

Abstract
Stimulating the immune system to attack cancer is a promising approach, even for the control of advanced cancers. Several cytokines that promote interferon-γ-dominated immune responses show antitumor activity, with interleukin 12 (IL-12) being of major importance. Here, we used an antibody-IL-12 fusion protein (NHS-IL12) that binds histones of necrotic cells to treat human sarcoma in humanized mice. Following sarcoma engraftment, NHS-IL12 therapy was combined with either engineered IL-7 (FcIL-7) or IL-2 (IL-2MAB602) for continuous cytokine bioavailability. NHS-IL12 strongly induced innate and adaptive antitumor immunity when combined with IL-7 or IL-2. NHS-IL12 therapy significantly improved survival of sarcoma-bearing mice and caused long-term remissions when combined with IL-2. NHS-IL12 induced pronounced cancer cell senescence, as documented by strong expression of senescence-associated p16INK4a and nuclear translocation of p-HP1γ, and permanent arrest of cancer cell proliferation. In addition, this cancer immunotherapy initiated the induction of myogenic differentiation, further promoting the hypothesis that efficient antitumor immunity includes mechanisms different from cytotoxicity for efficient cancer control in vivo.
Keywords: cancer-targeted IL-12, differentiation, humanized mice, immunocytokine, immunotherapy, M1/M2 macrophages, rhabdomyosarcoma, TH1-induced senescence, tumor-infiltrating lymphocytes, TH17 cells
Abbreviations: CIP1, CDK-interacting protein 1; DNAM-1, DNAX accessory molecule-1; KIR, killer-cell immunoglobulin-like receptor; MICA/B, MHC class I polypeptide-related sequence A/B; NKG, natural killer group; NSG, NOD SCID gamma chain knock out mouse; PCNA, proliferating cell nuclear antigen; pHP1γ, phosphorylated heterochromatin protein 1 gamma; PVR, poliovirus receptor; ROI, region of interest; RORC, RAR-related orphan receptor C; RMS, rhabdomyosarcoma, (eRMS: embryonal, aRMS: alveolar); SCT, stem cell transplantation; SPECT/CT, single-photon emission computed tomography; TRBV, T-cell receptor beta chain; ULBP, UL16 binding protein; WAF, wild-type activating fragment
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue tumor in children and an advanced stage of disease is associated with a very unfavorable prognosis.1 Despite considerable progress in defining the molecular, cellular, and environmental contributions to the pathophysiology of this tumor development, standard treatment is still confined to conventional surgery, radiotherapy, and/or chemotherapy. The clinical outcome of these strategies remains unsatisfactory.
Cytotoxic T cell and natural killer (NK) cell responses against RMS can be generated in vitro,2 with adoptive transfer as a potentially immunotherapeutic option, yet the antitumor activity of immune effector cells has many more facets. Beyond killing, combined signaling through interferon gamma (IFN-γ) and tumor necrosis factor (TNF) receptor 1-dependent pathways can result in tumor dormancy,3 therefore induced secretion of these cytokines may be an important way to locally control tumor growth. Indeed, several studies have shown improved survival of patients with increased frequencies of IFN-γ-producing T cells after allogeneic hematopoietic stem cell transplantation (SCT).4 In this regard, CD4+ T cells may be as important as CD8+ cytotoxic T lymphocytes.5 Interleukin 12 (IL-12), the major player in this network, can induce tumor regression and affects innate and adaptive immunity.6-8 Besides its role in T-cell priming, IL-12 efficiently reverts T helper (TH)17 cells back into a TH1 phenotype,9,10 restores M1 macrophage function,6,11-13 and mediates dendritic cell (DC)-NK interactions.14 Systemic administration of IL-12 has already shown efficacy against solid tumors but induced dose-limiting toxicity.15 Thus, targeted delivery of IL-12 to the tumor microenvironment seems a highly promising approach for tumor immunotherapy.
For direct delivery into tumor tissue, IL-12 was complexed with NHS76 (NHS-IL12), an antibody that binds to naked histone/DNA complexes in necrotic areas of solid tumors,16 thus improving the therapeutic utility of IL-12 by decreasing its systemic toxicity and increasing its plasma half-life.17 To enhance the required antitumoral response, NHS-IL12 was combined with IL-2 or IL-7. IL-2 was used complexed with the IL-2-specific monoclonal antibody MAB602 to increase its biological activity,18 enhance effector functions of low-avidity tumor-specific T cells,19 and reduce its availability to regulatory T cells.20
Because of the species specificity of IL-12, in order to test its efficacy we established a murine model of humanized hematopoiesis in NOD scid gamma−/− (NSG) mice (NSG plus huCD34+)21 with subsequent inoculation of human (hu)RMS tumors (A204). These mice developed local huRMS tumors within 3 weeks and received one of the following cytokine treatments (Fig. 1A): FcIL-7 (control), NHS-IL12 plus FcIL-7 (NHS-IL12/FcIL-7), or NHS-IL12 plus IL-2 complexed with MAB602 (NHS-IL12/IL-2MAB602). After a 5-week treatment period, mice were evaluated for the effects of cytokine administration on survival, tumor growth, cellular tumor infiltrates, and cellular differentiation. Long-term cohorts were treated at least until day 95 post tumor inoculation before evaluation.
Figure 1.

Study design, tumor growth and survival after RMS challenge and therapy. (A) NSG mice (4–6 weeks old) were sublethally irradiated and humanized with CD34+CD3− grafts. Fully engrafted mice were inoculated with 1 × 106 A204 cells at week 12. Immunotherapy began 18 d later when tumor volume reached 50–200 mm3. Mice were sacrificed after 5 weeks of treatment, when tumors of the FcIL-7 cohort had reached 20% of body weight. Four mice of the NHS-IL12/FcIL-7 and the NHS-IL12/IL-2MAB602 cohort were kept alive and treated at least until day 95 after tumor inoculation. (B) Effect of FcIL-7, NHS-IL12/FcIL-7, and NHS-IL12/IL-2MAB602 on survival. Survival curves were compared using log-rank test. Survival was highly significantly better for NHS-IL12 cohorts compared to FcIL-7 cohort. In the FcIL-7 control group, 4 animals died before day 52 and 3 were sacrificed on day 52 because of excessive tumor growth. In the NHS-IL12/FcIL-7 treatment cohort 2 mice died before day 52, 1 on day 56, and 1 on day 74 in the long-term treatment group. (C) Effect of FcIL-7, NHS-IL12/FcIL-7, and NHS-IL12/IL-2MAB602 on tumor growth. Mice bearing human RMS A204 were treated weekly with 20 µg FcIL-7 administered intravenously (rectangle), 20 µg NHS-IL12 ανδ 20 µg FcIL-7 (circle), or with 20 µg NHS-IL12 and 1.5 µg IL-2 complexed with 15 µg MAB602 (cross). Tumor sizes in mm3 are given as mean ± SD of 7 mice/group with short-term treatment (5 weeks). (D) Individual tumor sizes of 4 mice per NHS-IL12 group during long-term treatment (14 weeks, >95 days).
Our results show that administration of NHS-IL12/IL-2MAB602 to progressive huRMS successfully halted tumor growth and significantly improved survival without any additional cytostatic or cellular therapies. Importantly, in addition to enhancing distinct types of cellular immunity, this regimen halted cell proliferation, induced cellular senescence, and, puzzlingly, promoted myogenic differentiation in this otherwise aggressively growing RMS model.
