

Abstract
Extracellular vesicles (EVs) have been shown to transfer various molecules, including functional RNA between cells and this process has been suggested to be particularly relevant in tumor-host interactions. However, data on EV-mediated RNA transfer has been obtained primarily by in vitro experiments or involving ex vivo manipulations likely affecting its biology, leaving their physiological relevance unclear. We engineered glioma and carcinoma tumor cells to express Cre recombinase showing their release of EVs containing Cre mRNA in various EV subfractions including exosomes. Transplantation of these genetically modified tumor cells into mice with a Cre reporter background leads to frequent recombination events at the tumor site. In both tumor models the majority of recombined cells are CD45+ leukocytes, predominantly Gr1+CD11b+ myeloid-derived suppressor cells (MDSCs). In addition, multiple lineages of recombined cells can be observed in the glioma model. In the lung carcinoma model, recombined MDSCs display an enhanced immunosuppressive phenotype and an altered miRNA profile compared to their non-recombined counterparts. Cre-lox based tracing of tumor EV RNA transfer in vivo can therefore be used to identify individual target cells in the tumor microenvironment for further mechanistical or functional analysis.
Keywords: carcinoma, Cre/Lox, exosomes, extracellular vesicles, glioma, immunosuppression, MDSCs, miRNA
Abbreviations: GFAP, Glial fibrillary protein; PBS, phosphate buffered saline; NIH, National Institutes of Health; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis
Introduction
Cancer cells employ various mechanisms to compromise their microenvironment, thereby overcoming growth barriers or endogenous tumor suppressive processes.1 Recent data suggest a central role of tumor-derived EVs in numerous processes impacting tumor growth such as promoting metastasis,2,3 inducing drug resistance and to skew the differentiation of monocytes into MDSCs,4,5 that strongly suppress antitumor immunity promoting cancer progression. Conceptually, the interaction of tumor cells with the host tissue has been based on the interplay of individual pairs of ligands and their receptors impacting their gene regulation. In contrast, EVs enable the direct transfer of molecules that are normally not considered in cell-cell communication, particularly RNAs.6-8 Therefore, intercellular signaling based on the release of EVs by tumor cells and concomitant uptake by cells of the surrounding tissue has recently been introduced as a different paradigm.9 However, to our knowledge, it has not been possible to pinpoint transfer of EV content, including RNA, to individual target cells in vivo which presents a major obstacle to identify EV-induced cellular changes that may be of therapeutic value. We could recently establish the Cre/LoxP system to trace EV transfer of RNA from hematopoietic cells to neurons under inflammatory conditions.10 Now we addressed the question whether expression of Cre recombinase in different tumor cell lines could lead to the release of EVs containing functional Cre mRNA.
Results and Discussion
A schematic presentation of our overall experimental strategy is shown in Figure 1A. We stably transduced two murine tumor cell lines with a retroviral or lentiviral vector expressing Cre recombinase alone or in combination with GFP. We employed the cell line TU2449, derived from a spontaneously arising tumor in a murine glioma model, overexpressing v-src specifically in the astrocytic lineage.11,12 In addition, we used the Lewis lung carcinoma cell line LLC2. Cre and GFP expression was verified by immunocytochemistry and flow cytometry respectively. Cre mRNA could be detected in EV-containing pellets but not in the supernatant prepared from tumor cell conditioned culture medium after ultracentrifugation (Fig. 1B). Importantly, RNase treatment did not abolish the signal, indicating that the Cre mRNA is localized inside vesicles and thus protected from digestion.13 Interestingly, GFP mRNA could be detected in EVs from glioma but not carcinoma cells. To exclude the association of Cre mRNA with protein particles, we digested EV preparations from both cell lines with protease alone, or followed by RNase digestion. These treatments did not abolish detection of Cre in contrast to treatment of EV preparations with detergent to lyse membrane vesicles in combination with RNase digestion (Fig. 1C). EV preparations from both cell lines were additionally processed for ultrastructural analysis (Fig. 1D), showing predominantly vesicles of the shape and size typically described for exosomes.14
Figure 1.

