the left lung of C57BL/6 mice. One week following tumor inoculation, mice were individually treated with control isotype antibody (Ab) or DTA-1 Ab via i.p. route every 48 h for 4 weeks. (A) Five weeks following tumor implantation, mice were euthanized, lungs perfused and harvested for histological evaluation of tumor burden by H & E staining of tumor sections (arrows indicate tumor). (B) Tumor burden was also evaluated by cell surface staining for Epcam in a single cell suspension of tumor digests and quantified by fluorescence cytometry. Data are shown as the mean (n = 8 mice/group) ± SEM; statistical analysis was performed by Student's t-test; *P < 0.05 (DTA-1 vs control).
Anti-GITR antibody enhanced therapeutic vaccination responses
The impact of administering GITR agonist on therapeutic vaccination responses in vivo was evaluated utilizing 3LL OVA tumor cells in combination with autologous bone marrow-derived adherent (BMA) cells pulsed with OVA protein, which served as the vaccine. Fourteen-day BMA cell culture was evaluated by cell surface staining/flow cytometry analyses. BMA cells were comprised of a heterogeneous population of monocytes (51%-CD11b+), macrophages (14%-F480+CD11b+), DCs (10%-CD11c+, DEC205+), stromal cells (20%-CD45−/CD11b−/CD44+/CD34−) and B cells (8%-CD19+) and cell surface phenotype (MHC Class I (94%), MHC Class II (53%), CD80 (57%) and CD86 (37%). Mice bearing 3LL-OVA were treated with: (i) diluent, (ii) BMA-OVA, (iii) BMA-OVA + isotype and (iv) BMA-OVA + DTA-1. Compared to controls, therapeutic vaccination with BMA-OVA substantially inhibited tumor growth rate without eradicating the tumors (5-fold) (Fig. 5A). In comparison to controls, the combination of BMA-OVA + DTA-1 had the most pronounced tumor growth inhibition (20-fold) (Fig. 5A). H&E section revealed increased leukocytic infiltrates and increased tumor staining of cleaved caspase 3 (Fig. 5B). Among recipients of the combinatorial BMA-OVA plus DTA-1 bimodal therapy, 38% of mice rejected their primary tumors and 3LL-OVA tumor cells (2 × 105) on re-challenge (Table 1). Mice that had rejected tumors in the vaccination plus anti-GITR treatment had increased splenic production of IFNγ in response to OVA in comparison to naïve mice (Fig. 5C).
Figure 5.

Anti-GITR agonistic antibody enhanced therapeutic vaccination responses. To assess the ability of DTA-1 to enhance antitumor responses to therapeutic vaccination, 2.0 × 105 3LL-OVA tumor cells were injected s.c. in the right supra scapular area of C57BL/6 mice. The vaccine consisted of BMA cells that had been pulsed with OVA protein for 6 h. Prior to vaccination, BMA cells were washed in PBS twice, suspended in saline (0.1 × 106 in 200 μL/mouse) and administered by s.c. injection on the contra-lateral flank on days 7 and 14 post tumor-inoculation. Groups included: (i) Diluent, (ii) BMA-OVA, (iii) BMA-OVA + Isotype and (iv) BMA-OVA + DTA-1. Seven-days post tumor inoculation, the isotype or DTA-1 antibodies (100 μg/dose) were administered every 48 h for 2 weeks. (A) Tumor growth rate. (B) Histological examination by H & E staining and immunohistochemistry for cleaved caspase 3. The arrows are indicative of tumor infiltrates and cleaved caspase 3 staining. (C) Splenocytes of DTA-1 treated mice that rejected tumors on rechallenge showed increased production of IFNγ compared to naïve controls. Data are shown as the mean (n = 8 mice/group) ± SEM; statistical analysis was performed by Student's t-test; *P < 0.05.
Table 1.
