

Abstract
Despite the discovery and application of many parenteral (unfractionated and low-molecular-weight heparins) and oral anticoagulant vitamin K antagonist (VKA) drugs, the prevention and treatment of venous and arterial thrombotic phenomena remain major medical challenges. Furthermore, VKAs are the only oral anticoagulants used during the past 60 years. The main objective of this study is to present recent data on non-vitamin K antagonist oral anticoagulants (NOACs) and to analyze their advantages and disadvantages compared with those of VKAs based on a large number of recent studies. NOACs are novel direct-acting medications that are selective for one specific coagulation factor, either thrombin (IIa) or activated factor X (Xa). Several NOACs, such as dabigatran (a direct inhibitor of FIIa) and rivaroxaban, apixaban and edoxaban (direct inhibitors of factor Xa), have been used for at least 5 years but possibly 10 years. Unlike traditional VKAs, which prevent the coagulation process by suppressing the synthesis of vitamin K-dependent factors, NOACs directly inhibit key proteases (factors IIa and Xa). The important indications of these drugs are the prevention and treatment of deep vein thrombosis and pulmonary embolisms, and the prevention of atherothrombotic events in the heart and brain of patients with acute coronary syndrome and atrial fibrillation. They are not fixed, and dose-various strengths are available. Most studies have reported that more advantages than disadvantages for NOACs when compared with VKAs, with the most important advantages of NOACs including safety issues (ie, a lower incidence of major bleeding), convenience of use, minor drug and food interactions, a wide therapeutic window, and no need for laboratory monitoring. Nonetheless, there are some conditions for which VKAs remain the drug of choice. Based on the available data, we can conclude that NOACs have greater advantages and fewer disadvantages compared with VKAs. New studies are required to further assess the efficacy of NOACs.
Keywords: novel oral anticoagulants, direct IIa and Xa inhibitors, vitamin K antagonist, venous thromboembolism
Introduction
Thromboembolic diseases are of major clinical concern due to their high prevalence and consequences, which are often fatal. Venous thromboembolism (VTE) is estimated to be the third most common cardiovascular disorder after coronary heart disease and stroke.1 Treatment of venous and arterial thrombotic phenomena represents a major medical challenge, and the development of anticoagulant drugs represents a revolution in medicine. The route of administration of anticoagulant drugs can be either parenteral or oral.
During the last 60 years, vitamin K antagonists (VKAs), which include coumarin derivatives (eg, warfarin and acenocoumarol), have been the only oral anticoagulants used;2 however, new substances with anticoagulants effects, referred to as new oral anticoagulants, have recently been discovered. Compared with VKAs, this new generation of oral anticoagulants (non-vitamin K antagonist oral anticoagulants, NOACs) has more predictable anticoagulant responses, and NOACs have been shown to be effective in the prevention and treatment of VTE and in the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF).3,4 The VKA dose is determined on an individual basis (not fixed), whereas novel NOACs are administered in fixed doses, except when a patient has a functional disorder of the liver or kidney. NOACs are termed direct oral anticoagulants or target anticoagulants due to their direct inactivation of thrombin (FIIa) and factor X (FXa). Despite the various advantages of NOACs compared with VKAs, these drugs are not considered ideal because there are also some disadvantages compared with VKAs. The aim of this paper is to review new data from the literature regarding the advantages and disadvantages of these two types of oral anticoagulants.
Vitamin K anticoagulants
Oral anticoagulation was first established in 1941 by Karl Paul Link, who discovered dicumarol.5 VKA drugs are 4-hydroxycoumarin derivatives, which exert their anticoagulant effect by inhibiting vitamin K epoxide reductase and, possibly, vitamin KH2 reductase.6 These compounds act by reducing vitamin KH2 (reduced form of vitamin K) levels, thereby limiting the cofactor effect of vitamin K on the γ-carboxylation of the vitamin K-dependent coagulation factors II, VII, IX, and X. VKAs also limit the effect of anticoagulant proteins, protein C and protein S, resulting in an inhibition of these proteins3,7 because their synthesis depends on the presence of vitamin K. As VKAs inhibit protein C prior to its anticoagulant effect, it may be necessary to use bridging anticoagulation with low-molecular-weight heparins (LMWHs). Vitamin K acts as a cofactor in the post-translational carboxylation of glutamate residues to γ-carboxylglutamates in the N-terminal regions of the vitamin K-dependent proteins.8,9 For inhibition of this process, warfarin is the drug of choice in most countries, especially in the USA and Canada, whereas acenocoumarol and phenprocoumon are used in many European countries. Treatment with VKAs is indicated in various medical situations, such as for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and the prevention of recurrence, atrial fibrillation (AF) and stroke in patients with NVAF, acute myocardial infarction, and vasculopathy, as well as in patients with tissue heart valves or mechanical prosthetic cardiac valves. These drugs are also used as prophylaxis for VTE in high-risk patients (eg, post-orthopedic surgery, embolic peripheral, and arterial disease).7,10
Novel oral anticoagulants
The new oral anticoagulants represent novel direct-acting medications that are selective for one specific coagulation factor, either thrombin or activated factor Xa. These drugs have recently been approved for the prevention of VTE in patients after elective hip or knee arthroplasty in the European Union (EU) and many other countries worldwide.11 Several NOACs, such as dabigatran, rivaroxaban, apixaban, and edoxaban, have been used in many countries. The mechanisms of indirect (VKAs) and direct (NOACs) anticoagulants are presented in Figure 1. The main characteristics of VKAs and NOACs and their advantages and disadvantages are listed in Table 1.
Figure 1.
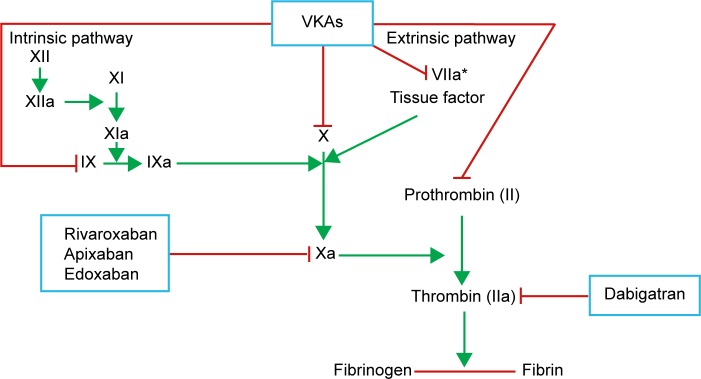
Mechanism of anticoagulants effect of indirect (VKAs) and direct anti-IIa and anti-Xa anticoagulants (NOACs).
Note: *VKA does not inhibit FVIIa, but prevents their synthesis, like other vitamin K-dependent factors (eg, II, IX, and X).
Abbreviations: NOACs, non-vitamin K antagonist oral anticoagulants; VKAs, vitamin K antagonists; FVIIa, activated factor VII.
Table 1.