Tumor cells expressing Cre secrete different vesicular subtypes containing Cre mRNA (A) Schematic presentation of the experimental strategy. Tumor cells are stably transduced to constitutively express Cre recombinase and GFP. After transplantation into a Cre reporter mouse, lateral transfer of Cre containing EVs leads to recombination events in the host. (B) Detection of Cre mRNA by RT-PCR in EV preparations from tumor cell conditioned medium. Cre is detectable in the pellet (lane 1) but not in the supernatant (lane 2) after ultracentrifugation. Cre expressing cells served as positive- (lane 3) and omission of reverse transcriptase (RT) from the reverse transcription step as negative controls (lane 4). GFP mRNA was detectable in EVs from glioma- but not carcinoma cells (lane 5), a positive control from GFP expressing cells (lane 6). (C) Cre mRNA is contained in vesicles. Cre mRNA can be detected in EVs (lane 1), also after treatment with Proteinase K (45 min, 2 mg/mL) (lane 2) or Proteinase K followed by RNase digestion (45 min) (lane 3), whereas treatment of EVs with detergent (Triton- X-100, 0.1%) followed by RNase digestion eliminated the signal for Cre (lane 4). RT- negative samples served as negative controls (lane 5). (D) Electron micrographs of vesicle preparations of glioma (TU2449) and carcinoma (LLC2) cell lines showing the cup shaped morphology and size of 50–100 nm typical for exosomes. (F and G) Pelleted (100.000 × g) EVs from cell culture supernatant were loaded on a continuous sucrose gradient and ultracentrifuged. Vesicle subpopulations were tested for all subfractions by blotting against the indicated proteins with cell lysates serving as controls. Fractions 2–7 typically contain exosomes whereas larger vesicles or membrane blebs are contained in fractions 9–12. Cre protein was not detectable in any of the fractions. Representative images of three (B–E) separate sample preparations (Scale bar D, 50 nm).
For further analysis, EV preparations from tumor cell supernatants were normalized according to their protein content and separated by sucrose density ultracentrifugation. The gradient fractions were processed for western blot analysis with the exosomal markers TSG101 and CD9 as well as calnexin to exclude putative ER contaminations. In line with previously published data, a signal for TSG101 and CD9 expression was strongest in the exosomal fractions 2–5 and very little to no signal in the high-density fractions 7–12.15 Calnexin reactivity was not detected. An aliquot from each fraction was processed separately for RT-PCR analysis for Cre mRNA. For the cell line TU2449, we found the strongest signal of Cre transcript in fractions 4–7, and a weaker but detectable signal in all the other fractions (Fig. 1D). Importantly, we could not detect Cre protein in any of the fractions. In separate experiments where up to 50 μg protein of total EV preparations were loaded per lane, we could not detect any signal, even after long exposure times. Cre protein was nearly undetectable in cellular detergents lysates and required nuclear extracts to become detectable (Fig. 1E). This is consistent with the notion that Cre contains an artificial nuclear translocation site (NLS) and therefore localizes to the nucleus.16,17
For LLC2 cells, Cre mRNA was found only in the exosomal fractions 3–7; Cre protein was also not detected in exosomes but was present in nuclear extracts of the cells (Fig. 1F). This shows that Cre mRNA expressed by tumor cells can become incorporated into EVs that are released from the cells with the highest amounts detected in exosomes. Interestingly, the Cre mRNA signal did not completely overlap with that of exosomal markers TSG101 and CD9. However, although efforts for a more precise classification are underway,8 EVs are not very well characterized in terms of their marker profile and putative biological functions. Our results may thus indicate possible subpopulations of vesicles that differ in their RNA cargo. The failure to detect Cre-protein in exosomes may be due to the localization of the protein in the nucleus rather than in the cytosol.
Using the murine glioma cell line TU2449, we investigated if recombination events could be observed after transplantation that would indicate a transfer of Cre mRNA via EVs. TU2449 cells show a very aggressive and highly infiltrative growth recapitulating many aspects typical for gliomas.12 Immunofluorescent staining for the marker protein β-galactosidase performed 10–12 d after tumor cell implantation revealed frequent recombination events predominantly along the edges of the tumor tissue but also deeper in the brain parenchyma (Fig. 2A). We did not observe any co-labeling of recombined cells with GFP endogenously expressed by the tumor cells, thereby excluding cell fusion as an alternative explanation for the observed recombination. To identify recombined cell types, we performed immunofluorescent co-staining of β-galactosidase with markers for neurons (NeuN, Fig. 2B), endothelial cells (von Willebrand Factor, vWF, Fig. 2C), microglia (Iba1, Fig. 2D) and leukocytes (CD45, Fig. 2E). Immune cell infiltrates can regularly be found within malignant gliomas although their influence on the progression of pathology is not very well understood. Analysis by flow cytometry revealed that >90% of all recombined cells in or around the tumor mass were CD45+ leukocytes and about 50% were CD11b+Gr1+ MDSCs (Fig. 2E). To test whether tumor-derived Cre mRNA-containing EVs are sufficient to induce reporter gene expression, we injected 2 μL total EV preparations as well as the subfractions 2–6 (exosomes) and 9–12 (membrane blebs) into the brains of Cre- reporter mice. As a control, we injected lysates from TU2449CreGfp cells to exclude transfer of Cre by cellular debris (2 separate EV preparations; n = 2 mice with 2 injections into each mouse). Recombined cells could be observed along the injection canal 3 d after injection of tumor EVs, exosomes and membrane blebs but not lysate. We observed cells expressing the neuronal marker NeuN, but not the astrocytic marker GFAP consistent with our findings in the transplantation model (Fig. 2F).
