

Abstract
Inhibition of JAK1 or JAK2 in human tumor cells was previously shown to increase susceptibility of these cells to NK cell lysis. In the present study, we examined the cellular mechanisms that mediate this effect in hematopoietic tumor cell lines and primary tumor cells. Incubation of tumor cells with supernatant from activated NK cells or interferon-gamma (IFNγ)-induced activation of pSTAT1 and increased expression of PD-L1 without altering expression of other activating or inhibitory NK cell ligands. These functional effects were blocked by chemical JAK inhibition or shRNAs targeting JAK1, JAK2 or STAT1. Inhibition of IFNγ signaling also prevented the upregulation of PD-L1 and blocking PD-L1 resulted in increased tumor lysis by NK cells. These results show that NK cell activation and secretion of IFNγ results in activation of JAK1, JAK2 and STAT1 in tumor cells, resulting in rapid up-regulation of PD-L1 expression. Increased expression of PD-L1 results in increased resistance to NK cell lysis. Blockade of JAK pathway activation prevents increased PD-L1 expression resulting in increased susceptibility of tumor cells to NK cell activity. These observations suggest that JAK pathway inhibitors as well as PD-1 and PD-L1 antibodies may work synergistically with other immune therapies by preventing IFN-induced inhibition of NK cell-mediated tumor cell lysis.
Keywords: IFNγ, JAK1/JAK2, NK cells, PD-1/PD-L1
Abbreviations: ADCC, Antibody dependent cellular cytotoxicity; AKT, Ak strain transforming; APC, Allophycocyanin; CTRL, Control; DMSO, Dimethyl sulfoxide; ERK, extracellular-signal-regulated kinases; MACS, Magnetic cell separation; MAPK, Mitogen-activated protein kinases; RAS, Rat sarcoma; STAT, signal transducer and activator of transcription
Introduction
NK cells play an important role in the elimination of cells that have undergone malignant transformation.1-3 NK cell functions are mediated in part through direct lysis of target cells.4,5 In addition, NK cells are an important source of immunoregulatory cytokines such as IFNγ, tumor necrosis factor-α (TNF-α) and granulocyte macrophage colony-stimulating factor (GM-CSF). These cytokines modulate responses of other immune cells and contribute to the overall effectiveness of the immune response. Despite the ability of NK cells to inhibit tumor cell growth, it is also evident that malignant cells can evade innate immune surveillance; however, the molecular basis for tumor cell resistance to NK cell-mediated lysis is not completely understood.6
To identify novel pathways that modulate tumor cell susceptibility to NK-mediated lysis, we previously screened a large lentiviral shRNA library to identify genes in tumor cells that promote NK resistance. Using this approach, we found that silencing JAK1 or JAK2 in various tumor targets resulted in significantly increased susceptibility to NK cell lysis. This effect was not noted when other members of the JAK family, JAK3 and TYK2, were silenced.7 JAK genes encode a family of non-receptor tyrosine kinases that are constitutively associated with a variety of cytokine receptors including type I and II interferons, GM-CSF, G-CSF and IL-6. After cytokine binding to these receptors, JAKs undergo tyrosine phosphorylation and initiate the phosphorylation of STAT proteins, which translocate to the nucleus and initiate gene transcription.8 JAK phosphorylation has also been shown to activate other important pathways such as PI3K, RAS, AKT and MAPK. JAK proteins thus play a pivotal role in many cellular functions such as cell growth, differentiation and survival, and activating mutations of these kinases have been associated with malignant transformation.8-10
Since JAK gene silencing had not previously been associated with tumor cell susceptibility to immune attack, we undertook a series of experiments to understand the mechanisms whereby JAK1 and JAK2 modulate tumor susceptibility to NK cells. Because JAK1 and JAK2 signal through the IFNγ receptor, we focused on the potential role of IFNγ when NK cells interact with tumor cell targets. These studies demonstrate that IFNγ triggers tumor cell resistance to NK cells and this resistance is mediated through increased expression of PD-L1 by tumor cells. PD-L1 expression inhibits NK cell activity, representing a novel mechanism whereby tumor cells can rapidly acquire resistance to both innate and adaptive immune responses.