Results
Establishing a fully functional immune system in NSG mice
To test the toxicity and potential therapeutic efficacy of the NHS-IL12 construct in the human system in vivo, we transplanted NSG mice with huCD34+ stem cells (Fig. 1A). Transplantation of >99.9% pure huCD34+ cells into NSG mice established all hematopoietic lineages within 12 weeks (Fig. S1A), as reported by us and others.21 In addition, we found complex T-cell receptor (TCR) repertoires in bone marrow, thymus, and spleen (Fig. S1B) and thymus equivalents (Fig. S1C). We also confirmed T-cell receptor excision circles and robust proliferative responses to mitogens (not shown). Together, these data show that the transplanted mice developed a human T-cell immune system that closely resembled the human in vivo situation. The mice also developed normal NK cells, as shown by NKp receptors and killer-cell immunoglobulin-like receptors (KIR) that reflected the donor's NK repertoire (Fig. S2A, B).
Tumor inoculation
Subcutaneous inoculation of 1 × 106 allogeneic A204 cells 12 weeks post stem cell transplantation (SCT) resulted in aggressively growing tumors in all mice within 3 weeks. Consistent with SCT of human patients with RMS, the immune system failed to reject the allogeneic A204. The tumors grew rapidly despite solid expression of surface HLA class I and II, MICA/B, Nectin-2 (CD112) and poliovirus receptor (PVR, CD155) but completely lacked UL16-binding proteins (ULBP) 1-4 (Fig. S2C).
Sarcoma therapy with histone-targeted IL-12 fusion proteins
Following tumor inoculation, solid A204 RMS tumors became established after 3 weeks. Subsequently, mice were treated weekly for 5 weeks with FcIL-7 alone (control), NHS-IL12/FcIL-7, or NHS-IL12/IL-2MAB602 (Fig. 1A). Intravenous injection of the constructs caused no visible systemic toxicity acutely or over an extended time (Fig. 1B: 4 mice/cohort, 100 d). In mice treated with FcIL-7 only, the sarcomas showed exponential growth. Four out of 7 mice died before week 5, and 3 mice reached endpoint criteria due to sarcoma burden at day 52. (Fig. 1C). In the FcIL7-group, we observed 6.5-fold tumor growth from day 27 to day 52, which was reduced to 1.8 fold in the NHS-IL12/IL-2MAB602 group (P ≤ 0.05, one-tailed t-test). Thus, the sarcomas remained significantly growth arrested in mice receiving NHS-IL12/IL-2MAB602 (n = 11) (Fig. 1C), resulting in survival of all mice (Fig. 1B, D). NHS-IL12/FcIL-7 protected 2 oυτoϕ 11 mice for a shorter period of time only (Fig. 1B, C); these mice died on day 43 and day 49 respectively. For analysis of all 3 groups, mice were sacrificed on day 52 (short-term treatment), except for 4 mice in the NHS-IL12 treatment groups that received therapy until day 100 (long-term treatment). Long-term NHS-IL12/FcIL-7 treatment successfully halted tumor growth in 1 oϕ 4 mice, delayed tumor growth in 2 mice, and eliminated the tumor in 1 mouse. Long-term NHS-IL12/IL-2MAB602 treatment eliminated tumors in 3 oϕ 4 mice and halted tumor growth in the remaining mouse (Fig. 1D).
Biodistribution of the NHS-IL12 construct
The monoclonal antibody NHS76 recognizes an intracellular antigen in necrotic cancer regions. We therefore investigated whether NHS-IL12 binds preferentially to sites of sarcomas. SPECT/CT biodistribution studies with 123I-labeled NHS-IL12 revealed significant in vivo enrichment of NHS-IL12 inside the sarcoma microenvironment (Fig. 2A). Quantification of 123I-labeled NHS-IL12 showed 4- to 6-fold radionuclide enrichment in the tumors compared with the contralateral muscle. 123I counts in the tumor region peaked 26 h after intravenous NHS-IL12 application, whereas in normal muscle tissue the 123I counts remained stable over time (Fig. 2B), confirming that NHS-IL12 preferentially binds to human sarcoma.
Figure 2.
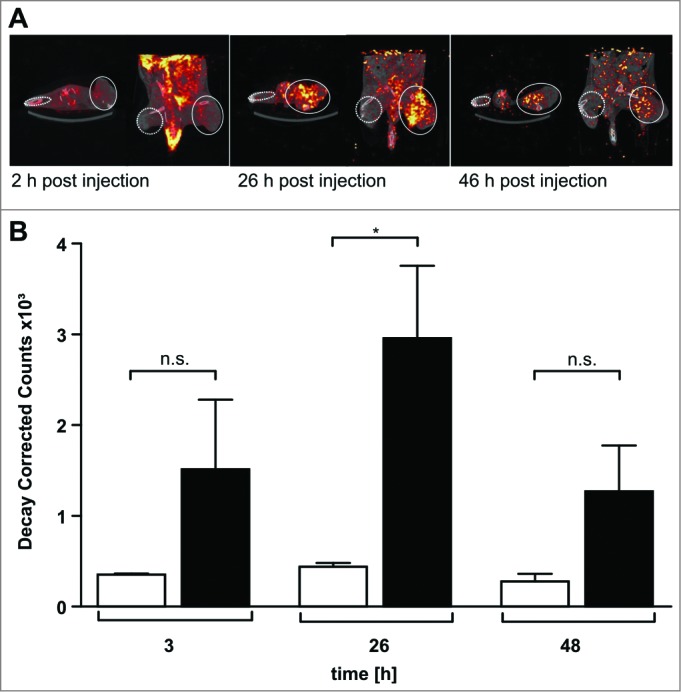
123I-labeled NHS-IL12 accumulates in the lesions of a human A204 tumor xenograft. (A) In vivo SPECT scans performed 2, 26, and 46 h after injection of a therapeutic dose (30 µg) of 123I-labeled NHS-IL12 show specific accumulation of NHS-IL12 in tumor (solid circles) compared to muscle tissue (dotted circles). (B) Uptake of 123I-NHS-IL12 reached its maximum in the tumor lesion 26 h after administration, whereas in muscle no specific signal could be detected over the entire scan time. Counts were decay-corrected to adjust for the radioactive decay of 123I between measurement time points (n = 2). * P ≤ 0.05.
Tumor-specific immune responses
To understand the differences underlying the therapeutic efficacy of the different treatment protocols, we performed histologic, immunohistochemical, and extensive molecular and functional characterization of the human immune cells infiltrating the A204 sarcomas. We considered cells representing both innate and adaptive immunity.
Strikingly, sarcomas of FcIL-7-treated mice only had a minor infiltrate containing exclusively macrophages (CD68+) and NK cells (CD56+) (Fig. S3). In sharp contrast, sarcomas of mice treated with either NHS-IL12 regimen showed a dense mononuclear infiltrate with NK cells, macrophages, and large numbers of CD4+ and CD8+ T cells (Fig. S3). The NK cells of all treatment groups expressed NKG2D mRNA and DNAM-1 (Fig. 3A), a ligand for the sarcoma-associated surface molecules Nectin-2 (CD112) and PVR (CD155) (Fig. S2C).