Figure 2.
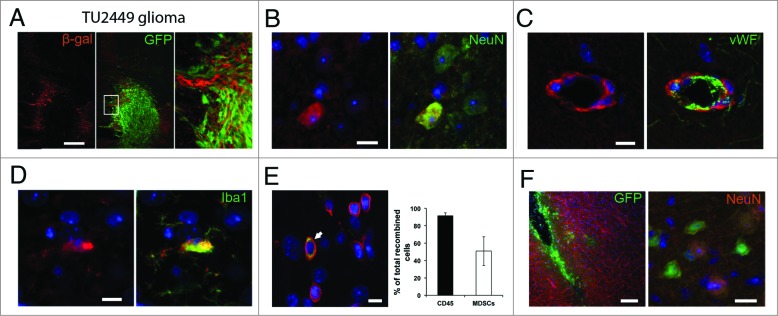
Recombination induced marker gene expression indicates EV uptake (A) Frequent recombination events indicated by immunohistochemical staining for β- galactosidase surrounding the GFP-positive tumor cell mass. Recombined cells included several lineages including neurons (B), endothelial cells (C) and microglia (D). Images are representative from stainings of 2–5 separate animals. (E) Most of recombined cells in the tumor are CD45+ leukocytes (white arrow in left panel) with CD11b+Gr1+ MDSCs as a major subpopulation (right panel). Mean (SD), n = 3. (F) Intracranial injection of EV preparations from Cre expressing glioma cells are sufficient to induce marker gene expression in neural cells (including neurons) in Cre reporter animals (n = 4 injections from 2 separate EV preparations). Scale bars A and E left panel 100 μm, B–E and F right panel, 10 μm.
Next, biologically distinct tumor cells (Cre-Gfp-expressing LLC2) were injected either in the tail vein or subcutaneously. LLC2 cells metastasize primarily to the lung, and we did not observe tumor formation in any other organ within the timeframe of our experiments (three weeks). Co-staining for the recombination marker β-galactosidase and GFP did not reveal any co-localization, again excluding cell fusion of Cre expressing tumor cells with host cells. Almost all β-galactosidase-positive cells co- expressed the leukocyte marker CD45 (Fig. 3A). This was confirmed by flow cytometry using Rosa26-EYFP reporter mice transplanted with LLC2Cre-Gfp tumor cells, showing that all recombined YFP-expressing cells were also positive for CD45 (data not shown). The percentage of recombined marker-positive cells within the total population of tumor infiltrating leukocytes ranged from 0.1% to 2.2% (Figs. 3B and C) with the tendency of an increased frequency of recombined cells in larger tumors. Analysis of tumor infiltrating leukocyte subpopulations by flow cytometry revealed that about 40% of all recombined leukocytes was represented by CD11b+Gr1+ MDSCs followed by a smaller proportion of Gr1-F4/80+ macrophages and Gr1-F4/80− CD11c+ dendritic cells (Fig. 2D). All three cell types have been described to be capable of exosome uptake in vitro.18,19 To analyze whether tumor-derived EVs could be detected systemically, we prepared EVs from the blood sera of four mice from each tumor model and tested for Cre mRNA by RT-PCR. In the Tu2449Cre-Gfp glioma model, all sera were negative, whereas with a carcinoma, two of four mice had detectable levels of Cre mRNA (Fig. 3E). In both tumor models, recombination events were detected only in the tumor lesions but not in distant organs such as spleen and liver. Taken together, our data from the lung carcinoma and glioma model demonstrates that EV transfer of functional RNA from tumor to host cells in the tumor microenvironment in vivo can be visualized by Cre-lox-mediated labeling. In the light of our findings in both tumor models, we found it particularly interesting that tumor-derived EVs have been shown to be sufficient to induce an immunosuppressive pattern in MDSCs in vitro.5 Consistent with these findings, comparing recombined MDSCs with their non-recombined counterparts in the LLC2 model, we found a significantly higher frequency of recombined cells expressing PD-L1 (Figs. 4A–C). This ligand has been described to be expressed on MDSCs and tumor cells20,21 and to induce T cell anergy by binding to its receptor PD-1.22,23 In addition, we sorted recombined and non-recombined MDSCs from tumor bearing Cre-reporter mice (n = 2) and analyzed the expression of ARG-1, TGF-β and inducible nitric oxide synthase (iNOS) that play a key role in the MDSC-mediated immunosuppression during the tumor progression.20,24 The data revealed an upregulation of the expression of ARG-1 and TGF-β (to a lesser extent), suggesting the stimulation of MDSC immunosuppressive functions (Fig. 4D). Interestingly, we also detected the stimulation of the iNOS expression in MDSCs, albeit on a very low absolute expression level (data not shown).