DTA-1 and therapeutic vaccination caused tumor rejection and immunological memory. 38% of mice in the BMA-OVA + DTA-1 Ab treatment group rejected primary tumors and 3LL-OVA tumor cells (2 × 105) on re-challenge
| Groups | Complete Regression |
|---|---|
| Diluent | 0/8 |
| BMA-Ova | 0/8 |
| BMA-Ova + Isotype | 0/8 |
| BMA-Ova + GITR | 3/8 |
Discussion
Lung cancer has a dismal prognosis despite existing clinical therapeutic avenues. Thus, novel therapeutic strategies must be developed to efficaciously combat this lethal malignant disease. Cancer immunotherapy, the harnessing of the immune system to fight cancer, holds promise for long-term benefit against lung cancer and immune-based strategies have recently ignited renewed interest in experimental cancer therapies. Based on advances in cellular and molecular immunology, sophisticated immunotherapeutic approaches are under development for the treatment of lung cancer, which traditionally has not been considered an immune-sensitive malignancy. For example, targeting the T-cell immune regulatory checkpoint mediated by PD-L1 has shown promise in lung cancer. Tumor cell-derived PD-L1 binds to PD-1 on the surface of T cells and prevents T cells from detecting and attacking the tumor. Relinquishing this immunosuppression by the treatment of cancer patients with MAbs that block either PD-1 or PD-L1 thereby leads to tumor shrinkage and improvement in long-term patient survival.9 In addition, vaccines have now reached Phase III clinical trials but active-specific immunotherapy has yet to become an element of standard therapy for patients with lung cancer.
The immune tumor microenvironment is tolerant to the tumor. Many facets of host APC-, NK- and T-cell activity are suppressed thus allowing tumors to progress in immune-competent hosts. The inadequate function of the host immune system through the downregulation of APC, NK and T-cell effectors, as well as the elaboration of immune suppressive molecules present major mechanisms by which tumors evade the immune system. Biological agents that stimulate the antitumor activity of innate and immune cells will be useful for therapeutic development and intervention in lung cancer. In this study, we evaluated the impact of the GITR agonist Ab on the antitumor activity and therapeutic vaccination responses against lung cancer. To stimulate innate (APC, NK) and T-cell immune responses, we utilized the anti-GITR agonistic Ab DTA-1. We hypothesized that stimulation of GITR would stimulate the antitumor activities of innate and immune cells and augment therapeutic vaccination responses against lung cancer. Our data demonstrate that APC-, NK- and T-cell activities were downregulated in the 3LL tumor-bearing host whereas anti-GITR agonist antibody treatment of 3LL tumor bearing mice led to increased activities of APCs, NK cells, and T cells. For example, we observed enhanced production of the immunostimulatory cytokines IFNγ, IL-12 and TNFα but reduced IL-10 in the tumor following GITR stimulation. This cytokine signature has been shown to improve APC activity and induce Type 1 CTL responses.4 NK activity was also increased following DTA-1 treatment. The increased activity of NK cell effectors is significant since these innate immune cells are the first line of defense against tumors and inhibit tumor growth in a non-MHC restricted manner and without prior sensitization to an antigen.21,22 We also found that DTA-1 treatment enhanced the frequency and activity of CD8+ T cells in the tumor with higher levels of IFNγ, perforin and granzyme B but reduced IL-10. DTA1 therapy also increased systemic T-cell activity against autologous 3LL tumor cells. CTL and NK cells possess similar cytolytic mechanisms including secretion of perforin and granzyme B. Our data shows that splenic activated NK and T cell effectors derived from DTA1-treated mice had increased expression of the cytolytic markers granzyme and perforin as well as elevated levels of IFNγ. Also, the DTA1-treated mice harbored activated T cells displayed reduced expression of IL-10. Consistent with this finding, there was increased IFNγ and reduced IL-10 levels in the tumor, a cytokine signature profile purported to promote antitumor activity. While IFNγ induces anti-angiogenesis through the induction of CXCL9 and CXCL10,20 reduction in IL-10 can improve APC activity and promote Type I responses.23