Figure 3.

Influence of FcIL-7, NHS-IL12/FcIL-7, and NHS-IL12/IL-2MAB602 on innate immunity. (A) Tumor homogenates of individuals in each cohort were subjected to RT-PCR–based fragment length analysis for the major triggering receptors NKG2C, -D, and -E, DNAM-1, and NK receptors NKp30, −44, and −46. Note the high congruity within a cohort. (B) TCR transcripts indicative of iNKT cells (invariant Vα24 and Vβ11), Vδ1 and −2 chains, and NKp46 at day 52. (C) TCRVα24 mRNA expression in A204 tumors detected as a single peak or in Gaussian distribution. (D) Expression of CD161 in homogenates of tumors and muscles. Quantitative values are given as mean fluorescence intensity. Each dot represents 1 individual tumor. **P ≤ 0.01, ***P ≤ 0.001.
mRNA expression of surface molecules that direct NK-cell differentiation and activation strictly required the NHS-IL12 construct. FcIL-7 or IL-2MAB602 modulated the effect of the NHS-IL12 construct on the infiltrating NK cell population. We found the activating receptors NKG2E, NKp44, and NKp46 only in tumors of mice treated with NHS-IL12/FcIL-7, whereas NKp30 expression was restricted to sarcomas of NHS-IL12/IL-2MAB602-treated and NHS-IL12/FcIL-7 long-term treated mice (Fig. 3A). As certain KIR molecules impair NK cell functions even in an MHC-I-deficient environment,22 we next analyzed KIR expression in sarcomas of mice treated with FcIL-7 or NHS-IL12/FcIL-7. qRT-PCR of total sarcoma revealed similar expression levels in both groups (Fig. S2B). As expected, KIR expression of normal mouse muscle tissue and tissue-infiltrating lymphocytes of human sarcoma xenografts differed: mouse muscle expressed KIR2DL3 and KIR2DL4 whereas the human sarcomas additionally expressed KIR2DL1 and KIR3DL1 (Fig. S2B). The NK cells proved to be functional, as NK cells freshly isolated from sarcoma tissue released IFN-γ after in vitro stimulation with NHS-IL12 (not shown).
NKp46+ NK cells, as well as TCRVα24-expressing iNKT cells and γδ T cells, all of which contribute to the innate TH17 compartment, were almost exclusively found in sarcomas of mice treated with NHS-IL-12/FcIL-7 (Fig. 3B, C).
Nevertheless, the NHS-IL-12/FcIL-7 cohort showed strong CD161 expression similar to that of controls (Fig. 3D) but significantly lower RAR-related orphan receptor C (RORC) expression than the control mice (Fig. 4A). Mice treated with NHS-IL12/IL-2MAB602 were devoid of CD161 mRNA (Fig. 3D) and RORC mRNA (Fig. 4A).
Figure 4.

Clonality analysis of αβ T cells treated with FcIL-7, NHS-IL12/FcIL-7, and NHS-IL12/IL-2MAB602. (A) Real-time PCR-based detection of various immune markers in tumor homogenates. Expression of the target gene was normalized to expression of human CD45. (B) Expression of 25 TRBV segments determined by CDR3-size spectratyping. Filled squares indicate expression of up to 12 fragments. Each vertical lane represents 1 mouse; dark squares indicate TRBV segments chosen for CDR3 sequence analysis. (C) CDR3 region protein sequences of selected TRBV segments; bold amino acid codes mark homologous sequences. (D) Expression ratios of T-bet/RORC and Foxp3/CD40L in tumor homogenates (n=4). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
In addition to innate lymphocyte populations, tumors of all NHS-IL12-treated cohorts contained a broad spectrum of CD3+ T cells (Fig. 4B, C, Fig. S3). These were absent in sarcomas of mice treated with FcIL-7, which showed scarce signals in Vβ spectratype analysis (Fig. 4B, C) and no infiltrating CD8+ T cells (Fig. S3). NHS-IL12-treated sarcomas showed a broad TCR repertoire, which was substantialized by oligoclonal or monoclonal peaks within various Vβ-families (Fig. 4C) as it occurs during preferential expansion of restricted T-cell clones. Cloning and sequencing of the CDR3 region confirmed that the peaks contained limited numbers of different T-cell clones with strongly amplified TRBV segments in the 2 treatment groups, such as TRBV29-1 in all individuals of the NHS-IL12/FcIL-7 cohort or TRBV5-5 and TRBV18 in the NHS-IL12/IL-2MAB602 cohort (Fig. 4B, C).
The relative expression of the transcription factors T-bet and RORC, which regulate IFN-γ and IL-17 respectively, mirrored the degree of TH1 bias in the tumor infiltrating lymphocytes of the respective cohorts. The T-bet/RORC ratio was <0.05 in the FcIL-7-only cohort, but was 19-fold higher (0.8) and 44-fold (2.2) higher in mice receiving NHS-IL12 with either FcIL-7 or IL-2MAB602 (Fig. 4D). In line with the T-bet/RORC expression, Foxp3 expression was approximately 10-fold lower in both NHS-IL12 groups than in the FcIL-7-only cohort (Fig. 4A). Accordingly, low Foxp3 expression inversely correlated with strong expression of the T-cell activation marker CD40L (Fig. 4A, D).
Senescence induction in NHS-IL12-treated sarcomas
The NHS-IL12 construct strongly suppressed sarcoma development in all treated mice (Fig. 1). Surprisingly, only sarcomas of the NHS-IL12/IL-2MAB602-group contained high amounts of perforin protein (Fig. S3) and granzyme K mRNA (Fig. 4A), whereas mice treated with NHS-IL12/FcIL-7 were virtually devoid of perforin protein (Fig. S3) and expressed low levels of granzyme K mRNA (Fig. 4A). This strongly suggests that the sarcoma-controlling immune response included mechanisms different from cytolysis. Moreover, the sarcomas did not contain sufficient numbers of CD4+ or CD8+ T cells to explain cancer control by killing or apoptosis (Fig. S3).
We therefore analyzed the effect of the immune response on sarcoma cell proliferation, as determined by the proliferation markers proliferating cell nuclear antigen (PCNA) and Ki67. In the rapidly growing FcIL-7-control tumors 50% of the sarcoma cells stained positive for PCNA or Ki67, showing that most of the cells were proliferating (Fig. 5A, B). In the NHS-IL12-treated groups, levels of both PCNA- (Fig. 5A) and Ki67-stained sarcoma cells were significantly lower than in the FcIL-7-control (Fig. 5A, B). To determine whether the immune response simply arrested the cell cycle or induced a stable growth arrest, as seen in cytokine-induced senescence,23 we double-stained the sarcomas for one proliferation marker and either senescence-associated phosphorylated heterochromatin protein 1 (p-HP1γ)24,25 or p16INK4a, also known as cell cycle regulator cyclin-dependent kinase inhibitor 2A (CDKN2A). Sarcomas treated with FcIL-7 only showed high PCNA/Ki67 expression and concurrent very low expression of p16INK4a/nuclear p-HP1γ, confirming that these sarcoma cells were rapidly proliferating (Fig. 5A, B). In sharp contrast, up to 70% of the sarcoma cells from mice treated with either NHS-IL12/IL-2MAB602 or NHS-IL12/FcIL-7 expressed the senescence marker p-HP1γ or p16INK4a (Fig. 5A, B), in the absence of PCNA (PCNA−/p-HP1γ+) (Fig. 5A, upper line inserts) and Ki67 (Ki67−/p16INK4a) (Fig. 5A, lower line inserts).