Figure 3.

MDCSs are the principal target for tumor-derived EVs. (A) Lung tumors were stained with antibodies for β-galactosidase and GFP and analyzed by fluorescent microscopy. Transplanted LLC2CreGfp cells as well as Alexa 568+ tumor- infiltrating CD45+ leukocytes are shown. (B) Representative dot plots showing YFP- expressing leukocytes in the tumor microenvironment and in the surrounding normal lung tissue; the gating strategy was based on the negative control, where splenocytes from the same mice were stained. (C) Data of four independent experiments are presented as the percentage of YFP+ leukocytes among total CD45+ leukocytes. *p = 0.0279. (D) YFP+ leukocytes were analyzed by flow cytometry using markers for MDSCs (CD11b+Gr1+), macrophages (MF; Gr-1-CD11b+F4/80+) and dendritic cells (DC; Gr-1-F4/80-CD11b+CD11c+). Data of four independent experiments are shown (mean (SD); nine mice per group). (E) The presence of Cre mRNA containing EVs was tested in the peripheral blood serum of tumor bearing mice. EVs prepared from the respective tumor cell line culture supernatant served as positive controls (Scale bars A, left image, 100 μm; right image 10 μm).
Figure 4.

Marker gene-positive MDSCs display enhanced immunosuppressive properties and differing miRNA profiles. (A–C) Leukocytes from mice bearing LLC2 analyzed by flow cytometry were gated on live MDSCs. Representative dot plots show CD11b+Gr1+ MDSCs (indicated in the black box) in lung tumors (A). (B) Recombined (left panel) or non-recombined (right panel) PD-L1-expressing MDSCs in tumors are presented. The gating for PD-L1 was done on the basis of the Fluorescence-Minus-One (FMO) control. (C) Cumulative data for PD-L1+ tumor- infiltrating MDSCs that took up EVs (YFP+) or failed to take up EVs (YFP-) are expressed as the percentage within all YFP+ or YFP- cells in the tumor and the measurement from the each individual mouse is represented as a single dot on the graph. Mean (SD); five mice per group, p = 0.0007 by one-tailed unpaired t test. (D) YFP+ and YFP- MDSCs were sorted from tumors of two reporter mice (#1 and #2, 2,500 cells). The expression of arginase-1 and TGF-β was quantified by TaqMan qPCR. Relative expression levels were set at the value one for recombined cells (empty bars), filled bars represent non-recombined MDSCs. The reaction was performed in triplicate, results are depicted as mean (SEM). (E) MDSCs from the same two mice (#1 and #2, 5,000 cells) and from one additional mouse (#3, 2,000 cells) were sorted, and their miRNA profile was measured by qPCR array. miRNAs listed in the box were present in LLC2 derived EVs and in all recombined but not in non-recombined MDSCs.
The RNA carried by tumor-derived EVs includes short non-coding RNA and miRNA.6 miRNAs are increasingly recognized to play an important role in the immune regulation and have been suggested to be transferred from tumor to stroma cells through exosomes.25 Therefore, we first analyzed exosomes isolated from LLC2 tumor cell supernatants for their miRNA content using miRNA qPCR arrays. We detected a total number of 161 miRNAs (Cp <36 ) (Table S1). Next, MDSCs were sorted from LLC2-Cre lung tumors from 3 individual Cre reporter mice based on the presence or absence of reporter gene expression. Analysis of their miRNA profile based on qPCR arrays demonstrated a general increase in the number of miRNAs detectable in all three sets of recombined MDSCs compared to their non-recombined counterparts (Fig. 4E). Combining the overlapping populations of miRNAs that were only present in recombined MDSCs with the list of miRNAs found in tumor-derived exosomes yielded four miRNAs; miR-126-3p, miR- 27b, miR-320 and miR-342-3p (box Fig. 4E). Interestingly, all these miRNAs have been reported in the context of tumor progression,26-29 although their role in the regulation of immune cell functions is not known.