In accord with the increased NK- and T-cell activities, there was an increase in the frequency of apoptotic tumor cells and a concomitant inhibition in tumor burden and migration of tumor cells from the primary tumor site to the lung. To prospectively determine the significance of increased activated T and NK cells following DTA-1 treatment, these effectors were individually depleted with mAbs administered to tumor-bearing mice. We found that depletion of NK cells, CD4+ or CD8+ T cells attenuated the antitumor activity of DTA-1. In comparison to the effects of CD4+ T and CD8+ T-cell depletion, NK-cell depletion more potently inhibited the antitumor activity of DTA-1 treatment. Based on these results, we surmise that the reduction in tumor growth following DTA-1 treatment could be accounted for by cooperation between activated NK- and T-cell effectors synergistically mediating antitumor activity in this lung cancer model. Based on the increased T and NK cell IFNγ production in the tumor, we also evaluated the angiogenic profiles in the tumor. The expression of transcripts encoding anti-angiogenic (CXCL9, CXCL10) and pro-angiogenic markers (VEGFa, CXCL2, CXCL5, Angiopoietin 1 and Angiopoietin 2) were quantified in the tumor by RT-qPCR. Following GITR stimulation, the anti-angiogenic chemokine transcripts Cxcl9 and Cxcl10 were present in relatively higher levels but the pro-angiogenic cytokine transcripts Vegfa, Angpt1, Angpt2, Cxcl2 and Cxcl5 were reduced. Accompanying this profile was a reduction in the transcript (Plvap) encoding the endothelial marker MECA 32 in the tumor that suggests that GITR stimulation promoted angiostasis in the tumor microenvironment by altering the balance of pro and anti-angiogenic chemokines. Thus, stimulation of the GITR pathway by DTA-1 Ab not only improves APC-, NK- and T-cell activities but promotes anti-angiogenesis chemokine signature in the tumor. In our studies, the GITR agonist Ab treatment of tumor-bearing mice did not change the frequency or activity of Treg cells (data not shown). However, the frequency of MDSCs was reduced following DTA-1 treatment that correlated with the decrease in activity of this suppressor cell population (data not shown).
Despite the significant tumor inhibitory effects of DTA-1 treatment, as a monotherapy it was insufficient to eradicate tumors. We next sought to evaluate the impact of DTA-1 treatment on therapeutic vaccination responses in vivo. To initiate resident antigen-specific immune responses, we utilized a cellular vaccine consisting of BMA cells pulsed with the model OVA antigen expressed by the OVA modified 3LL tumor cells. The BMA cells were pulsed with the OVA antigen to allow for antigen processing and presentation and then injected s.c. on the contra-lateral flank of the established tumor to initiate antigen specific antitumor immune responses. The BMA cells displayed a cell surface phenotype endowing them with APC activity consisting of efficiently processed and presented antigens capable of eliciting antigen-specific CD8+ T cells in vitro (data not shown) and the induction of antitumor responses in vivo. We selected this vaccine approach because of the relative ease of preparation and potential for wide applicability in cancer immunotherapy. For example the next-generation sequencing technologies enable efficient identification of activating molecular alterations that can be personally designed for tumor peptide antigens against molecular alterations in the coding sequences of oncogenic genes in a given patients’ tumor. This will provide broad applicability of this vaccination strategy for a wide variety of solid tumors. The bone marrow approach to generate the cellular vaccine can serve as an off-the-shelf reagent, MHC-matched for individual patients. This approach is also amenable to stimulation with adjuvants prior to administration. Furthermore, this vaccination approach will have great utility in the developing countries because of production, delivery and cost. In comparison to controls, therapeutic vaccination with BMA-OVA led to decreased tumor burden without complete eradication of the tumors. However, DTA-1 administration in combination with BMA-OVA vaccination led to the most substantial reduction in tumor burden with 38% of the mice completely eradicating the tumors and the remainder mice showing a 20-fold reduction in tumor burden in comparison to control. Our data demonstrates that therapeutic vaccination is more effective when combined with GITR agonist treatment. Finally, durable therapeutic benefit is supported by immunological memory induced in mice that rejected primary tumors following therapy, as these mice were efficient in rejecting a secondary tumor challenge and had enhanced splenic T-cell secretion of IFNγ in response to OVA stimulation in vitro.
Taken together, our data demonstrates that GITR stimulation alters the activities of the inflammatory infiltrates in the tumor microenvironment making the tumor permissive for immune destruction. In addition, GITR stimulation potently augmented therapeutic vaccination responses in vivo. We anticipate that the agonist GITR antibody will be beneficial to lung cancer patients and most effective when combined with therapeutic vaccination. Our data suggests that targeting the GITR pathway in combination with therapeutic vaccination could have broad applicability against solid malignancies. The results of this study are encouraging and warrant further evaluation of this combined approach in the treatment of other malignancies.