Figure 5.

Induction of senescence markers and antiproliferative effect of NHS-IL12/FcIL-7 and NHS-IL12/IL-2MAB602 treatment. (A) Cellular senescence and proliferation within tumor sections were determined by immunofluorescent double-staining for nuclear p-HP1γ or p16INK4a (red) in combination with PCNA or Ki67 (blue), respectively (1:100). Nuclei are shown in green. The inserts show a higher magnification (1:300) to visualize nuclear dots of p-HP1γ or p16INK4a staining. Scale bars represent 100 µm (1:100) and 30 µm (1:300). (B) Mean percentage of p-HP1γ-positive cells (i.e., cells with more than 5 nuclear dots), p16INK4a-positive cells (as determined by higher magnification [1:300]), or Ki67-positive cells after treatment with FcIL-7, NHS-IL12/FcIL-7, or NHS-IL12/IL-2MAB602 (n=3). ** P ≤ 0.01, *** P ≤ 0.001. (C) Cytokine-induced growth arrest in primary human RMS cancer cell preparations. CCA cells (eRMS, passage 7), SRH (eRMS, passage 8), or ZCRH cells (aRMS, passage >9) were seeded at a density of 2 × 104 cells/9.6 cm2. On days 3 and 4 the cells were treated with 100 ng/mL IFN-γ and 10 ng/mL TNF or with medium alone (control). On day 7, the cytokines were removed and the cells were trypsinized, counted, and reseeded at 2 × 104 cells/9.6 cm2. After incubation for another 4 d (ZCRH and SRH) or 10 d (CCA) living cells were counted. Growth curves of the responder cells CCA and SRH and the non-responder cells ZCRH in the absence (Co.) or presence of interferon gamma (IFN-γ) plus tumor necrosis factor (TNF) (n=3).
Since IFN-γ and TNF are the 2 major effector cytokines of IL-12-driven TH1-immunity and as these 2 cytokines can induce senescence,23 we incubated various patient-derived human RMS cell lines of very early passage with increasing doses of IFN-γ and TNF. Either cytokine alone caused no or only moderate growth inhibition (not shown). However, when combined they caused a permanent senescence-defining growth arrest in 2 of 3 sarcomas (Fig. 5C). Importantly, the senescence-resistant sarcomas did not express the cell cycle regulator p16INK4a (not shown), confirming that senescence induced by IFN-γ and TNF strictly required the activation of p16INK4a.
Induction of differentiation in A204 rhabdomyosarcomas
As the IFN-γ- and TNF-dominated immune response caused a permanent cell cycle arrest in RMS and skeletal muscle differentiation depends on early withdrawal of myoblasts from the cell cycle to allow expression of muscle-specific genes and cell fusion into multinucleate myotubes,26-28 we wondered whether this growth arrest might also affect the differentiation of A204 sarcomas. Desmin is a reliable marker for rhabdomyoblastic differentiation29 that is absent in either undifferentiated or poorly differentiated RMS.30 Accordingly, A204 sarcomas showed neither the cross-striation that characterizes myocytes nor expression of desmin prior to transplantation (not shown). Following transplantation into humanized mice, the FcIL-7-treated proliferating sarcomas remained poorly differentiated, with a few single desmin+ cells diffusely distributed within the tumors but no cross-striation (Fig. 6A, B). In sharp contrast, NHS-IL12 therapy not only inhibited tumor growth and induced senescence, but also gradually restored the myogenic structure and marker expression in A204 sarcomas. NHS-IL12/FcIL-7 induced randomly distributed, linear areas of maturated RMS cells that clearly expressed desmin and had areas with cross-striation. This differentiation toward functional muscle tissue was even more pronounced in sarcomas from mice treated with NHS-IL12/IL-2MAB602. Such growth-arrested sarcomas showed restiform/rope-like propagation of differentiation zones with tube-like/cribriform structures, extensively penetrating the tumor (Fig. 6A, B). Thus, growth arrest of sarcomas in vivo induced senescence and was associated with restoration of the cell-fate specific markers of myocytes, the origin of A204 RMS.
Figure 6.

In vivo expression and organization of desmin as a marker of myogenic differentiation in A204 RMS. (A) Histologic slides from tumors (n = 3/cohort) of all cohorts (FcIL-7, NHS-IL12/FcIL-7, or NHS-IL12/IL-2MAB602 treated) were stained for desmin (scale bars: 200 µm) and analyzed by a pathologist (B) in a blinded manner. LT: long-term treatment; ST: short-term treatment.
In vitro, growth arrest of A204 sarcoma cells induced by IFN-γ and TNF was accompanied by an immediate and highly significant increase (10-fold compared to untreated control) of cyclin dependent kinase inhibitor p21CIP1/WAF1,27,28,31,32 which in skeletal muscle intrinsically withdraws the cells from cycling26 (Fig. 7A).
Figure 7.

Multinucleate, senescent A204 cells and expression of p21 and myogenic markers in native and cytokine-treated A204 cells. (A) Relative expression of p21 before and after treatment with interferon gamma (IFN-γ) plus tumor necrosis factor (TNF) (++) or medium (–) as a control, measured by quantitative RT-PCR (n = 3). (B) IFN-γ and TNF treatment (blue triangles) terminates cancer cell proliferation but does not kill sarcoma cells (n = 3). For comparison, normal cell cultures (black dot) show unimpeded proliferation (n = 3). (C) Upper lane: Cytokine-treated A204 cancer cells are senescent (black arrow: bluish-gray staining) and multinucleate (white arrows, DAPI staining). Lower lane: A204 sarcoma cells treated with medium as a negative control are negative for SA-β-Gal and mononuclear (DAPI staining). Scale bars: 100 µm. (D) Cytokine-induced elongated, multinucleate, and syncytial morphology in A204 cells (middle and right) compared to A204 cells in standard culture (left), imaged by transmission microscopy. Scale bars: 200 µm.
Strikingly, within 5 d IFN-γ and TNF also massively induced the number of bi- and polynucleated cells in sarcoma cultures (Fig. 7B, C), suggesting a p16INK4a-mediated irreversible block of cytokinesis.33 Additionally, sarcoma cells morphed into β-galactosidase+ elongated multinucleate cells and formed multinucleate syncytia (Fig. 7A, B), a step in advanced muscle cell differentiation. Growth arrest and a multinucleated phenotype were not restricted to sarcoma cell lines; IFN-γ and TNF provoked the same effects in vitro in tumor cell lines including 3 glioblastomas, 2 neuroblastomas, and 1 each of breast cancer, hepatocellular cancer, and colorectal cancer, representing malignancies derived from diverse origins (Fig. S4). Our data suggest the potential of NHS-IL12 therapy for powerful induction of TH1 immunity and release of the TH1 cytokines IFN-γ and TNF. This strategy seems highly promising for implementation of the TH1-induced senescence pathway in the treatment of aggressive solid malignancies.