Main characteristics of the VKAs and NOACs, advantages and their disadvantages
| Drug | Vitamin K antagonists (VKAs) | Dabigatran | Rivaroxaban | Apixaban | Edoxaban |
|---|---|---|---|---|---|
| Mechanism of action | Indirect through inhibition of vitamin K epoxide reductase (VKOCR1), lower levels of vitamin KH2, and consequently and vitamin K-dependent coagulation factors | Direct anti-IIa | Direct anti-Xa | Direct anti-Xa | Direct anti-Xa |
| Bioavailability | Warfarin 99% | ||||
| R-enantiomer acenocoumarol 100% | 6%–7% | 60%–80% | 66% | 58.3% | |
| tmax | 1.5–2 hours | 2.5–4 hours | 3 hours | 1.5 hours | |
| Half-life | S- and R-warfarin 32 and 42 hours | 12–14 hours | 7–13 hours | 8–15 hours | 9–11 hours |
| S- and R-acenocoumarol 2 and 8 hours | |||||
| Onset of action | 36–72 hours | 0.5–2 hours 2–4 hours | 1–3 hours | 1–2 hours | |
| Route of elimination | Hepatically metabolized | 80% renal | 70% renal (30% unchanged, 40% inactive, and 30% fecal) | 25% renal | 35% renal |
| Advantages | High bioavailability Test monitoring with PT (INR), dose adjustment dependent on INR value They have antidote (vitamin K) Can use in all group ages Long clinical experience with VKAs (these drugs have been used as anticoagulants for over 60 years) Not expensive |
Predictable pharmacokinetics and pharmacodynamics Low drug–drug and food interactions No dietary restriction Rapid onset and offset Short half-life In general no need for laboratory monitoring, although in some cases it is required Wide therapeutic window Switching patient from LMWH and VKAs to NOACs |
|||
| Disadvantages | Unpredictable pharmacokinetics and individual (great variability of individual dose) Great drug–drug interactions Dietary restriction Need for frequent monitoring of INR Narrow therapeutic window Slow onset and offset Long half-life (this is a problem when required emergency surgery and in cases of bleeding due to accumulation of the drug in the blood) VKAs-induced skin necrosis if started without LMWH |
Do not exist standardized test for monitoring of NOACs, when it is necessary for monitoring of these drugs, eg, in hepatic and renal disease Sometimes rapid offset and short half-life may be considered as disadvantages Currently lack of antidote High cost Not enough experience NOACs therapy can be initiated without LMWH (no risk for induced skin necrosis) due to their rapid onset |
|||
Abbreviations: NOACs, non-vitamin K antagonist oral anticoagulants; INR, international normalized ratio; PT, prothrombin time; LMWH, low-molecular-weight heparin; tmax, time maximum plasma concentration.
Dabigatran
Dabigatran was the first approved NOAC; it was approved in 2008 by the EU and by the Food and Drug Administration (FDA) in 2010 based on the results of the Randomized Evaluation of Long-Term Anticoagulant Therapy (RE-LY) trial for warfarin, which was compared with dabigatran.12 A new oral, direct thrombin (FIIa) inhibitor that prevents the conversion of fibrinogen to fibrin and thereby prevents clot formation, dabigatran is indicated to reduce the risk of stroke and systemic embolism in patients with NVAF.13 Dabigatran is a synthetic small molecule, hirudin analog that exhibits univalent binding to only one of the two key thrombin sites. It is the product of the prodrug (dabigatran etexilate) of dabigatran, which is rapidly transformed to dabigatran after oral ingestion and hepatic processing.14 The trade name of dabigatran etexilate is Pradaxa (Boehringer Ingelheim, Ingelheim, Germany). The drug can be administered with or without food and is rapidly absorbed, but its absorption after oral administration (oral bioavailability) is low (6%–7%) and is independent of the dose of the prodrug.13 Some studies have shown that the plasma concentration of dabigatran increases in a dose-dependent manner such that the peak plasma concentration (Cmax) is achieved 1.5–2 hours after oral administration and is not related to age or sex.15 The mean plasma terminal half-life of dabigatran is 12–14 hours and is independent of dose.16 The absorption and bioconversion of dabigatran occur in enterocytes, hepatocytes, and the portal vein. Dabigatran does not inhibit cytochrome P450 (CYP); therefore, its potential for drug–drug interactions is low. Unlike VKA, dabigatran exhibits a predictable dose response and, therefore, does not require routine coagulation monitoring.17 The primary route for dabigatran elimination in humans is renal (80%).13,15 There are several comparisons of the effects of dabigatran with those of warfarin, and the RE-LY trial showed that dabigatran is not inferior to warfarin with respect to the prevention of stroke or systemic embolism in patients with NVAF. In this trial, dabigatran administered at a dose of 110 mg was associated with rates of stroke and systemic embolism that were similar to those associated with warfarin and also showed lower rates of major hemorrhage. Additionally, the same authors found that at a dose of 150 mg, dabigatran was associated with lower rates of stroke and systemic embolism compared with warfarin, but with similar rates of major hemorrhage.12
Hohnloser et al in the RE-LY trial found that myocardial infarction (MI) occurred at annual rates of 0.82% with dabigatran 110 mg twice daily compared with 0.64% with warfarin (hazard ratio [HR] 1.29, 95% confidence interval [CI], 0.96–1.75, and P=0.09 for dabigatran 110 mg). In the same study, it was found that MI occurred at annual rates of 0.81% with dabigatran 150 mg twice daily compared with 0.64% with warfarin (HR 1.27, 95% CI, 0.94–1.71, P=0.12 for dabigatran 150 mg).18 However, according to Artang et al dabigatran is associated with a greater risk of MI than warfarin (odds ratio [OR] 1.35, 95% CI, 1.10–1.66, P=0.005).19 Indeed, a statistically significant difference between dabigatran and warfarin in relation to risk of MI was reported. Graham et al showed that dabigatran reduced the risk of ischemic stroke, intracranial hemorrhage as well as death but increased the risk of major gastrointestinal bleeding compared with warfarin in elderly patients with NVAF.20 The RE-COVER trial showed a rate of recurrent VTE in patients treated with dabigatran (150 mg twice daily) of 2.4% compared with those treated with warfarin international normalized ratio (INR 2–3), for which the rate was 2.1%. These results demonstrated the noninferiority of dabigatran compared to warfarin for the prevention of recurrent VTE (difference risk 0.4%, 95% CI, 0.8–1.5, P<0.001 for the prespecified noninferiority margin). According to this trial, the rate of major bleeding episodes in the dabigatran group was lower (1.6%) compared to the warfarin group (1.9%) (HR 0.82, 95% CI, 0.45–1.48, P=0.53). The same parameters for episodes of any bleeding were HR 0.71, 95% CI, 0.59–0.85, P=0.0002, revealing the superiority of dabigatran.21 The data from the RE-COVER II trial confirmed the results of the RE-COVER trial with regard to recurrent VTE, indicating the noninferiority of dabigatran (2.3%) compared to warfarin (2.2%) (HR 1.08, 95% CI, 0.64–1.80, absolute risk difference 0.2%, 95% CI, 1.0–1.3, P,0.001 for the prespecified noninferiority margin). Additionally, the results of the RE-COVER II trial showed that the risk for clinically relevant bleeding (dabigatran 1.2% vs warfarin 1.7%, HR 0.69, 95% CI, 0.36–1.32, P=0.259) or any bleeding (dabigatran 15.6% vs warfarin 22.1%, HR 0.67, 95% CI, 0.56–0.81, P<0.001) is significantly lower with dabigatran.22