Conclusions
In summary, we showed for the first time the Cre-lox-based tracing of EV-mediated RNA transfer from tumor to host cells in the tumor microenvironment in vivo and show changes in immunosuppressive phenotype and miRNA profiles after EV uptake. The possibility to identify and isolate cells that are the target of tumor EV RNA will be crucial for the identification and analysis of novel pathways in stromal cells that support tumor growth. In addition, our data demonstrate that in tumor models of carcinoma and glioma, MDSCs represent a major cell population targeted by tumor released EVs. Together with other soluble factors, tumor EVs could thus be responsible for the MDSC expansion and activation of their immunosuppressive functions in a wide range of tumors.
Materials and Methods
Mice and animal surgery
Permission for all animal experiments was obtained by the regional Ethical Commission for Animal Experimentation of the state of Hessen, Germany (ethical permission Gen. Nr. F94/19). Cre reporter mice were Rosa26-LacZ (JAX- mice stock number 003309), Rosa26-EGFP (JAX-mice stock number 004077) or Rosa26-EYFP (JAX-mice stock number 005130). Genotyping was performed using particular PCR- protocols provided by Jackson Laboratory (http://www.jax.org). Mice 6–12 weeks of age were kept in the animal facility of the University Hospital Frankfurt (ZFE) under a 12 h day/night cycle with free access to food and water. For brain tumor inoculation, 104 TU2449CreGfp or TU2449Cre cells in 1 μL PBS were injected intracranially in deeply anesthetized mice. Animals were killed 10–12 d after injection. For the carcinoma model, 106 LLC2 cells in 200 μL PBS were injected into the tail veins, or 2.5 × 105 LLC2Cre-Gfp cells in 200 μL PBS were injected subcutaneously into Cre reporter mice. For intracranial EV injection, 2 μL EV preparations in PBS were injected into the forebrains of Cre reporter mice (n = 2). Three days after injection, mice were killed and analyzed. As a control experiment, 2 μL lysate generated from 105 TU2449CreGfp cells by repeated freeze-thaw cycles was injected (n = 2). At least 14 animals were analyzed for each model. Animal numbers for experiments with statistical analysis were based on expected incidence based on qualitative observations from tissue sections as indicated in the figure legends.
Cell lines
Cell line TU2449 was generated as described.11,12 Lewis lung carcinoma cell line LLC2 was a gift from Yvonne Reiss (Frankfurt University Medical School). Stable expression of Cre was achieved by transduction with a commercial lentivirus (Allelebiotech) or Cre GFP with a retrovirus resulting in the cell lines TU2449Cre-Gfp and LLC2Cre-Gfp. Cell lines were routinely tested for mycoplasma infection.
Antibodies
Rat anti-mouse directly conjugated mAbs (CD45-PerCP, clone 30-F11; CD45.2- PerCP-Cy5.5, CD11b-V450, clone M1/70; CD11b-PE and APC, Gr-1-APC, clone RB6-8C5, Gr1-PE-Cy7, PD-L1-PE, and CD11c-APC) were from BD Biosciences (Heidelberg, Germany). Purified rat IgG2 CD16/CD32 (BD PharMingen, clone 2.4G2) was used prior staining to block Fcγ III/II receptors. Antibodies for immune staining were anti-β-Gal (mouse monoclonal, Promega; rabbit polyclonal, MP Biomedicals, anti-Iba1 (rabbit polyclonal, Wako), anti-NeuN (rabbit polyclonal, PE-Cy7 conjugated, Bioss Antibodies and mouse monoclonal Alexa-488 conjugated, Chemicon International), anti-vWF (rabbit polyclonal, DAKO), anti-GFP (Alexa-488 conjugated, rabbit polyclonal, Invitrogen), anti-human CD68, CD15 and leukocyte common antigen LCA (all mouse monoclonal, Dako).
Tissue processing
Deeply anesthetized mice were perfused transcardially with PBS followed by 4% cold paraformaldehyde (PFA) in PBS. All organs were post-fixed in 4% PFA in PBS for 12–24 h. For cryosectioning, organs were cryoprotected in 15% sucrose for additional 24 h before they were embedded and sectioned (10–12 μm) as a frozen block. Coronal brains sections (40–50 μm) were cut on a vibratome (Leica, Germany) and kept in PBS at 4°C.