Discussion
Advanced RMS is a highly aggressive cancer, and ≥75% of patients die within 5 years despite intensive radiochemotherapy.1 In line with this, both untreated NSG (not shown) and NSG mice reconstituted with a human immune system were killed by huRMS within 52 days. Here, we show that tumor-targeted IL-12 significantly abrogated cancer growth in NSG mice reconstituted with a human immune system. Surprisingly, we found that the IL-12-driven human immune system not only killed cancer cells but also used alternative mechanisms to control cancer, specifically by inducing senescence and/or differentiation in these cancer cells.
Originally, we sought to eliminate all sarcoma cells by allogeneic T and NK cells after stimulation with IL-12. To this end, we compared the growth of RMS in an allogeneic human immune system, supported by either the T-cell survival factor IL-7, or by NHS-IL12 combined with either IL-7 or IL-2. IL-7 alone neither halted tumor growth nor induced an effective antitumor immune response. Stimulating the allogeneic human immune system with IL-12 was essential to efficiently control tumor growth in humanized mice. The NHS-IL12/IL-2MAB602 combination showed significant superiority regarding overall survival and reduced tumor volume.
Examination of the intratumoral immune compartments with histologic methods suggested that NHS-IL12 was not needed for cancer infiltration by NK cells and CD68+ macrophages. In sharp contrast, the IL-12 construct was needed to drive macrophages into the MHC-class-II-expressing M1-type, which efficiently produces effector molecules and inflammatory cytokines (TNF, IL-12, IL-6, IL-1β).13 NHS-IL12 induced strong expression of natural killer receptors NKp30 or NKp44 and NKp46 on NK cells. In the absence of IL-12, tumor-associated macrophages were MHC class-II-deficient M2 macrophages and NK cells were devoid of activating NK receptors.
Moreover, high levels of CD161 and RORC, as seen in controls, point to a IL-17-producing phenotype,34 whereas CD161 expression coupled with a low level of RORC, as observed in the NHS-IL12/FcIL-7 cohort, indicates TH1 polarization of αβ and γδ T cells as well as iNKT and NK cells into effector and central memory phenotypes that secrete high levels of IFN-γ and TNF but lack lytic activity.35 NHS-IL12/IL-2MAB602 tumors devoid of CD161 and RORC mRNA strongly suggest that IL-17-producing NK cell or T-cell phenotypes were suppressed in these mice by IL-2.36,37
As expected, IL-12 also promoted cancer infiltration by CD4+ and CD8+ T cells and perforin was only detected after stimulation with NHS-IL12 and IL-2MAB602. The IL-12 construct strongly increased the T-bet/RORC ratio and significantly dampened Foxp3 expression. These data might lead to the suggestion that IL-12 promoted the allogeneic immune system to kill RMS cells in this particular system. However, mice treated with IL-12 and IL-7 constructs did not show an increase in cells expressing granzyme or perforin. Additionally, immunohistology showed no evidence for RMS killing, apoptosis, or necrosis in efficiently treated mice in both IL-12 cohorts, suggesting that IL-12 drives critical mechanisms of cancer control independent from killing.3,23,38-41 Most importantly, however, growth-arrested cancers started to express desmin in a steric cross-striated configuration.
While investigating the underlying mechanism, we found that IL-12-associated TH1-immunity induced a senescence-defining growth arrest, mirrored by abrogated expression of the proliferation marker Ki67, nuclear p-HP1γ,23-25 and p16INK4a23 in up to 75% of all tumor cells. In humans, p16INK4a-established stable G1 cell cycle arrest42,43 can be enforced by the irreversible block of cytokinesis, as the p16INK4a/Rb-pathway synergizes with mitogenic signals in reducing the level of a mitotic exit network kinase required for cytokinesis.33,44 In accordance with this mechanism, in vitro culture of A204 sarcoma cells with IFN-γ and TNF induced a senescent (growth-arrested, β-galactosidase+) phenotype displaying a multinucleate morphology within 5 days. p21CIP1/WAF, an effector molecule downstream of p16INK4a, collaborates with p16INK4a in establishing growth arrest,45 and functions as a coordinator of cell cycle exit and differentiation when induced independently from p53, such as by IFN-γ.46,47 Despite the low expression of p21CIP1/WAF, untreated A204 RMS cells continued to proliferate and did not undergo differentiation. In sharp contrast, aggressive RMS cells exposed in vitro to IFN-γ and TNF—cytokines that are produced by TH1-polarized T cells and NK cells in vivo8—showed an immediate dramatic increase in p21CIP1/WAF characteristic of a p53-independent response to differentiation inducers,48 suggesting that p53-independent p21 expression is essential for coordinating cell cycle exit and induction of myogenic differentiation. Thus, these in vitro experiments further substantiate our in vivo observation of 3D cross-striated muscle cell configuration in growth-arrested tumors by showing that TH1-induced p16INK4a-Rb-mediated senescence is enforced through a block of cytokinesis and myogenic differentiation accomplished in the presence of TH1-cytokine-induced p21CIP1/WAF.49 Growth arrest and block of cytokinesis induced by a single dose of IFN-γ/TNF in diverse cell lines from multiple cancers, including glioblastoma, neuroblastoma, breast cancer, hepatocellular carcinoma, and colorectal carcinoma, strongly suggests senescence and differentiation as a new but ubiquitous mechanism of immune-regulated cancer control.
In summary, our data show that the targeted delivery of IL-12 to the tumor microenvironment efficiently arrested the growth of tumors and induced differentiation beyond its cytolytic function, and provide the first in vivo evidence that the previously described p16INK4a pathway and p53-independent p21CIP1/WAF are operational in therapeutically induced cancer control. This adds an important new effector mechanism to TH1 immunity and redefines our understanding of immune-mediated tumor control in humans.
Methods
Mice
NSG mice (JAX mouse stock name NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; Jackson Laboratory, USA) were housed in single airflow cages under specific pathogen-free conditions in the Research Animal Facility at the Children's Hospital, University of Tübingen, Germany. All animal procedures were reviewed by the animal care committee of the University of Tübingen (Nr. K01/07 and K06/11).
Humanization of NSG mice
HuCD34+ stem cells were derived from a surplus of granulocyte-colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells from parental donors that had been T-cell depleted by CD34+ selection (CliniMACS, Miltenyi, Germany). Cells were suspended at a 1:2 ratio in a solution of DMSO/5% HSA (20%/80%) and cryopreserved with a Sylab Icecube device and a controlled freezing rate. After thawing, cells were stained with trypan blue and counted in a Neubauer cell count chamber. Informed consent regarding the scientific use of surplus cells was obtained from all donors in accordance with the Declaration of Helsinki. Purity of the CD34+ population was further increased to >99.99% by a second round of CD3+ depletion after thawing (LS MACS, Miltenyi). Stem cell donors were all HLA-mismatched to the RMS A204 cell line. huCD34+ cells (1 × 106 cells in 100 µl prewarmed PBS) were injected in the tail vein of sublethally irradiated (250 cGy) NSG mice. Engraftment was supported by weekly applications of 20 µg FcIL-7 (Merck, Germany). In each of the NHS-IL12 treatment groups, 4 animals received long-term NHS-IL12 cytokine treatment with FcIL-7 or IL-2MAB602 for a maximum of 15 weeks (100 days).