Rivaroxaban
Rivaroxaban is the second NOAC approved in many countries, in 2008 by the European Medicine Agency and by the FDA based on the results of the rivaroxaban versus warfarin in non-valvular atrial fibrillation (ROCKET AF) trial.23 Rivaroxaban, an oxazolidinone derivative,24 is a selective direct inhibitor of FXa.25 Activated FXa plays an important role in the coagulation cascade because it links the intrinsic and extrinsic coagulation pathways and acts as a rate-limiting step in thrombin formation. Inhibition of FXa and the prevention of thrombin generation from NOACs can be achieved directly, but FXa inhibition can also be attained indirectly through the use of parenteral anticoagulant drugs, such as fondaparinux, idraparinux, unfractionated heparin, and LMWHs.26 The trade name of rivaroxaban is Xarelto (Bayer HealthCare, Leverkusen, Germany). It is a non-basic compound that is rapidly absorbable and has a high bioavailability (60%–80%) after oral administration.27 It is known that the pharmacokinetics (PK) of rivaroxaban are dose dependent, with Cmax occurring 2.5–4 hours after oral administration.27,28 Approximately 30% of rivaroxaban is excreted unchanged in the urine and through fecal elimination.29 Metabolism of this drug occurs in the liver, primarily via the CYP isozyme CYP3A4.30 According to Eriksson et al and Turpie et al rivaroxaban can be administered with food or within 2 hours of eating.31,32 Several PK studies have demonstrated that after typical doses of rivaroxaban, its half-life elimination is approximately 7 hours (4–7 hours).26 For daily exposure, the area under the curve (AUC0–24) is 1.094 ng h/mL. The coefficient of variation for exposure parameters (AUC0–24, Cmax, Cmin) for rivaroxaban is 29%–49%.
The concentration of rivaroxaban in blood during 24 hours and its anti-factor Xa activity are 10 ng/mL and 0.17 IU/mL, respectively.33 Therefore, one-dose administration of rivaroxaban is sufficient to provide daily anticoagulant activity. Only the 2.5 mg twice daily dose of rivaroxaban is licensed for secondary prevention in acute coronary syndrome in combination with standard antiplatelet therapy in adults with elevated cardiac biomarkers in patients with creatinine clearance (CrCL) >15 mL/min.34 According to Mega et al a twice daily 2.5-mg dose of rivaroxaban reduced the rates of death from cardiovascular causes (2.7% vs 4.1%, P=0.002) and from any cause (2.9% vs 4.5%, P=0.002); however, these authors did not find a survival benefit from twice daily 5-mg dose of rivaroxaban.35
Although there is a lack of information in the literature regarding the use of rivaroxaban in elderly patients, our opinion is that 5 mg doses of rivaroxaban can be used twice daily in patients ≥75 years to prevent thromboembolic events.
Regarding the PK and pharmacodynamic (PD) properties of rivaroxaban, there are no differences with respect to sex or race. Nonetheless, renal and hepatic insufficiency impacts the PK and PD properties of rivaroxaban, depending on the degree of hepatic and renal failure; thus, mild hepatic disease (Child-Pugh Class A) does not result in any clinically relevant differences in the PK and PD of rivaroxaban.36 The role of rivaroxaban in the treatment of VTE was investigated in three large randomized trials in the EINSTEIN programs (EINSTEIN-DVT, EINSTEIN-PE, and EINSTEIN-extension study).37 In the EINSTEIN-DVT and EINSTEIN-PE, rivaroxaban was equivalent to the standard treatment in the overall population, as well as in older adults and those with renal insufficiency and fragility.38 The clinical approval of rivaroxaban for the treatment of DVT and for the prevention of DVT or PE was based on the results of the randomized Phase III open-label EINSTEIN-DVT trial. In this study, 3,449 patients with acute symptomatic DVT were treated with either rivaroxaban 15 mg twice daily for 3 weeks, followed by once daily rivaroxaban 20 mg for 3, 6, or 12 months, or with enoxaparin according to body weight twice daily for a minimum of 5 days, followed by a dose-adjusted VKA. The study showed that recurrent VTE occurred in the patients treated with rivaroxaban (2.1% vs 3.0% of cases treated with enoxaparin plus VKA) (P<0.001 for noninferiority).37 In the EINSTEIN-PE, 4,832 patients with acute PE with or without DVT were similarly treated with either rivaroxaban or enoxaparin, followed by VKAs. The event rates for the primary efficacy endpoint of symptomatic VTE were 2.1% versus 1.8% (P=0.003 for noninferiority) in the rivaroxaban- and enoxaparin plus VKA-treated patient groups, respectively. Cases of major and non-major bleeding were similar in both groups: there was no difference between rivaroxaban and enoxaparin in the VKA group, but a significant reduction in major bleeding occurred in 1.1% of the patients in the rivaroxaban group compared with 2.2% in the enoxaparin group (HR 0.49, 95% CI, 0.31–0.79, P=0.003).39
Apixaban
Apixaban is another NOAC that is a reversible direct Xa antagonist.40 It exerts a similar anticoagulant activity as rivaroxaban, by the direct inhibition of factor Xa, which is formed by both intrinsic and extrinsic coagulation pathways. This prevention of thrombin formation from prothrombin is needed to prevent the conversion of fibrinogen to fibrin. Apixaban is the third NOAC that was approved by the FDA and by the European Medicine Agency, in 2011 based on the results of ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation).41 The trade name of apixaban is Eliquis (Bristol-Myers Squibb, New York, NY, USA). Apixaban is rapidly absorbed after oral administration; its bioavailability is approximately 66% and is not affected by food. It has a time maximum plasma concentration (short tmax), similar to other NOACs, with a half-life of 8–15 hours; however, compared with other new NOACs, apixaban has the smallest renal clearance (25%).42 Similar to rivaroxaban, apixaban is metabolized in the liver in the CYP-dependent isozyme pathway (CYP3A4).30 As noted above, the ARISTOTLE trial was a randomized double-blind trial that compared apixaban with the dose-adjusted warfarin INR of 2.0–3.0 (5 mg twice daily). In this study, 18,201 patients were included and divided into two groups: 9,120 patients in the apixaban group and 9,081 patients in the warfarin group. The primary outcome was the rate of stroke (both ischemic or hemorrhagic or systemic embolism) in patients who were treated with warfarin compared with those receiving apixaban. The authors concluded that apixaban was superior to warfarin for preventing stroke and systemic embolism in patients with AF (1.27% per year compared with 1.60% in the warfarin group). The results were P<0.001 for noninferiority and P=0.01 for superiority (HR with apixaban 0.79, 95% CI, 0.66–0.95) to warfarin. Additionally, the authors found a hemorrhagic stroke rate of 0.24% per year in the apixaban group compared with 0.47% per year in the warfarin group (OR 0.51, 95% CI, 0.35–0.75, P<0.001). Therefore, a lower mortality rate was observed in the apixaban group compared with the warfarin group.41