Material and Methods
Cell lines and reagents
The murine Lewis lung carcinoma (3LL, H-2b, also known as LLC, (ATCC CRL-1642) obtained from American Type Culture Collection and the β galactosidase reporter T cell hybridoma (B3Z which recognizes the kb Class I molecule and an ovalbumin (OVA) peptide, SL8 (SIINFEKL) obtained from N Shastri 18 were used in the studies. 3LL-OVA cells were generated by transfecting 3LL parental cells with the OVA constructs obtained from Dr. Frelinger. The expression vectors encoding either the full-length Ova or truncated Ova-(138-386) (Ova non-secretory) were transfected using Lipofectin (GIBCO/BRL) according to the manufacturer's instructions. Selection with the appropriate drug was performed as described.19 Stable 3LL-OVA transfectants were selected following OVA ELISA of cell lysates and cloned by limited dilution in 96-well plates. For the experiments described in this study we used the 3LL-OVA that expressed the truncated Ova-(138-386). The culture medium (CM) contained RPMI 1640 supplemented with 10% fetal bovine serum (Gemini Bioproducts), 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 2 mmol/L glutamine (JRH Biosciences). Fluorescein isothiocyanate-, phycoerythrin-, allophycocyanin-, PerCP- or APC-Cy7-conjugated anti-mouse mAbs to CD3 (145-2C11), CD4 (RM4-5), CD69 (H1.2F3) and CD8a (53-6.7) were purchased from BD Biosciences. Fluorescent-conjugated anti-mouse antibodies for flow cytometry: anti-CD49b, anti-CD11c, F4/80, DEC205, anti-CD44, anti-CD45, anti-CD34, anti-CD19, anti-perforin, anti-granzyme, anti-IL10, anti-IFNγ anti-EpCam and anti-CD107a were from eBioscience and anti-Gr1, anti-CD11b, anti-CXCR3 and Meca 32 were from Biolegend. Agonistic Ab to GITR (DTA-1), anti-CD4 (L3T4), anti-CD8 (YTS169.4), and anti-NK (NK1.1-PK136) were from BioXCell. Isotype control Ab was purchased from Sigma. IL-2, IFNγ, IL-12, IL-10 and TNFα were quantified with cytokine specific ELISA kits (eBioScience). Sensitivity: 3 pg/mL IL-2, 3 pg/mL IFNγ, 3-5 pg/mL IL-12, 30 pg/mL IL-10 and 8 pg/mL TNFα. Ovalbumin protein and Bradford protein quantification dye was obtained from Sigma. Tissue digestion buffer consisted of [0.2 mg/mL of Collagenase A (Boehringer Mannheim/Roche), 25 U/mL DNase (Sigma), and 0.3 U/mL of Dispase (Invitrogen) in RPMI. T cell purification columns were purchased from R&D Systems and Tregs and MDSCs were purified using Miltenyi kits. RNA isolation kit was from Qiagen, cDNA kit from BioRad and real time PCR primers were from IDT. Carboxyfluorescein succinimidyl ester (CFSE) was obtained from Invitrogen.
Cell culture
Cells (3LL, DC 2.4, B3Z and 3LL-OVA) were routinely cultured in Corning T75 cm2 tissue culture flask in humidified atmosphere containing 5% CO2 in air in culture medium (CM). The cell lines were mycoplasma and murine viral pathogen free. The cell lines were used up to the 10th passage. For bone marrow adherent (BMA) cell culture, bone marrow was harvested by flushing the femurs of C57BL/6 mice with RPMI supplemented with 20% fetal bovine medium (RP-20). The pooled marrow cells were plated in RP-20 supplemented with 1% penicillin-streptomycin and cultured for 72 h on flasks coated with 2% gelatin (Sigma). Non-adherent cells were washed off and adherent cells expanded in RP-20. Following the culture period (11–14 days), single cell suspensions of cultured BMA cells were stained for cell surface markers for: monocytes (CD11b+), macrophages (CD11b+/F4/80+), stromal cells (CD45−/CD11b−/CD44+/CD34−), DCs (CD11c+, DEC205+) and B cells (CD19+) and evaluated by flow cytometry analyses.