Tumor implantation and measurement
Embryonal pediatric RMS A204 cells were obtained from ATCC (HTB 82). Cells were subcultured for 1 passage in RPMI 1640 plus 10% FBS, washed 3 times with sterile saline, and tested for mycoplasma with the PCR Mycoplasma Test Kit (AppliChem, Germany) before implantation into engrafted NSG mice. For each mouse, 1 × 106 tumor cells were transplanted in the right flank by subcutaneous injection. Tumor volume was determined with the following equation: VT = a*b*d*π/6, where a, b, and d describe length, width, and depth of the tumor. Mice were sacrificed 52 d after tumor inoculation, the time point at which the tumors of animals in the FcIL-7 cohorts had reached ≥20% of body weight. Four mice in each NHS-IL12 group were kept alive for long-term treatment.
Administration of anti-human histone/humanIL-12 fusion protein and IL-2/anti-IL-2 antibody complexes
Once per week, 20 µg of the NHS-IL12 fusion protein together with 20 µg FcIL-7 (both from Merck, Germany) was administered to engrafted humanized NSG mice via puncture of the tail vein after the tumor had grown to ≥150 mm3. As stated above, one additional cohort of mice received a mixture of 1.5 µg IL-2 plus 15 µg anti-IL-2 mAb MAB602 per week. Recombinant human IL-2 (PROLEUKIN, Aldesleukin, Chiron) and MAB602 (anti-hIL-2 mABCD122, clone 5355, R&D Systems) were co-incubated for 15 min at room temperature before injection.
In Vivo SPECT/CT Imaging
In vivo imaging of inoculated mice was carried out using an Inveon Multimodality SPECT/CT (Siemens Healthcare, Knoxville, TN). Carrier-free sodium iodide (123I) was purchased from GE Healthcare and radioiodination of NHS-IL12 was performed using Pierce® Iodination Reagent (Thermo Scientific). Mice were injected with 30 μg NHS-IL12 labeled with 18 MBq 123I via the tail vein and in vivo SPECT/CT images were acquired 2, 26, and 46 hours after tracer administration. During injection and measurement, mice were anesthetized with 1.5% isoflurane in oxygen (0.5 L/min). Regions of interest (ROI) were contoured on reconstructed SPECT images based on CT information obtained over several slices to cover the entire tumor. For reference, ROIs of equivalent size were placed on unaffected muscle tissue at the left hind leg of the same animal. For evaluation of 123I-NHS-IL12 uptake, we used decay corrected counts.
Six-color flow cytometry
Immune reconstitution was evaluated for several individuals 10-12 weeks after transplantation and peripheral blood, spleen, bone marrow, and thymus were analyzed using the following mouse mAbs that were specific for human epitopes and not cross-reactive with murine protein: CD62L(Dreg56)-FITC, CD25(2A3)-APC, CD3(SK7)-PerCP, CD8(SK1)-PerCP, CD8(SK1)-APC-H7, CD4(SK3)-FITC, CD4(SK3)-PerCP, CD14(M5E2)-PE, CD56(My31)-PE, HLA-DR(L243)-PerCP, NKp30(P30-15)-PE, NKp44(P44-8)-APC, NKp46(9E2)-PE and their corresponding IgG isotypes (BD PharMingen, Germany); CD45(MEM-28)-Pacific Blue, CD19(HIB19)-PerCP, CD3(MEM-57)-Alexa Fluor 700, and CD4(MEM-241)-Alexa Fluor 700 (Exbio, Czechoslovakia); CD45(HI30)-PE with respective isotype IgG (Biolegend, Germany). Engraftment was routinely checked 12-14 weeks after transplantation by retro-orbital bleeding and FACS staining. The A204 cell line was characterized with antibodies against ULBP-1(Z-9), ULBP-2(2F9), ULBP-3(F16), ULBP-4(6E6) (Santa Cruz Biotechnology, USA); MICA/B(6D4)APC CD112(R2.525)PE, CD155(SKII.4)PE (Biolegend, Germany); HLA-ABC(W6/32)PE (DAKO Cytomation, Germany); secondary antibody RAM-PE(X56) (BD PharMingen, Germany); and isotype controls (Beckman Coulter and R&D Systems, Germany). Flow cytometry was performed on an LSR II (BD Biosciences) using Diva© software.
Immunohistologic staining
Frozen tissue slides (5 µm thickness) were incubated in 4% buffered formalin for 2 min, washed in distilled water, boiled in citrate buffer pH 6.0 in a pressure chamber for 4 min, and washed in Tris-NaCl-Tween. Sample slides were transferred to a wet chamber and stained with Zytochem-Plus AP Polymer-Kit (Zytomed Systems, Germany). Primary antibodies against the following proteins were used: CD3 (SP7, 1:50; DCS Innovative Diagnostic Systeme GmBH, Germany); monoclonal rabbit anti human CD4 (SP35, 1:50, Zytomed Systems); CD8 (C8/144B, 1:100), CD56 (123C3-D5, 1:20), CD68 (PG-M1, 1:150), HLA-DR-α (TAL.1B5, 1:200), desmin (D33, 1:100) (DAKO, Germany); perforin (5810, 1:200, Novocastra/Leica, Germany). Final staining was performed with a Permanent AP Red Kit (Zytomed Systems). Analysis of immunohistologic slides was performed in a single-blinded manner.
Immunofluorescence
Fresh frozen cryostat sections of human xenografted A204 tumors were stained as described previously.25 Briefly, cryostat sections were fixed with periodate-lysine-paraformaldehyde and blocked with donkey serum (1:20) for 30 min at room temperature (RT). Slides were then incubated with rabbit anti-p-HP1γ (1:80; Abcam, UK), mouse-anti-PCNA (1:50; Cell Signaling Technology, MA), mouse anti-p16INK4a (1:50; Santa Cruz Biotechnology, Germany), or rabbit anti-Ki67 (1:100; Abcam, USA) for 1 h at RT. After 3 washes, the sections were incubated with Cy3- or Cy5-conjugated donkey anti-rabbit antibody and Cy3- or Cy5-conjugated donkey anti-mouse antibody (all from Dianova, Germany). Nuclei were stained with Yopro (1:2000; Invitrogen, Germany) for 5 min before mounting the slides with Mowiol (Hoechst, Germany). The sections were analyzed with a Leica TCS-Sp/Leica DM RB confocal laser scanning microscope (Leica Microsystems, Germany). Images were processed with the Leica Confocal Software LCS (Version 2.61).
Treatment of patient-derived primary RMS cell lines with TNF and IFN-γ
Permanent growth arrest of different sarcoma cell lines after cytokine treatment was determined using the procedure described previously.23 Short-term culture to study early differentiation events in RMS was accomplished by seeding sarcoma cells at low numbers (4,000 or 8,000 cells/cm2) and incubating with DMEM with and without 10 ng/mL tumor necrosis factor (TNF) and 100 ng/mL interferon gamma (IFN-γ).
Molecular methods
Total RNA was isolated with the RNeasy Mini Kit and RNase-Free DNAse Set (both Qiagen, Germany). cDNA was synthesized using Superscript III First Strand Synthesis Super Mix (Life Technology, Germany).