Edoxaban
Edoxaban (DU-176b) is an oral direct, specific inhibitor of FXa with an approximate 10,000-fold selectivity for FXa over thrombin.43 The compound was developed by Daiichi Sankyo (Daiichi Sankyo Company, Ltd., Tokyo, Japan) and approved in July 2011 in Japan for the prevention of VTE following lower-limb orthopedic surgery. Trade names of edoxaban are Savaysa and Lixiana.44 In addition, edoxaban was approved by the FDA in January 2015 for the prevention of stroke and non-central-nervous-system systemic embolisms.45 Edoxaban is rapidly absorbed, and it was estimated that its absolute bioavailability is 58.3%.46 In this study, after administration of a single dose of 60 mg, the Cmax of edoxaban occurred at 1.5 hours in healthy subjects, whereas its half-life was 9–11 hours. This drug has dual mechanisms of elimination; approximately one-third is eliminated via the kidney and the remainder via feces.47 Similar to dabigatran and rivaroxaban, edoxaban is also a substrate for the efflux transporter P-glycoprotein (P-gp). For this reason, in the ENGAGE AF-TIMI 48 trial, reduction of the edoxaban dosage by 50% was required when used in combination with strong P-gp inhibitors, such as verapamil.3 It should mentioned that in a Phase II trial involving 523 patients undergoing total knee replacement surgery, administration of DU-176b resulted in with a dose-dependent decrease in VTE, without increases in blending events.48 In another reported Phase II trial in which DU-176b was investigated at 30 mg and 60 mg either once a day or twice daily compared to dose-adjusted warfarin in 1,146 patients with AF, doses of 60 mg twice daily was accompanied by increased bleeding events.49 In the HOKUSAI trial, a statistically significant reduction in recurrent VTE was observed in edoxaban-treated patients, at 3.2% compared to 3.5% for the warfarin group (HR 0.89, 95% CI, 0.70–1.3, P<0.001 for noninferiority).50 DU-176b was also tested in a Phase III study for prophylaxis in major orthopedic surgery in comparison to enoxaparin at doses of 20 mg twice daily; edoxaban was superior to enoxaparin at this dosage, and the results for safety were similar.51 In a recent paper, Fuji et al found major or clinically relevant non-major bleeding in 6.7%, 3.5%, and 5.0% of patients with mild renal impairment at edoxaban 30 mg, severe renal impairment at edoxaban 15 mg, and the fondaparinux group at 1.5 mg once daily subcutaneous, respectively. At these doses, there were no major bleeding events and no thromboembolic events.52 According to Rognoni et al edoxaban is not inferior to warfarin for preventing stroke and systemic embolisms in patients with NVAF, with a lower rate of intracranial bleeding. Therefore, according these authors, edoxaban proved to be a cost-effective alternative to warfarin in these patients.53
Clinical indications for NOACs
NOACs have been approved for various thromboembolic indications, such as the prevention of stroke and systemic embolism in adult patients with NVAF with one or more risk factors.3,13 The important indications for these drugs are the treatment of DVT and PE, and the prevention of recurrent DVT and PE in adults.54 Rivaroxaban was the first NOAC that received European approval for the prevention of atherothrombotic events in patients with acute coronary syndrome; however, similar to apixaban and dabigatran, it is not often used in clinical practice. Apixaban, dabigatran, and rivaroxaban are approved in the EU for the prevention of VTE after elective hip or knee replacement surgery. These drugs were approved based on the results of Phase III trials in which each of the abovementioned direct inhibitors was compared with standard thromboprophylaxis with subcutaneous LMWHs (enoxaparin).55 In addition, edoxaban is approved in Japan for the prevention of VTE following lower-limb orthopedic surgery, whereas EU countries are seeking its approval for the prevention of stroke and systemic embolic events.
Contraindications
There are many contraindications for the use of NOACs, such as clinically significant active bleeding, conditions that may be associated with major bleeding, hepatic disease with coagulopathy (severe hepatic impairment in cirrhotic patients), and additional risk factors that can increase the risk of bleeding, such as other anticoagulants, platelet inhibitors, and non-steroidal anti-inflammatory drugs.4 Additionally, hypersensitivity to NOACs is contraindicated.56 For some conditions, NOACs should be described with caution in different dosages according to age, weight, and renal function based on summary of product characteristics for each compound. Dabigatran is contraindicated in severe renal impairment (CrCL <30 mL/min), whereas rivaroxaban and apixaban are not recommended in patients with CrCL <15 mL/min. Edoxaban is contraindicated in patients with CrCL >95 mL/min (increased ischemic stroke) but should administered 30 mg once a day in those with CrCL >15–50 mL/min. Patients who are greater than or less than 80 years and those with body weight greater than or less than 60 kg should receive a reduced dose of apixaban of 2.5 mg twice daily.34,57–59
There are also contraindications for VKAs, which may be relative and absolute. Some relative contraindications are uncontrolled hypertension, severe liver disease, recent surgery, and procedures involving the nervous system, spine, or eye. Absolute contraindications involve the presence of severe or active bleeding diathesis, non-adherence to medication and INR monitoring, pregnancy, allergy, or intolerance to VKAs.60,61 Based on these contraindications, some reports in the literature suggest that the risks do not outweigh the benefits of warfarin.62
Advantages of NOACs over VKAs
NOACs have various advantages in the prevention and treatment of patients with a predisposition toward AF, DVT, PE, stroke, and other conditions that are related to inherited or acquired thrombophilia.14,31 Below, we describe the main advantages of NOACs compared with VKAs in preventing various factors that are responsible for thromboembolic disorders and in the treatment of thromboembolic diseases, such as the absence of food interactions, few strong drug interactions,63 predictable PK and PD, a rapid onset and offset of action, a short half-time, and the absence of the need for laboratory monitoring.