Tumorigenesis model
Pathogen- free C57BL/6 mice (6–8wk old; Jackson Lab) were maintained in the West Los Angeles Veterans Affairs Animal Research vivarium in accord with the institution's animal review board guidelines. All animal work was conducted in accord with the Veterans Affairs Institutional Animal care and Use Committee guidelines: id A3002-01. The Veterans Affairs Institutional Animal care and Use Committee review board approved all the studies involving animals in this manuscript. Animals exhibiting signs of pain or meeting the endpoint criteria were euthanized immediately according to the accepted institution-based protocol.
Mice were monitored daily for signs of distress from the tumor burden and to ameliorate pain and suffering mice were euthanized if the animals exhibited any clinical signs of distress, such as loss of appetite, 10% cachexia weight loss, loss of mobility, restlessness, depression, respiratory distress, tumor/skin breakdown, or failure to groom. Two × 105 3LL tumor cells were injected s.c. in the right supra scapular area of C57BL/6 mice. Tumor volumes were monitored by measuring 2 bisecting diameters of each tumor with calipers. Tumor volumes were calculated using the formula: V = 0.4ab,2 a = large diameter and small diameter. One week following tumor inoculation, mice with palpable tumors were injected i.p. with anti-GITR-specific (100 μg/dose) or isotype IgG2b antibody (100 μg/dose) every 48 h for 2 weeks. DTA-1 is a non-depleting agonistic antibody for GITR. Two or 3 weeks post tumor implantation, tumors and spleen were harvested for evaluation of the frequency and activity of leukocytic populations by specific staining/flow cytometric analyses. For NK, CD4+ and CD8+ T-cell depletion, 7- day tumor bearing mice were individually treated with the respective antibody (200 μg/dose via i.p. injection) or isotype control every 48 h for 2 weeks. This schedule depleted the respective effector cell population as determined by staining/flow cytometry analyses of spleens of the treated mice (data not shown).
CFSE based cytolysis assay
Total cytolytic T cell activity in the spleen against parental 3LL tumor cells or syngeneic B16 melanoma cells were evaluated following treatment with anti-GITR or Isotype control Ab on day 21 post tumor inoculation. Tumor targets were labeled with CFSE at a concentration of 1 μM in PBS for 15–20 min according to the manufacturer's instructions. After washing, the labeled targets were incubated with T cells purified from spleens using T cell columns (R&D Systems), and cytolytic activities were evaluated against the autologous 3LL tumor cell line and the syngeneic control B16 melanoma tumor cell line. The purified splenic T cell effectors were co-cultured with tumor cell targets (E:T of 10:1–5:1) for 4 h in quadruplet wells in a 96-well plate. Following a co-culture, cells were washed and analyzed by flow cytometry. Decreases in the frequency and intensity of CFSE labeled cells were used to calculate % of cytolysis in tumor targets.
Orthotopic model
Implantation of the tumors in the lung was performed as previously described.20 Briefly, 104 3LL cells (in 25 μL sterile NS diluent) were injected by the transthoracic route of C57BL/6 mice utilizing a tuberculin syringe with a 30-gauge needle in the left lung under ketamine/xylazine anesthesia. One week following tumor inoculation, a group of mice were sacrificed to confirm and determine the baseline tumor burden before initiation of therapy. One week following tumor inoculation, mice were individually treated with control isotype Ab or anti-GITR Ab (100 μg/dose) via i.p. route every 48 h for 4 weeks. Five weeks following tumor implantation, lungs were perfused and harvested for evaluation of tumor burden by H&E staining of tumor sections. Tumor burden was also evaluated in a single cell suspension of lung tumor digests by EpCam staining of tumor cells followed by cytofluorimetric evaluation. A total of 20,000 events were acquired on the FACSCanto flow cytometer and data analyzed by the FCS Express 3 software.