KIR expression analysis
Expression analysis was performed as previously described.50 NKp30, NKp44, NKp46, DNAM-1, and CD161 transcripts were determined with specific primers in end-point PCR using 5´FAM-labeled reverse primers (primers and protocols can be obtained on request). PCR products were analyzed for fragment length in an ABI sequencer with Gene Scan-600 LIZ for length standard and GeneMapper software (both Applied Biosystems, Germany); mean fluorescence intensity (MFI) was used for semiquantitative analysis.
Expression of T-bet, RORC, Foxp3, CD40L, granzyme B, granzyme K
Intratumoral gene expression was determined with real-time PCR on a BioRad C1000 Thermal cycler/CFX96 real-time System (Munich, Germany) using cDNA-specific primers (sequences available on request) and the iQ SYBR Green Supermix (BioRad). Expression was normalized to that of human CD45. RORC expression was detected with a specific primer set (Search LC, Germany) and FastStart DNA SYBR Green I (Roche, Germany).
Vα-24 chain identification
Vα-24 chain identification was performed as published previously.51
Vβ spectratyping analysis
Diversity of the TCR Vβ-chain expression and complexity of the TCR repertoire was analyzed according to Gorski et al.52 with minor modifications. We used 5´FAM-labeled Cβ-primer and an ABI sequencer with Gene Scan-600 LIZ and GeneMapper software for detection of PCR amplicons (Applied Biosystems).
γδ immunoscope
The immunoscope was determined as published previously.53
Identification of TCR-CDR3 regions
Vβ PCR products were cloned into pGEMTeasy (Promega, Germany) and amplified in XL1-Blue competent cells (Stratagene, USA) using standard procedures. Insert-positive clones were transferred directly to a reamplifying Vβ PCR. PCR products (5 µl aliquots) were analyzed on a 2.5% agarose gel and products of relevant length were sent to Seqlab (Germany) for sequence analysis. Translation of cDNA into protein sequence was conducted with EMBOSS Transeq free software.
Statistical analysis
In all figures, error bars represent SEM and center values represent mean of data unless indicated otherwise. Statistical significance was analyzed with one-tailed Student t-tests unless indicated otherwise, and the data met the assumptions of this test. Experiments with mice included 11 individuals per cohort, which represents a sufficient cohort size for statistical analysis. Long-term treatment was applied to 4 animals per NHS-IL12 cohort. Mice were randomly allocated to the different treatment groups after their tumors reached the predetermined tumor size. Survival was evaluated with Kaplan-Meier curves and compared with log-rank test. Only tumor-related deaths were evaluated for survival analysis.
Acknowledgments
The excellent technical assistance of M. Dierstein and B. Fehrenbacher is gratefully acknowledged.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
KS designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. MA established the mouse model and molecular methods, and performed experiments. CW established methodology, performed research, prepared the figures, and edited the manuscript. HZ, MS, FM, AE, GB, FE, KSo, SS performed experiments and analyzed data. JS, BJP performed in vivo imaging SPECT/CT, interpretation of data, and wrote the manuscript. MR, TW, HB analyzed data, and designed and performed immunofluorescence staining for detection of senescence. MR developed the concept of cytokine-induced senescence in cancers. HM, CS, ME, RT, WS analyzed data. AL provided primers. SDG designed NHS-IL12 and FcIL-7. RH provided ideas for the study, co-designed the NSG mouse, and provided mice and stem cells for the study.
Funding
The fusion proteins NHS-IL-12 and FcIL-7 were a kind gift of Merck Serono (Merck, Germany). This research was supported by the European Union FP7 program as part of the project NANOII, grant agreement number 229289. KS was supported by the Deutsche Leukämie Forschungshilfe Projekt A2008/5(2) DKS 2010.10, CW, HZ and MS were supported by the Jürgen Manchot-Foundation, JS by the Werner Siemens-Foundation, and ME by a grant of the IZKF Würzburg (B-107). MR was supported by the Sander Stiftung (2005.043.2, 2005.043.3 and 2012.056.1), the Deutsche Krebshilfe (No. 109037), and the Deutsche Forschungsgemeinschaft (SFB 685). RH was supported by grants from the Stiftung für krebskranke Kinder Tübingen e.V., the Jose-Carreras Leukaemia Foundation, and the Stefan-Morsch-Stiftung.
References
- 1.Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WH, Carli M, Anderson JR. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol 2008; 26:2384-9; PMID:; http://dx.doi.org/ 10.1200/JCO.2007.14.7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Broeke LT, Pendleton CD, Mackall C, Helman LJ, Berzofsky JA. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res 2006; 66:1818-23; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2549 [DOI] [PubMed] [Google Scholar]
- 3.Muller-Hermelink N, Braumuller H, Pichler B, Wieder T, Mailhammer R, Schaak K, Ghoreschi K, Yazdi A, Haubner R, Sander CA, et al.. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008; 13:507-18; PMID:; http://dx.doi.org/ 10.1016/j.ccr.2008.04.001 [DOI] [PubMed] [Google Scholar]
- 4.Wiegering V, Eyrich M, Rutkowski S, Wolfl M, Schlegel PG, Winkler B. TH1 predominance is associated with improved survival in pediatric medulloblastoma patients. Cancer Immunol Immunother 2011; 60:693-703; PMID:; http://dx.doi.org/ 10.1007/s00262-011-0981-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007; 109:5346-54; PMID:; http://dx.doi.org/ 10.1182/blood-2006-10-051318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, et al.. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest 2011; 121:4746-57; PMID:; http://dx.doi.org/ 10.1172/JCI58814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, Gately MK, Wolf SF, Schreiber RD, Storkus WJ, et al.. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol 1994; 153:1697-706; PMID: [PubMed] [Google Scholar]
- 8.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133-46; PMID:; http://dx.doi.org/ 10.1038/nri1001 [DOI] [PubMed] [Google Scholar]
- 9.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity 2009; 30:92-107; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T, Radbruch A, Chang HD. IFN-gamma and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur J Immunol 2010; 40:3017-27; PMID:; http://dx.doi.org/ 10.1002/eji.201040539 [DOI] [PubMed] [Google Scholar]
- 11.Wigginton JM, Kuhns DB, Back TC, Brunda MJ, Wiltrout RH, Cox GW. Interleukin 12 primes macrophages for nitric oxide production in vivo and restores depressed nitric oxide production by macrophages from tumor-bearing mice: implications for the antitumor activity of interleukin 12 and/or interleukin 2. Cancer Res 1996; 56:1131-6; PMID: [PubMed] [Google Scholar]
- 12.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol 2009; 9:259-70; PMID:; http://dx.doi.org/ 10.1038/nri2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, et al.. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med 2010; 362:875-85; PMID:; http://dx.doi.org/ 10.1056/NEJMoa0905680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l'union fait la force”. Blood 2005; 106:2252-8; PMID:; http://dx.doi.org/ 10.1182/blood-2005-03-1154 [DOI] [PubMed] [Google Scholar]
- 15.Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, Atkins MB. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res 2000; 6:1678-92; PMID: [PubMed] [Google Scholar]