Drug–drug interactions of NOACs
In general, there are few drug–drug interactions between NOACs and other drugs, which enable the concurrent use of other drugs in patients who are being treated with NOACs. However, it is important to mention some of the important mechanisms of drug–drug interactions. A significant interaction mechanism for NOACs (except rivaroxaban) consists of the re-secretion of a P-gp transporter after absorption in the gut. It is known that the P-gp transporter may be involved in renal clearance, including that of rivaroxaban.64 Most rivaroxaban (two-thirds) is metabolized by the CYP system, especially CYP3A4. Many drugs used in patients with AF are P-gp substrates, such as verapamil, dronedarone, and amiodarone. Therefore, the concomitant use of NOACs and inhibitors or inducers of CYP3A4 is not recommended due to increases or decreases in plasma concentrations.65 According to Wang et al strong CYP3A4 inhibition or induction may affect the rivaroxaban plasma concentration. Most apixaban is hepatically cleared as an unchanged molecule, with only a minor portion being metabolized by CYP3A4; therefore, CYP3A4 drug interactions are less important.66 However, based on the summary of product characteristics, apixaban should be used with caution if co-administered with strong inducers of both CYP3A4 and P-gp.4 Dabigatran has few clinically significant drug–drug interactions, but it (similar to rivaroxaban) is a P-gp substrate. Therefore, its concomitant use with ketoconazole, verapamil, and amiodarone, which may increase its anticoagulant effects, should be avoided, whereas concomitant use with rifampicin may decrease its effect.24 Erythromycin, ketoconazole, and amiodarone are CYP3A4 inhibitors, which can increase the serum concentration of rivaroxaban and, therefore, increase the risk for bleeding; clarithromycin is a strong CYP3A4 inhibitor and a moderate P-gp inhibitor.67 Another group of drugs, such as phenytoin and rifampicin, are known as CYP3A4 inducers and may increase the metabolism of rivaroxaban and, consequently, decrease the degree of anticoagulation. Compared with VKAs, the number of interactions of NOACs with other drugs is very small because VKAs react with a wide range of drugs, which manifests as significant changes in their PK and PD.
Food interactions of NOACs
Unlike VKAs, which are affected by the intake of various types of food, especially food products that contain vitamin K, the actions of NOACs are not associated with food. This is very important, as patients who receive these drugs do not need to avoid any food products because there is no difficulty in balancing anticoagulant therapy.25 Under some circumstances, patients exhibit disturbed vitamin K metabolism, such as inadequate intake of vitamin K from food, biliary obstruction, and digestive disorders, which can manifest as maldigestion and malabsorption in the gut, as well as disorders of the normal intestinal flora as a result of antibiotic intake or intestinal infection.
Predictable PK and PD
Most authors agree that NOACs are characterized by predictable PK, which is an important advantage over VKAs. Observations in Phase I and Phase II trials have revealed that rivaroxaban has predictable PK properties, with absolute bioavailability after oral dosing. The rivaroxaban dose is proportional to its PK with respect to its anticoagulant effect, which increases in a linear manner with increasing plasma concentration.28 Other NOACs may exhibit similar predictable profiles, but some PK properties differ in various ways, and this variation may be important in a given clinical situation. However, in most cases, the PK and PD profiles of rivaroxaban and dabigatran remain within acceptable limits. Some studies have indicated that relevant PK and PD parameters are consistent independent of body weight,68 age, and sex.69 The above data suggest the possibility of using a fixed dose of these drugs, regardless of demographic variations, with no requirement for anticoagulant monitoring.70
Rapid onset and offset of NOAC action
The most important advantage of NOACs over VKAs is the rapid onset of action, as this characteristic enables rapid action (~1.5–3 hours) of the drug after oral administration; rapid offset is also important in some conditions if patients require surgical treatment. Additionally, rapid onset and offset actions eliminate the need for initial treatment with a parenteral anticoagulant in patients with acute thrombosis. These properties of NOACs reduce the need for “bridging” patients at high risk of thrombosis with a parenteral anticoagulant.63
Lack of need for laboratory monitoring
As another important feature of NOACs, along with their minimal drug–drug and food interactions and predictable relevant PK and PD parameters, routine monitoring is not required, regardless of body weight,68 age, sex,69 race, and demographic variations. Additional advantages of NOACs over VKAs include the wide therapeutic windows, greater efficacy in AF, and lower risk of intracranial hemorrhage,63 except for dabigatran, which at doses of 150 mg has an intracranial hemorrhage rate equal to that of warfarin.12
Disadvantages of NOACs over VKAs
Despite the aforementioned advantages of NOACs over VKAs, these drugs are not ideal because their use is limited or contraindicated under some circumstances. For example, none of the direct NOACs are approved to use drugs during pregnancy or in babies and children.3 Additionally, NOACs have not yet been applied in patients with mechanical mitral valve issues (with increased rates of thromboembolic and bleeding complications),71 patients with malignant disease, and those with antiphospholipid syndrome, which is associated with a greater risk of thrombophilic states.25
Chronic kidney disease
Although a main advantage of NOACs is the lack of monitoring requirement, NOACs are not appropriate in some patients, such as who have liver or kidney disease.72 Approximately 80% of dabigatran, but less rivaroxaban and apixaban (33% and 25%, respectively), is eliminated through the kidneys as an active drug. These values suggest that renal function must be assessed before applying any of the NOAC drugs. Indeed, the Cockroft–Gault formula should be used to calculate creatinine clearance by considering body weight.63 Therefore, the application of NOACs in chronic kidney disease should be performed with caution, especially in elderly patients, as this group generally has moderate (creatinine clearance 30–50 mL/min) or severe (10–30 mL/min) renal insufficiency, with the area under the concentration–time curve (AUC) increasing 2.7- and 6-fold and the plasma elimination half-life increasing at least twofold. Furthermore, dabigatran is not recommended in patients with severe renal insufficiency73 because 80% of the drug is eliminated by the kidney, whereas apixaban and rivaroxaban should be used with caution, and dosage adjustment is necessary.74
Hepatic disease
Apixaban and rivaroxaban are contraindicated in hepatic disease associated with coagulopathy and clinically relevant risk. However, NOACs can be used in patients with moderate liver insufficiency, though dosage adjustment is necessary. In cases of severe hepatic impairment (eg, Child-Pugh Class C) and cirrhotic patients with Child-Pugh Class B or C, rivaroxaban should not be administered,74 whereas in cases of mild or moderate hepatic impairment, patients may be administered apixaban with caution, and dose adjustments are required.