Vaccination model
3LL-OVA (2.0 × 105) tumor cells were injected s.c. in the right supra scapular area of C57BL/6 mice. The vaccine consisted of BMA cells that had been pulsed with OVA protein. Briefly, BMA cells were pulsed with 2.5 mg/mL OVA protein in CM at 37°C in an incubator with a humidified atmosphere containing 5% CO2 in air for 6 h. Cells were washed in PBS twice and suspended in normal saline (0.1 × 106 in 200 μL/mouse) and administered by s.c. injection (days 7 and 14 post tumor inoculation) on the contra-lateral flank of the flank with tumor. Groups included: (i) Diluent, (ii) BMA-OVA, (iii) BMA-OVA + isotype and (iv) BMA-OVA + anti-GITR. Seven-days post tumor inoculation, the isotype control or GITR agonist antibodies (100 μg/dose) were administered every 48 h for 2 weeks. Tumor burden was monitored as described above and H&E or staining for apoptotic tumor cells was performed on the tumor sections. Treated mice that had completely rejected the 3LL-OVA tumors were re-challenged with 2 × 105 3LL-OVA tumor cells on the left flank and monitored for tumor growth. Splenocytes from mice that had rejected the secondary tumor challenge were monitored for IFNγ secretion (IFNγ ELISA, R&D Systems) by plating 5 × 106 splenotytes in an overnight culture in the presence or absence of the 2.5 mg/mL OVA protein.
Antigen processing and presentation assay
A single cell suspension of tumor digest (5–10 × 104 cells/well) from controls or anti-GITR treated tumor bearing mice were co-cultured with 2.5 mg/mL OVA protein and the MHC Class I restricted CD8+ T cell line B3Z (105 cells/well) plated in CM in triplicate wells of a 96-well plate for 24 h. IL-2 secreted by the activated CD8+ T cells in the supernatant was quantified by ELISA.
Flow cytometry
Cytofluorimetric analysis was performed for the following T cell surface markers CD3, CD4, CD8, CD69 on single cell suspension of tumor digests following treatment as described above. Tumor cell digests were evaluated for the NK cells with the surface marker CD49b. CD4+ T cells, CD8+ T cells, and NK cells were individually evaluated for intracytoplasmic perforin, granzyme B, IFNγ or IL-10. CD107a on NK and T cells were evaluated by cell surface staining/flow cytometry. For analyses of leukocytic infiltrates, tumors were mechanically dissociated on a wire mesh by crushing with a 10 mL syringe and incubated in tissue digestion buffer at 37°C for 25 min. The cells were filtered through 70 μm nylon strainers (BD Biosciences), stained with specific markers and analyzed by flow cytometry. Samples were acquired on a FACSCanto (BD Biosciences/FACSCalibur flow cytometer (Becton Dickinson) in the University of California, Los Angeles, Jonsson Cancer Center Flow Cytometry Core Facility. A total of 10,000 to 25,000 live cell gated events were analyzed using FCS Express 3 (De Novo Software). Cells incubated with irrelevant isotype-matched antibodies and unstained cells served as controls.
Cytokine ELISA
The cytokines (IFNγ, TNFα, IL-10 and IL-12) in supernatants of splenocyte cultures in vitro or from spleen or tumor homogenates following in vivo experiments were determined by ELISA and the plates read at the specified wavelengths with a Microplate Reader (Amersham Biosciences).
Tumor tissue sectioning and immunohistochemistry
To determine the extent of lymphocytes infiltrating the tumors in the various treatment groups, C57BL/6 mice bearing 7-day tumors were injected i.p. with GITR-specific (100 μg/dose) or isotype IgG2b antibody (100 μg/dose) every 48 h for 2 weeks. Non-necrotic tumors were isolated and embedded in paraffin and serially sectioned to 5-μm thickness. Sections were H&E or immune stained for CD3+ T lymphocytes. To determine apoptotic tumor cells, tumor sections were stained for cleaved caspase-3. Antigen retrieval was accomplished with sodium citrate (10 mmol/L, pH 6.0). Sections were blocked with 10% normal goat serum, and probed with an antibody against CD3 or cleaved caspase 3. Primary antibodies were incubated overnight at 4°C. After incubation with secondary antibody (Vector Laboratories), staining was developed using DAB Substrate kit for Peroxidase (SK-4100, Vector Laboratories). Counter-stain was achieved with hematoxylin. The slides were observed under 1X71 Olympus Fluorescence microscope attached to a CCD camera. The images were acquired with 10X, 20X and 60X objectives using the Image Pro software.