Immunohistochemical visualization and microscopy
Fluorescent images were taken on a confocal LSM Nikon TE2000-E microscope. Images were processed using the EC-C1 3.60 software and ImageJ (NIH). Figures were mounted in Adobe Photoshop CS4.
Preparation of EVs
The tumor cell supernatant or peripheral blood samples were collected and processed by differential centrifugation as described.14 Exosome preparation were analyzed for protein content (Bradford assay, Biorad) and by nanoparticle tracking analysis. EVs isolated by differential ultracentrifugation were resuspended 1:1,000 to 1:2,000 in PBS and assayed using the Nanoparticle Tracking Analysis (Nanosight, Malvern Instruments, Amesbury, UK). Particles were measured at 23°C, for 60 s and number of particles was determined using NTA Software 2.2. Total preparations of EVs used in the present study had particle:protein ratios of 1 × 1010 to 4 × 1010 according to published procedures.30,31
Electron microscopic analysis
Vesicle preparations were fixed and stained as described14 with minor modifications. The uranyl-oxalate (step 6) was substituted with 2% of uranyl-acetate in dH2O (Dehydrate, Ted Pella, Inc.) and the ratio of methylcellulose (Sigma-Aldrich) to uranyl-acetate was changed to 1% each (step 7). Grids were analyzed using Tecnai Spirit BioTWIN electron microscope (FEI, Netherlands) at 120 kV. Pictures were taken with a FEI Eagle 4 K bottom-mount CCD camera.
RT-PCR analysis
RNA was purified using Qiagen RNeasy Micro Kit, according to manufacturer's instructions. cDNA synthesis was performed with SuperScipt III reverse transcriptase (RT) (Invitrogen) and OligodT18 primers (Fermentas) or with Cre primers. Residual DNA contamination was excluded by controls in which RT was omitted during cDNA synthesis. For Cre mRNA detection, a heminested PCR was performed with 5 μL of vesicle cDNA preparation and Primers FORWARD-out and REVERSE (FORWARD- out: 5′-GTGGGAGAATGCTGATCCACA-3′; REVERSE 5′- ACACCATTCTTTCTGACCCG-3′) in the first reaction followed by the second PCR using 2 μL of the amplificate and FORWARD-in (5′GGTTACCAAGCTGGTGGAGA - 3′) and REVERSE. PCR products were visualized on a 3% agarose gel. For quantitative PCR, TaqMan probes were used (AppliedBiosystems). qPCR was performed on a BioRad iCycler. Quantitation of the products was performed using the dt/ddt method against the housekeeping genes β-actin and Rps13 using the BioQuant Software.
Biochemical analysis
SDS-PAGE analysis and western blot have been described before.32 Cells were lysed in 1% NP40/PBS with protease inhibitor cocktail a described. Cell lysates were prepared by centrifugation for 20 min in a table-top centrifuge at 13.000 × rpm. The supernatant representing the cell lysates was processed for SDS-PAGE by diluting with SDS-sample buffer. The residual pellet containing intact nuclei was used to prepare nuclear extracts. For this, it was resuspended in SDS-sample buffer and the DNA was sheared by sonication as described.17
Sucrose gradient analysis
For sucrose density gradient fractionation, vesicles were loaded on top of a stepwise sucrose gradient at the following concentrations: 2 M, 1.3 M, 1.16 M, 0.8 M, 0.5 M and 0.25 M in TBS. The gradient was centrifuged for 2.5 h at 100.000 × g, 4°C using a Beckman SW40 Rotor (k-factor: 137). 300 g of total protein were applied to each sucrose gradient and twelve 1 mL fractions were collected from the top of the gradient, diluted with TBS (1:10) and ultra-centrifuged for 2,5 hrs at 100.000 × g. The resulting pellet was treated with RNAseA to eliminate any free RNA. For protein analysis, gradient fractions were subjected to protein precipitation using Chloroform/Methanol as described.33 Precipitates were dissolved in SDS-sample buffer and used for SDS-page and western blot analysis. Blots were probed in a sequential fashion with the various antibodies. The following antibodies were used: anti-mouse CD9 (R&D Systems, clone 139712), anti-Cre-recombinase (Covance, NMS-106P), anti-TSG101 (Genetex, clone 4A10) and anti-Calnexin (Ab-583, Biomol, Hamburg).