Absence of a specific test
In general, NOAC therapy monitoring is not necessary. However, in some situations, such as the need for urgent surgical intervention, intravenous thrombolysis in acute ischemic stroke patients, intracerebral bleeding, and overdose, anti-coagulation assessment is necessary. Finally, a new thrombolysis decision-making protocol for the standardized use of NOACs in acute ischemic stroke patients potentially eligible for intravenous thrombolysis has been recently developed and is under further investigation in a larger study.75
Sensitive tests, such as the thrombin clotting time and ecarin clotting time tests, can be used to quantify the anticoagulant effects of dabigatran. The activated partial thromboplastin time is less sensitive than the thrombin clotting time and ecarin clotting time tests.76,77 Recently, other options, such as the heparin chromogenic assay, have been suggested for the indirect measurement of apixaban levels. Additionally, the plasma concentration of rivaroxaban can be assessed using a chromogenic FXa assay, whereas the plasma concentration of dabigatran can be quantified using the HEMOCLOT dilute time assay.78,79
Additional disadvantages
Additional disadvantages of NOACs compared with VKAs are related to cost and the importance of compliance; some patients cannot afford NOACs, and poor compliance with short-acting oral anticoagulants (NOACs) increases the risk for thromboembolic events. In these cases, VKAs remain the drugs of choice.3 The short half-lives of NOACs can be considered both an advantage and a disadvantage under various circumstances. For example, the advantage of the short half-life of an NOAC may be relevant for emergency surgery and in cases of bleeding due to accumulation of the drug in the blood, whereas the short half-life is a disadvantage if the patient forgets to take the drug, which could put the patient at risk. The lack of a specific antidote is a problem in the case of spontaneous bleeding from overdose or in the case of traumatic injury requiring urgent surgical intervention.55 The pharmaceutical company Boehringer Ingelheim is conducting clinical studies of a dabigatran antidote.80 In such cases, patients should be given blood plasma products, such as fresh frozen plasma, concentration of prothrombin complex, or recombinant factor Xa.81 However, all of these products pose a problem, either because they can cause thrombotic complications or they are expensive. Indeed, there are financial issues due to the high price of NOACs, which is a significant concern for many patients.40
Conclusion
Because the application of oral anticoagulant therapy with VKAs presents some difficulties related to major drug and food interactions as well as other problems, such as great individual variability in the effect and the need for continuous monitoring, additional anticoagulant drugs need to be developed. Novel anticoagulant drugs called new oral anticoagulants or direct oral anticoagulants have been introduced in the past 7 years. The advantages of NOACs over VKAs are their high efficacy in preventing stroke in AF and NVAF, lower incidence of major bleeding, convenience of use, minor drug and food interactions, predictable PK and PD, rapid onset and offset of action, short half-life, and lack of the need for laboratory monitoring. However, some disadvantages of NOACs should be mentioned, such as their higher cost, the absence of specific antidotes, and limited experience with these drugs. In addition, NOACs should not be used in patients with severe renal and hepatic disease (absence of validated monitoring test), patients with mechanical heart valves, individuals younger than 18 years of age, and elderly patients. Future studies will further clarify the role of NOACs in preventing and treating thromboembolic disease.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol. 1992;19(2):246–247. doi: 10.1016/0735-1097(92)90473-z. [DOI] [PubMed] [Google Scholar]
- 2.Gómez-Outes A, Suárez-Gea ML, Calvo-Rojas G, et al. Discovery of anticoagulant drugs: a historical perspective. Curr Drug Discov Technol. 2012;9(2):83–104. doi: 10.2174/1570163811209020083. [DOI] [PubMed] [Google Scholar]
- 3.Holy EW, Beer JH. Update on the status of new oral anticoagulants for stroke prevention in patients with atrial fibrillation. Cardiovasc Med. 2013;16:103–114. [Google Scholar]
- 4.Heidbuchel H, Verhamme P, Alings M, et al. European Heart Rhythm Association Practical Guide on the use of new oral anticoagulants in patients with non-valvular atrial fibrillation. Europace. 2013;15(5):625–651. doi: 10.1093/europace/eut083. [DOI] [PubMed] [Google Scholar]
- 5.Campbell HA, Roberts WL, Smith WK, Link KP. Studies of the hemorrhagic sweet clover disease. I. The preparation of hemorrhagic concentrates. J Biol Chem. 1940;136:47–55. [Google Scholar]
- 6.Ferlund P, Stenflo J, Roepstorff P, Thomsen J. Vitamin K and the biosynthesis of prothrombin. V. Gamma-carboxyglutamic acids, the vitamin K-dependent structures in prothrombin. J Biol Chem. 1975;250(15):6125–6133. [PubMed] [Google Scholar]
- 7.Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 1998;114:445S–469S. doi: 10.1378/chest.114.5_supplement.445s. [DOI] [PubMed] [Google Scholar]
- 8.Fasco MJ, Hildebrandt EF, Suttie JW. Evidence that warfarin anticoagulant action involves two distinct reductase activities. J Biol Chem. 1982;257(19):11210–11212. [PubMed] [Google Scholar]
- 9.Choonara IA, Malia RG, Haynes BP, et al. The relationship between inhibition of vitamin K1 2,3-epoxide reductase and reduction of clotting factor activity with warfarin. Br J Clin Pharmacol. 1988;25(1):1–7. doi: 10.1111/j.1365-2125.1988.tb03274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ickx BE, Steib A. Perioperative management of patients receiving vitamin K antagonists. Can J Anaesth. 2006;53(6 Suppl):S113–S122. doi: 10.1007/BF03022258. [DOI] [PubMed] [Google Scholar]
- 11.Klauser W, Dütsch M. Partial management of new oral anticoagulants after total hip or total knee arthroplasty. Musculoskelet Surg. 2013;97(3):189–197. doi: 10.1007/s12306-013-0306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 13.da Silva RM. Novel oral anticoagulants in non-valvular atrial fibrillation. Cardiovasc Hematol Agents Med Chem. 2014;12(1):3–8. doi: 10.2174/187152571201141201091848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gayle JA, Kaye AD, Kaye AM, Shah R. Anticoagulants: newer ones, mechanisms, and perioperative updates. Anesthesiol Clin. 2010;28(4):667–679. doi: 10.1016/j.anclin.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos. 2008;36(2):386–399. doi: 10.1124/dmd.107.019083. [DOI] [PubMed] [Google Scholar]
- 16.Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007;64(3):292–303. doi: 10.1111/j.1365-2125.2007.02899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-Outes A, Terleira-Fernández AI, Calvo-Rojas G, Suárez-Gea ML, Vargas-Castrillón E. Dabigatran, rivaroxaban, or apixaban versus warfarin in patients with nonvalvular atrial fibrillation: a systematic review and meta-analysis of subgroups. Thrombosis. 2013;2013:640723. doi: 10.1155/2013/640723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohnloser SH, Oldgren J, Yang S, et al. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-term Anticoagulation Therapy) trial. Circulation. 2012;125(5):669–676. doi: 10.1161/CIRCULATIONAHA.111.055970. [DOI] [PubMed] [Google Scholar]
- 19.Artang R, Rome E, Nielsen JD, Vidaillet HJ. Meta-analysis of randomized controlled trials on risk of myocardial infarction from the use of oral direct thrombin inhibitors. Am J Cardiol. 2013;112(12):1973–1979. doi: 10.1016/j.amjcard.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Graham DJ, Reichman ME, Wernecke M, et al. Cardiovascular, bleeding, and mortality risk in elderly Medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation. 2015;131(2):157–164. doi: 10.1161/CIRCULATIONAHA.114.012061. [DOI] [PubMed] [Google Scholar]