Apoptosis
To determine the extent of apoptosis in the tumors following treatment, a single cell suspension of the tumor tissues were stained using a propidium iodide/Annexin V-FITC apoptosis detection kit (BD PharMingen) according to the manufacturer's instruction and the percentage of apoptotic CD45− tumor cells were analyzed by flow cytometry.
Total RNA preparation, cDNA synthesis and real-time qPCR
Mice bearing 7-day old tumors were treated with isotype or anti-GITR antibodies and 2 week following treatment, tumor tissues were quantified for Angpt1 (Ang1), Angpt2 (Ang2), Vegfa, Cxcl2, Cxcl5, Cxcl9, Cxcl10 and Plvap/Meca 32 gene expression using a SYBR Green Quantitative PCR Kit in the iCycler (Bio-Rad) and corrected with the β-actin housekeeping control gene. For qPCR analyses, RNA was isolated using a Qiagen kit. The cDNA was prepared with a kit (BioRad) according to the manufacturer's instructions. Amplifications were done in a total volume of 25 μL for 40 cycles of 15 s at 95°C, 20 s at 60°C, and 30 s at 72°C. Primer sequences were as follows: β-actin F, 5′- CCACAGCTGAGAGGGAAATC -3′ and R, 5′- TCTCCAGGGAGGAAGAGGAT -3′; Caspase 8 F, 5′- TGCTTGGACTACATCCCACAC-3′ and R, 5′- GTTGCAGTCTAGGAAGTTGACC -3′; Ang-1 F, 5′- TCTCATGCTAACAGGAGGTTGGTG -3′ R, 5′-GGATCATCATGGTGGTGGAACGTA-3′; Ang-2 F, 5′- CAAGAGCTCGGTTGCTATCCGTAA-3′ R, 5′ GTCCATGTCACAGTAGGCCTTGAT3′; VEGF-a F, 5′- TGTACCTCCACCATGCCAAGT-3′ R, 5′- CGCTGGTAGACGTCCATGAA-3′; CXCL5 F, 5′- GGTCCACAGTGCCCTACG-3′ R, 5′- GCGAGTGCATTCCGCTTA-3′; CXCL2 F, 5′- AGTGAACTGCGCTGTCAATG -3′ R, 5′- GAGAGTGGCTATGACTTCTGTCTG-3′; CXCL9 F, 5′- GCACGATCCACTACAAATCCC-3′ R, 5′- GGTTTGATCTCCGTTCTTCAGT-3′; CXCL10 F, 5′- CCAAGTGCTGCCGTCATTTTC-3′ R, 5′- TCCCTATGGCCCTCATTCTCA-3′.
Statistical analyses
All data are presented as mean ± SE. Statistical analysis was performed using Prism (GraphPad Software). We used analysis of variance for data with multiple groups, unpaired Student's t-test for dual comparison. P values <0.05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the: University of California Los Angeles Lung Cancer Program, Department of Veterans Affairs Medical Research Funds, NIH Grants (RO1 CA95686, RO1 CA126944 and P50 CA90388), National Center for Research Resources, Grant UL1RR033176, and is now at the National Center for Advancing Translational Sciences, Grant UL1TR000124 and Tobacco Related Disease Program Award Program of University of California (15RT-0207 and 20FT0082). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. Ca; 63:11–30; PMID: [DOI] [PubMed] [Google Scholar]
- 2.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263–74; PMID: [DOI] [PubMed] [Google Scholar]
- 3.Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res 2000; 6:1755–66; PMID: [PubMed] [Google Scholar]
- 4.Andersson A, Srivastava MK, Harris-White M, Huang M, Zhu L, Elashoff D, Strieter RM, Dubinett SM, Sharma S. Role of CXCR3 ligands in IL-7/IL-7R{alpha}-Fc-mediated antitumor activity in lung cancer. Clin Cancer Res 2011; 17:3660–72; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson SK, Kerr KM, Chapman AD, Kennedy MM, King G, Cockburn JS, Jeffrey RR. Immune cell infiltrates and prognosis in primary carcinoma of the lung. Lung Cancer 2000; 27:27–35; PMID:; http://dx.doi.org/ 10.1016/S0169-5002(99)00095-1 [DOI] [PubMed] [Google Scholar]
- 6.Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, Itoh T, Ohbuchi T, Kondo S, Katoh H. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer 2006; 94:275–80; PMID:; http://dx.doi.org/ 10.1038/sj.bjc.6602934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, et al.. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 2008; 26:4410–7; PMID:; http://dx.doi.org/ 10.1200/JCO.2007.15.0284 [DOI] [PubMed] [Google Scholar]