Flow cytometry
All tumor-bearing animals were included in the analysis. Subpopulations of cells derived from individual animals were compared to each other, therefore no randomization or blinding method was applied. Lung tumors were separated from the surrounding lung tissue. Single-cell suspensions were prepared by mechanical disaggregation in cold PBS. Brain tumors were dissected out, minced with fine scissors and dissociated in Accutase (PAA Laboratories GmbH, Austria) for 30 min with repeated tituration. After dissociation, cell suspensions were filtered through 40 μm cell strainers (BD Biosciences) and treated with Fc-block for 10 min followed by incubation with mAbs for 30 min at 4°C. Acquisition and sorting was performed by multicolor flow cytometry using a FACSCanto II or a FACSAria with FACSDiva software (both from BD Biosciences) with dead cell exclusion. FlowJo software (Tree Star, Ashland, OR) was used to analyze at least 100,000 events.
Screening of miRNA by quantitative real-time PCR
miRNA was isolated from MDSCs sorted by FACS or from exosomes purified from tumor cell supernatant using the miRNeasy Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer´s protocol. The extracted miRNA was diluted in 15-L H2O. Quantitative miRNA expression analysis was performed using real-time PCR (LightCycler®480 Real-Time PCR System, Roche Applied Science, Mannheim, Germany). To screen for miRNA expression, the miRCURY LNA™ Universal microRNA Ready-to-Use PCR panel (Exiqon, Vedbaek, Denmark) was used according to the manufacturer's instructions. Prior to the analysis, plate-specific effects were normalized using the reference RNA included. Cp values from recombined MDSCs were then compared to the Cp values measured in non-recombined MDSCs.
Statistical analysis
Results were assessed with Student's t-test using GraphPad Prism software (San Diego, CA). Differences in values at p < 0.05 were considered significant. Supplemental Material: Table with the list of miRNAs detected in tumor exosomes as well as isolated MDSCs.
Acknowledgments
We thank Jakob Weissenberger for the gift of the cell line TU2449.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the DFG (individual grant MO2211/1-1 to S.M.), the Edinger foundation, LOEWE OSF B3 (S.M.), Dr. Mildred Scheel Foundation for Cancer Research (grant 108992, to V.U.), and by the Initiative and Networking Fund of the Helmholtz Association within the Helmholtz Alliance on Immunotherapy of Cancer (to V.U.). M.T. was supported by LOEWE OSF B6 and BMBF.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar CM et al.. Melanoma exosomes educate bone marrow progenitor cells toward a pro- metastatic phenotype through MET. Nat Med 2012; 18(6):883-91; PMID:; http://dx.doi.org/ 10.1038/nm.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M, Wrana JL. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012; 151:1542-56; PMID:; http://dx.doi.org/ 10.1016/j.cell.2012.11.024 [DOI] [PubMed] [Google Scholar]
- 4.Filipazzi P, Bürdek M, Villa A, Rivoltini L, Huber V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin Cancer Biol 2012; 22:342-9; PMID:; http://dx.doi.org/ 10.1016/j.semcancer.2012.02.005 [DOI] [PubMed] [Google Scholar]
- 5.Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res 2007; 67:2912-5; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0520 [DOI] [PubMed] [Google Scholar]
- 6.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9:654-9; PMID:; http://dx.doi.org/ 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- 7.Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10:1470-6; PMID:; http://dx.doi.org/ 10.1038/ncb1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borràs FE, Breakefield X, Budnik V et al.. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol 2012; 10:e1001450; PMID:; http://dx.doi.org/ 10.1371/journal.pbio.1001450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rak J, Guha A. Extracellular vesicles–vehicles that spread cancer genes. Bioessays 2012; 34:489-97; PMID:; http://dx.doi.org/ 10.1002/bies.201100169 [DOI] [PubMed] [Google Scholar]
- 10.Ridder K, Keller S, Dams M, Rupp A-K, Schlaudraff J, Turco DD, Starmann J, Macas J, Karpova D, Devraj K et al.. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol 2014; 12:e1001874; PMID:; http://dx.doi.org/ 10.1371/journal.pbio.1001874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weissenberger J, Steinbach JP, Malin G, Spada S, Rülicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997; 14:2005-13; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1201168 [DOI] [PubMed] [Google Scholar]