- 21.Schulman S, Kearon C, Kakkar AK, et al. RE-COVER Study Group Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361(24):2342–2352. doi: 10.1056/NEJMoa0906598. [DOI] [PubMed] [Google Scholar]
- 22.Schulman S, Kakkar AK, Goldhaber SZ, et al. RE-COVER II Trial Investigators. Treatment of acute venous thromboembolism with dabigatran or warfarin and pooled analysis. Circulation. 2014;129(7):764–772. doi: 10.1161/CIRCULATIONAHA.113.004450. [DOI] [PubMed] [Google Scholar]
- 23.Patel MR, Mahaffey KW, Garg J, et al. ROCKET AF Investigators Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–891. doi: 10.1056/NEJMoa1009638. [DOI] [PubMed] [Google Scholar]
- 24.Little JW. New oral anticoagulants: will they replace warfarin? Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(5):570–580. doi: 10.1016/j.oooo.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman R, Brenner B. The promise of novel direct oral anticoagulants. Best Pract Res Clin Haematol. 2012;25(3):351–360. doi: 10.1016/j.beha.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Gulseth MP, Michaud J, Nutescu EA. Rivaroxaban: an oral direct inhibitor of factor Xa. Am J Health Syst Pharm. 2008;65(16):1520–1529. doi: 10.2146/ajhp070624. [DOI] [PubMed] [Google Scholar]
- 27.Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-3939 – an oral, direct Factor Xa inhibitor – after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61(12):873–880. doi: 10.1007/s00228-005-0043-5. [DOI] [PubMed] [Google Scholar]
- 28.Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY-59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78(4):412–421. doi: 10.1016/j.clpt.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Turpie AG. Oral, direct factor Xa inhibitors in development for the prevention and treatment of thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2007;27(6):1238–1247. doi: 10.1161/ATVBAHA.107.139402. [DOI] [PubMed] [Google Scholar]
- 30.Weinz C, Radke M, Sshmreer R. In vitro metabolisem of BAY 5979-39 and oral, direct factor Xa inhibitor. Drug Metab Rev. 2004;36(Supp 1):98. [Google Scholar]
- 31.Eriksson BI, Borris LC, Dahl OE, et al. ODIXa-HIP Study Investigators A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement. Circulation. 2006;114(22):2374–2381. doi: 10.1161/CIRCULATIONAHA.106.642074. [DOI] [PubMed] [Google Scholar]
- 32.Turpie AG, Fisher WD, Bauer KA, et al. OdiXa-Knee Study Group BAY 59-7939: an oral, direct factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost. 2005;3(11):2479–2486. doi: 10.1111/j.1538-7836.2005.01602.x. [DOI] [PubMed] [Google Scholar]
- 33.Frost C, Song Y, Barrett YC, et al. A randomized direct comprasion of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin Pharmacol. 2014;6:179–187. doi: 10.2147/CPAA.S61131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayer Pharma AG. Xarelto (Rivaroxaban) Xarelto®: Summary of Product Characteristics–EU. 2013. [Accessed May 1, 2014]. Available from: http://www.xarelto.com/en/information-on-xarelto/summary-of-product-characteristics/
- 35.Mega JL, Braunwald E, Wiviott SD. ATLAS ACS 2-TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9–19. doi: 10.1056/NEJMoa1112277. [DOI] [PubMed] [Google Scholar]
- 36.Halabi A, Kubitza D, Zuehlsdorf M. Effect of hepatic impairment on the pharmacokinetics and tolerability of rivaroxaban, and oral, direct factor Xa inhibitor. J Thromb Haemost. 2007;5(Suppl 2):P-M-635. [Google Scholar]
- 37.EINSTEIN Investigators Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499–2510. doi: 10.1056/NEJMoa1007903. [DOI] [PubMed] [Google Scholar]
- 38.Hirschl M, Kundi M. New oral anticoagulants in the treatment of acute venous thromboembolism – a systematic review with indirect comparisons. Vasa. 2014;43(5):535–564. doi: 10.1024/0301-1526/a000373. [DOI] [PubMed] [Google Scholar]
- 39.EINSTEIN-PE Investigators Oral rivaroxaba for the treatment symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287–1897. doi: 10.1056/NEJMoa1113572. [DOI] [PubMed] [Google Scholar]
- 40.Gallego P, Roldán V, Lip GY. Novel oral anticoagulants in cardiovascular disease. J Cardiovasc Pharmacol Ther. 2014;19(1):34–44. doi: 10.1177/1074248413501392. [DOI] [PubMed] [Google Scholar]
- 41.Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992. doi: 10.1056/NEJMoa1107039. [DOI] [PubMed] [Google Scholar]
- 42.Shantsila E, Lip GY. Apixaban, an oral, direct inhibitor of activated factor Xa. Curr Opin Investig Drugs. 2008;9(9):1020–1033. [PubMed] [Google Scholar]
- 43.Furugohri T, Isobe K, Honda Y, et al. DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost. 2008;6(9):1542–1549. doi: 10.1111/j.1538-7836.2008.03064.x. [DOI] [PubMed] [Google Scholar]
- 44.Daiichi Sankyo Europe GmbH . First market approval in Japan for LIXIANA (Edoxaban) [press release] Tokyo: Daiichi Sankyo Europe GmbH; 2011. Apr 22, [Accessed December 1, 2013]. Available at: http://www.daiichisankyo.com/media_investors/media_relations/press_releases/detail/005784.html. [Google Scholar]
- 45.O’Riordan M. FDA Approves Edoxaban for Stroke Prevention in AF and DVT/PE Prevention. Medscape. Jan 9, 2015. [Accessed January 10, 2015]. Available from: http://www.medscape.com/viewarticle/837837.
- 46.Yin OQ, Tetsuya K, Miller R. Edoxaban population pharmacokinetics and exposure-response analysis in patients with non-valvular trial fibrillation. Eur J Clin Pharmacol. 2014;70(11):1339–1351. doi: 10.1007/s00228-014-1736-4. [DOI] [PubMed] [Google Scholar]
- 47.Eikelboom JW, Weitz JI. New anticoagulants. Circulation. 2010;121(13):1523–1532. doi: 10.1161/CIRCULATIONAHA.109.853119. [DOI] [PubMed] [Google Scholar]
- 48.Fuji T, Fujita S, Tachibana S, Kawai Y. Randomized, double-blind, multi-dose efficacy, safety and biomarker study of the oral factor Xa inhibitor DU-176b compared with placebo for prevention of venous thromboembolism in patients after total knee arthoplasty. Blood. 112:34. abstract. [Google Scholar]
- 49.Weitz JI, Connolly SJ, Patel I. Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation. Thromb Haemost. 2010;104:633–641. doi: 10.1160/TH10-01-0066. [DOI] [PubMed] [Google Scholar]
- 50.Hokusai-VTE Investigators. Büller HR, Décousus H, Grosso MA, et al. Edoxapan versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406–1415. doi: 10.1056/NEJMoa1306638. [DOI] [PubMed] [Google Scholar]
- 51.Fuji T, Wang CJ, Fujita S, et al. Edoxaban versus enoxaparin for thrombophylaxis after total knee arthoplasty: the STARS E-3 trial. Pathophysiol Haemost Thromb. 2010;37:A20. (OC297) [Google Scholar]
- 52.Fuji T, Fujita S, Kawai Y, et al. A randomized, open-label trial of edoxaban in Japanese patients with severe renal impairment undergoing lower-limb orthopedic surgery. Thromb J. 2015;13(1):6. doi: 10.1186/s12959-014-0034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rognoni C, Marchetti M, Quaglini S, Liberato NL. Edoxaban versus warfarin for stroke prevention in non-valvular trial fibrillation: a cost-effectiveness analysis. J Thromb Thrombolysis. 2015;39(2):149–154. doi: 10.1007/s11239-014-1104-3. [DOI] [PubMed] [Google Scholar]
- 54.Cohen AT, Spiro TE, Büller HR, et al. Rivoroxaban for thrombophylaxis in acutely ill medical patients. N Engl J Med. 2013;368(6):513–523. doi: 10.1056/NEJMoa1111096. [DOI] [PubMed] [Google Scholar]
- 55.Cowell RP. Direct oral anticoagulants: integration into clinical practice. Postgrad Med J. 2014;90(1067):529–539. doi: 10.1136/postgradmedj-2013-132474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yates J, Choudhry M, Keys G. A case report describing a suspected riraroxaban hypersensibility reaction in a surgical patient. J Clin Pharm Ther. 2013;38(2):159–161. doi: 10.1111/jcpt.12013. [DOI] [PubMed] [Google Scholar]
- 57.European Medicines Agency Pradaxa® – Summary of Product Characteristics. 2014. [Accessed March 13, 2012]. Available from: www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000829/WC500041059.pdf.