- 8.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443–54; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robert C, Thomas L, Bondarenko I, O'Day S, M DJ, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al.. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517–26; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1104621 [DOI] [PubMed] [Google Scholar]
- 11.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 2002; 3:135–42; PMID:; http://dx.doi.org/ 10.1038/ni759 [DOI] [PubMed] [Google Scholar]
- 12.Nocentini G, Riccardi C. GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur J Immunol 2005; 35:1016–22; PMID:; http://dx.doi.org/ 10.1002/eji.200425818 [DOI] [PubMed] [Google Scholar]
- 13.Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, Ayroldi E, Riccardi C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol 2004; 34:613–22; PMID:; http://dx.doi.org/ 10.1002/eji.200324804 [DOI] [PubMed] [Google Scholar]
- 14.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol 2004; 173:5008–20; PMID:; http://dx.doi.org/ 10.4049/jimmunol.173.8.5008 [DOI] [PubMed] [Google Scholar]
- 15.Esparza EM, Arch RH. Glucocorticoid-induced TNF receptor, a costimulatory receptor on naive and activated T cells, uses TNF receptor-associated factor 2 in a novel fashion as an inhibitor of NF-kappa B activation. J Immunol 2005; 174:7875–82; PMID:; http://dx.doi.org/ 10.4049/jimmunol.174.12.7875 [DOI] [PubMed] [Google Scholar]
- 16.Esparza EM, Arch RH. Glucocorticoid-induced TNF receptor functions as a costimulatory receptor that promotes survival in early phases of T cell activation. J Immunol 2005; 174:7869–74; PMID:; http://dx.doi.org/ 10.4049/jimmunol.174.12.7869 [DOI] [PubMed] [Google Scholar]
- 17.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al.. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–33; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shastri N, Gonzalez F. Endogenous generation and presentation of the ovalbumin peptide/Kb complex to T cells. J Immunol 1993; 150:2724–36; PMID: [PubMed] [Google Scholar]
- 19.McAdam AJ, Pulaski BA, Storozynsky E, Yeh KY, Sickel JZ, Frelinger JG, Lord EM. Analysis of the effect of cytokines (interleukins 2, 3, 4, and 6, granulocyte-monocyte colony-stimulating factor, and interferon-gamma) on generation of primary cytotoxic T lymphocytes against a weakly immunogenic tumor. Cell Immunol 1995; 165:183–92; PMID:; http://dx.doi.org/ 10.1006/cimm.1995.1204 [DOI] [PubMed] [Google Scholar]
- 20.Andersson A, Yang SC, Huang M, Zhu L, Kar UK, Batra RK, Elashoff D, Strieter RM, Dubinett SM, Sharma S. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol 2009; 182:6951–8; PMID:; http://dx.doi.org/ 10.4049/jimmunol.0803340 [DOI] [PubMed] [Google Scholar]
- 21.Zamai L, Ponti C, Mirandola P, Gobbi G, Papa S, Galeotti L, Cocco L, Vitale M. NK cells and cancer. J Immunol 2007; 178:4011–6; PMID:; http://dx.doi.org/ 10.4049/jimmunol.178.7.4011 [DOI] [PubMed] [Google Scholar]
- 22.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity 2007; 26:798–811; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2007.04.010 [DOI] [PubMed] [Google Scholar]
- 23.Sharma S, Stolina M, Lin Y, Gardner B, Miller PW, Kronenberg M, Dubinett SM. T cell-derived IL-10 promotes lung cancer growth by suppressing both T cell and APC function. J Immunol 1999; 163:5020–8; PMID: [PubMed] [Google Scholar]