- 12.Pohl U, Wick W, Weissenberger J, Steinbach JP, Dichgans J, Aguzzi A, Weller M. Characterization of Tu-2449, a glioma cell line derived from a spontaneous tumor in GFAP-v-src-transgenic mice: comparison with established murine glioma cell lines. Int J Oncol 1999; 15:829-34; PMID:; http://dx.doi.org/ 10.3892/ijo.15.4.829 [DOI] [PubMed] [Google Scholar]
- 13.Keller S, Ridinger J, Rupp A-K, Janssen JWG, Altevogt P. Body fluid derived exosomes as a novel template for clinical diagnostics. J Transl Med 2011; 9:86; PMID:; http://dx.doi.org/ 10.1186/1479-5876-9-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3:Unit 3.22; PMID:; http://dx.doi.org/ 10.1002/0471143030.cb0322s30 [DOI] [PubMed] [Google Scholar]
- 15.Keller S, König A-K, Marmé F, Runz S, Wolterink S, Koensgen D, Mustea A, Sehouli J, Altevogt P. Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett 2009; 278:73-81; PMID:; http://dx.doi.org/ 10.1016/j.canlet.2008.12.028 [DOI] [PubMed] [Google Scholar]
- 16.Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP- mediated gene targeting. Cell 1993; 73:1155-64; PMID:; http://dx.doi.org/ 10.1016/0092-8674(93)90644-6 [DOI] [PubMed] [Google Scholar]
- 17.Schwenk F, Sauer B, Kukoc N, Hoess R, Müller W, Kocks C, Kühn R, Rajewsky K. Generation of Cre recombinase-specific monoclonal antibodies, able to characterize the pattern of Cre expression in cre-transgenic mouse strains. J Immunol Methods 1997; 207:203-12; PMID:; http://dx.doi.org/ 10.1016/S0022-1759(97)00116-6 [DOI] [PubMed] [Google Scholar]
- 18.Mignot G, Roux S, Thery C, Ségura E, Zitvogel L. Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med 2006; 10:376-88; PMID:; http://dx.doi.org/ 10.1111/j.1582-4934.2006.tb00406.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol 2002; 3:1156-62; PMID:; http://dx.doi.org/ 10.1038/ni854 [DOI] [PubMed] [Google Scholar]
- 20.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:; http://dx.doi.org/ 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012; 24:207-12; PMID:; http://dx.doi.org/ 10.1016/j.coi.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1a, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014; 211:781-90; PMID:; http://dx.doi.org/ 10.1084/jem.20131916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 2009; 10:1185-92; PMID:; http://dx.doi.org/ 10.1038/ni.1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:; http://dx.doi.org/ 10.4049/jimmunol.0802740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada H, Kohanbash G, Lotze MT. MicroRNAs in immune regulation– opportunities for cancer immunotherapy. Int J Biochem Cell Biol 2010; 42:1256-61; PMID:; http://dx.doi.org/ 10.1016/j.biocel.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F et al.. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol 2012; 14:159-67; PMID:; http://dx.doi.org/ 10.1038/ncb2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin L, Wessely O, Marcusson EG, Ivan C, Calin GA, Alahari SK. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-a in breast cancer. Cancer Res 2013; 73:2884-96; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van der Auwera I, Limame R, van Dam P, Vermeulen PB, Dirix LY, Van Laere SJ. Integrated miRNA and mRNA expression profiling of the inflammatory breast cancer subtype. Br J Cancer 2010; 103:532-41; PMID:; http://dx.doi.org/ 10.1038/sj.bjc.6605787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth P, Wischhusen J, Happold C, Chandran PA, Hofer S, Eisele G, Weller M, Keller A. A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem 2011; 118:449-57; PMID:; http://dx.doi.org/ 10.1111/j.1471-4159.2011.07307.x [DOI] [PubMed] [Google Scholar]
- 30.Webber J, Clayton A. How pure are your vesicles? J Extracell vesicles 2013; 2:1-6; PMID: 24009896; http://dx.doi.org/ 10.3402/jev.v2i0.19861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, Dyrskjøt L, Orntoft TF, Howard KA, Ostenfeld MS. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell vesicles 2014; 3:25011; PMID:; http://dx.doi.org/ 10.3402/jev.v3.25011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rupp A-K, Rupp C, Keller S, Brase JC, Ehehalt R, Fogel M, Moldenhauer G, Marmé F, Sültmann H, Altevogt P. Loss of EpCAM expression in breast cancer derived serum exosomes: role of proteolytic cleavage. Gynecol Oncol 2011; 122:437-46; PMID:; http://dx.doi.org/ 10.1016/j.ygyno.2011.04.035 [DOI] [PubMed] [Google Scholar]
- 33.Riedle S, Kiefel H, Gast D, Bondong S, Wolterink S, Gutwein P, Altevogt P. Nuclear translocation and signalling of L1-CAM in human carcinoma cells requires ADAM10 and presenilin/gamma-secretase activity. Biochem J 2009; 420:391-402; PMID:; http://dx.doi.org/ 10.1042/BJ20081625 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.