- 58.European Medicines Agency Eliquis® – Summary of Product Characteristics. 2014. [Accessed June 2, 2013]. Available from: Available from http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf.
- 59.SAVAYSA™ (edoxaban) tablets [prescribing information] Parsippany, NJ: Daiichi Sankyo, Inc.; 2015. [Accessed February 9, 2015]. Available from: http://dsi.com/prescribing-information-portlet/getPIContent?productName=Savaysa&inline=true. [Google Scholar]
- 60.Tadros R, Shakib S. Warfarin-indications, risks and drug interactions. Aust Fam Physician. 2010;39(7):476–479. [PubMed] [Google Scholar]
- 61.Abadi S, Einarson A, Koren G. Use of warfarin during pregnancy. Can Fam Physician. 2002;484(4):695–697. [PMC free article] [PubMed] [Google Scholar]
- 62.Garwood CL, Corbett TL. Use of anticoagulation in elderly patients with atrial fibrillation who are at risk for falls. Ann Pharmacother. 2008;42(4):523–532. doi: 10.1345/aph.1K498. [DOI] [PubMed] [Google Scholar]
- 63.Bauer KA. Pros and cons of new oral anticoagulants. Hematology Am Soc Hematol Educ Program. 2013;2013:464–470. doi: 10.1182/asheducation-2013.1.464. [DOI] [PubMed] [Google Scholar]
- 64.Gnoth MJ, Buetehorn U, Muenster U, Schwarz T, Sandmann S. In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther. 2011;338(1):372–380. doi: 10.1124/jpet.111.180240. [DOI] [PubMed] [Google Scholar]
- 65.Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood. 2008;111(10):4871–4879. doi: 10.1182/blood-2007-10-120543. [DOI] [PubMed] [Google Scholar]
- 66.Wang L, Zhang D, Raghavan N, et al. In vitro assessment of metabolic drug–drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–458. doi: 10.1124/dmd.109.029694. [DOI] [PubMed] [Google Scholar]
- 67.O’Connell JE, Stassen LF. New oral anticoagulants and their implications for dental patients. J Ir Dent Assoc. 2014;60(3):137–143. [PubMed] [Google Scholar]
- 68.Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of riraroxaban (BAY 59-7939) in healthy subjects. J Clin Pharmacol. 2007;47(2):218–226. doi: 10.1177/0091270006296058. [DOI] [PubMed] [Google Scholar]
- 69.Kubitza D, Becka M, Roth A, Mueck W. The influence of age and gender on the pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct Factor Xa inhibitor. J Clin Pharmacol. 2013;53(3):249–255. doi: 10.1002/jcph.5. [DOI] [PubMed] [Google Scholar]
- 70.Mueck W, Schwers S, Stampfuss J. Rivaroxaban and other novel oral anticoagulants: pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb J. 2013;11(1):10. doi: 10.1186/1477-9560-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eikelboom JW, Connolly SJ, Brueckmann M, RE-ALGN Investigators Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med. 2013;369(13):1206–1214. doi: 10.1056/NEJMoa1300615. [DOI] [PubMed] [Google Scholar]
- 72.Douxfils J, Tamigniau A, Chatelain B, Goffinet C, Dogné JM, Mullier F. Measurement of non-VKA oral anticoagulants versus classic ones: the appropriate use of hemostasis assays. Thromb J. 2014;12:24. doi: 10.1186/1477-9560-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brighton T. New oral anticoagulant drugs – mechanisms of action. Aust Prescr. 2010;33:38–41. [Google Scholar]
- 74.Wang Y, Bajorek B. New oral anticoagulants in practice: pharmacological practical considerations. Am J Cardiovasc Drugs. 2014;14(3):175–189. doi: 10.1007/s40256-013-0061-0. [DOI] [PubMed] [Google Scholar]
- 75.Kepplinger J, Barlinn K, Gehrisch S, et al. Are the recommendations for the emergency management of acute ischemic stroke patients on novel oral anticoagulants sufficient? Stroke. 2014;45:ATMP57. [Google Scholar]
- 76.van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate – a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2012;103(6):1116–1127. doi: 10.1160/TH09-11-0758. [DOI] [PubMed] [Google Scholar]
- 77.Blann AD. Non-vitamin K antagonist oral anticoagulants (NOACs): a view from the laboratory. Br J Biomed Sci. 2014;71(4):158–167. doi: 10.1080/09674845.2014.11669981. [DOI] [PubMed] [Google Scholar]
- 78.Samama MM, Contant K, Spiro TE, et al. Evaluation of the anti-factor Xa chromogenic assay for the measurement of rivaroxaban plasma concentrations using calibrators and controls. Thromb Haemost. 2012;107:379–387. doi: 10.1160/TH11-06-0391. [DOI] [PubMed] [Google Scholar]
- 79.Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis. 2012;23:138–143. doi: 10.1097/MBC.0b013e32834f1b0c. [DOI] [PubMed] [Google Scholar]
- 80.Boehringer Ingelheim’s Investigational Antidote for Pradaxa® (dabigatran etexilate mesylate) receives FDA breakthrough therapy designation [press release] Ridgefield, CT: Boehringer Ingelheim; 2014. Jun 26, [Accessed July 26, 2014]. Available from: http://us.boehringer-ingelheim.com/news_events/press_releases/press_release_archive/2014/06-26-14-boehringer-ingelheim-investigational-antidote-pradaxa-dabigatran-etexilate-me-sylate-fda-breakthrough-therapy-designation.html. [Google Scholar]
- 81.Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. 2013;19(4):446–451. doi: 10.1038/nm.3102. [DOI] [PubMed] [Google Scholar]