- 16.Sharifi J, Khawli LA, Hu P, King S, Epstein AL. Characterization of a phage display-derived human monoclonal antibody (NHS76) counterpart to chimeric TNT-1 directed against necrotic regions of solid tumors. Hybrid Hybridomics 2001; 20:305-12; PMID:; http://dx.doi.org/ 10.1089/15368590152740707 [DOI] [PubMed] [Google Scholar]
- 17.Chames P, Van RM, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 2009; 157:220-33; PMID:; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006; 311:1924-7; PMID:; http://dx.doi.org/ 10.1126/science.1122927 [DOI] [PubMed] [Google Scholar]
- 19.Verdeil G, Marquardt K, Surh CD, Sherman LA. Adjuvants targeting innate and adaptive immunity synergize to enhance tumor immunotherapy. Proc Natl Acad Sci U S A 2008; 105:16683-8; PMID:; http://dx.doi.org/ 10.1073/pnas.0805054105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 2004; 4:665-74; PMID:; http://dx.doi.org/ 10.1038/nri1435 [DOI] [PubMed] [Google Scholar]
- 21.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al.. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005; 174:6477-89; PMID:; http://dx.doi.org/ 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- 22.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495-502; PMID:; http://dx.doi.org/ 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braumuller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al.. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:; http://dx.doi.org/ 10.1038/nature11824 [DOI] [PubMed] [Google Scholar]
- 24.Saretzki G. Cellular senescence in the development and treatment of cancer. Curr Pharm Des 2010; 16:79-100; PMID:; http://dx.doi.org/ 10.2174/138161210789941874 [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle 2007; 6:784-9; PMID:; http://dx.doi.org/ 10.4161/cc.6.7.4079 [DOI] [PubMed] [Google Scholar]
- 26.Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol 1996; 132:657-66; PMID:; http://dx.doi.org/ 10.1083/jcb.132.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 1995; 267:1018-21; PMID:; http://dx.doi.org/ 10.1126/science.7863327 [DOI] [PubMed] [Google Scholar]
- 28.Walsh K, Perlman H. Cell cycle exit upon myogenic differentiation. Curr Opin Genet Dev 1997; 7:597-602; PMID:; http://dx.doi.org/ 10.1016/S0959-437X(97)80005-6 [DOI] [PubMed] [Google Scholar]
- 29.Paulin D, Li Z. Desmin: a major intermediate filament protein essential for the structural integrity and function of muscle. Exp Cell Res 2004; 301:1-7; PMID:; http://dx.doi.org/ 10.1016/j.yexcr.2004.08.004 [DOI] [PubMed] [Google Scholar]
- 30.Carter RL, McCarthy KP, Machin LG, Jameson CF, Philp ER, Pinkerton CR. Expression of desmin and myoglobin in rhabdomyosarcomas and in developing skeletal muscle. Histopathology 1989; 15:585-95; PMID:; http://dx.doi.org/ 10.1111/j.1365-2559.1989.tb01624.x [DOI] [PubMed] [Google Scholar]
- 31.Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Curr Opin Oncol 2013; 25:52-8; PMID:; http://dx.doi.org/ 10.1097/CCO.0b013e32835b639e [DOI] [PubMed] [Google Scholar]
- 32.Schneider JW, Gu W, Zhu L, Mahdavi V, Nadal-Ginard B. Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science 1994; 264:1467-71; PMID:; http://dx.doi.org/ 10.1126/science.8197461 [DOI] [PubMed] [Google Scholar]
- 33.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8:1291-7; PMID:; http://dx.doi.org/ 10.1038/ncb1491 [DOI] [PubMed] [Google Scholar]
- 34.Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, Flint J, Willberg CB, Bengsch B, Seigel B, et al.. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci U S A 2010; 107:3006-11; PMID:; http://dx.doi.org/ 10.1073/pnas.0914839107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi T, jbakhsh-Jones S, Strober S. Expression of CD161 (NKR-P1A) defines subsets of human CD4 and CD8 T cells with different functional activities. J Immunol 2006; 176:211-6; PMID:; http://dx.doi.org/ 10.4049/jimmunol.176.1.211 [DOI] [PubMed] [Google Scholar]
- 36.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al.. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26:371-81; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2007.02.009 [DOI] [PubMed] [Google Scholar]
- 37.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al.. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 2010; 467:967-71; PMID:; http://dx.doi.org/ 10.1038/nature09447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res 2002; 62:5069-75; PMID: [PubMed] [Google Scholar]
- 39.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007; 450:903-7; PMID:; http://dx.doi.org/ 10.1038/nature06309 [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest 2008; 118:1398-404; PMID:; http://dx.doi.org/ 10.1172/JCI33522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001; 410:1107-11; PMID:; http://dx.doi.org/ 10.1038/35074122 [DOI] [PubMed] [Google Scholar]
- 42.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432:307-15; PMID:; http://dx.doi.org/ 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- 43.Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech Ageing Dev 2006; 127:16-24; PMID:; http://dx.doi.org/ 10.1016/j.mad.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 44.Bothos J, Tuttle RL, Ottey M, Luca FC, Halazonetis TD. Human LATS1 is a mitotic exit network kinase. Cancer Res 2005; 65:6568-75; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0862 [DOI] [PubMed] [Google Scholar]
- 45.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7:667-77; PMID:; http://dx.doi.org/ 10.1038/nrm1987 [DOI] [PubMed] [Google Scholar]
- 46.Hobeika AC, Etienne W, Torres BA, Johnson HM, Subramaniam PS. IFN-gamma induction of p21(WAF1) is required for cell cycle inhibition and suppression of apoptosis. J Interferon Cytokine Res 1999; 19:1351-61; PMID:; http://dx.doi.org/ 10.1089/107999099312812 [DOI] [PubMed] [Google Scholar]
- 47.Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene 2000; 19:5712-9; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1203963 [DOI] [PubMed] [Google Scholar]
- 48.Steinman RA, Hoffman B, Iro A, Guillouf C, Liebermann DA, el-Houseini ME. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene 1994; 9:3389-96; PMID: [PubMed] [Google Scholar]
- 49.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400-14; PMID:; http://dx.doi.org/ 10.1038/nrc2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X, Knowles J, Barfield RC, Kasow KA, Madden R, Woodard P, Srivastava DK, Horwitz EM, Handgretinger R, Hale GA. A novel approach for quantification of KIR expression in healthy donors and pediatric recipients of hematopoietic SCTs. Bone Marrow Transplant 2009; 43:525-32; PMID:; http://dx.doi.org/ 10.1038/bmt.2008.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han M, Harrison L, Kehn P, Stevenson K, Currier J, Robinson MA. Invariant or highly conserved TCR alpha are expressed on double-negative (CD3+CD4-CD8-) and CD8+ T cells. J Immunol. 1999; 163:301-11; PMID: [PubMed] [Google Scholar]
- 52.Gorski J, Yassai M, Zhu X, Kissela B, Kissella B, Keever C, Flomenberg N. Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 size spectratyping. Correlation with immune status. J Immunol 1994; 152:5109-19; PMID: [PubMed] [Google Scholar]
- 53.Dechanet J, Merville P, Lim A, Retière C, Pitard V, Lafarge X, Michelson S, Méric C, Hallet MM, Kourilsky P, et al.. Implication of gammadelta T cells in the human immune response to cytomegalovirus. J Clin Invest 1999; 103:437-49; PMID:; http://dx.doi.org/ 10.1172/JCI5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.