Results
Effects of JAK1/JAK2 silencing or inhibition on basal activation of JAK signaling pathways in tumor cell lines and primary tumor cells
To understand the role of JAK1 and JAK2 in modulating susceptibility of tumor cells to NK cells, we first characterized the basal activation of JAK signaling pathways in tumor cell lines. JAK kinases are associated with cytokine receptors and ligand binding of these receptors rapidly induces JAK phosphorylation, which in turn activates STAT transcription factors.11 JAK kinases have also been reported to activate other kinases such as PI3K/AKT and ERK.12,13 Using antibodies specific for phosphorylated proteins, we examined the activation status of STAT1(pY701), STAT1(pS727), STAT3(pY705), STAT3(pS727), STAT4(pY693), STAT5(pY694), STAT6(pY641), AKT(pS473) and ERK1/2(pT202/pY204) in the following cell lines; KM12BM, IM-9, K562, U266, U937, RPMI8226 and MM1S. As shown in representative examples in Figure 1 and Supplemental Figure 1A, STAT1(pY701), STAT1(pS727), STAT3(pY705), STAT4(pY693) and STAT6(pY641) showed no evidence of basal activation when compared to IgG CTRL staining controls. In contrast, STAT3(pS727) was phosphorylated in all cell lines while phosphorylation of STAT5(pY694), AKT(pS473) and ERK1/2(pT202/pY204) was detected at different levels depending on the tumor cell line analyzed. We then tested primary samples from patients with multiple myeloma (MM), acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL). Primary cells exhibited similar results with constitutive phosphorylation of STAT3(pS727), variable levels of phosphorylation of STAT5(pY694), AKT(pS473) and ERK1/2(pT202/pY204) and little evidence of activation of other STAT proteins (Fig. 1).
Figure 1.
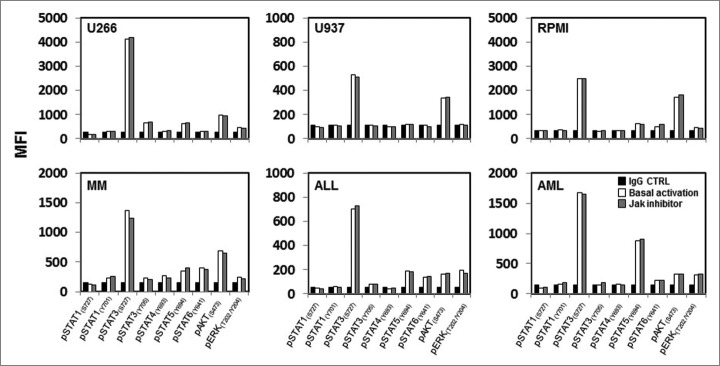
Baseline phosphorylation of STAT proteins, AKT and ERK in hematopoietic tumor cell lines and primary tumor cells. Representative examples of hematopoietic tumor cell lines or primary tumor cells analyzed for expression of several pSTAT proteins, pAKT and pERK at their basal level and after pre-treatment with a JAK inhibitor. Bar graphs indicate mean fluorescence intensity (MFI) expression determined by flow cytometry.
Our previous studies showed that silencing JAK1 or JAK2 resulted in increased tumor susceptibility to NK-mediated lysis.7 To determine whether JAK inhibition affected the constitutive phosphorylation of STAT3(pS727), STAT5(pY694), ERK1/2(pT202/pY204) and AKT(pS473), we examined various tumor cell lines and primary tumor cells after in vitro treatment with 40 nM JAK inhibitor 1. This concentration can also inhibit other members of the JAK family but was chosen based on our previous experience of tumor pre-treatment with different concentration and subsequent co-culture with NK cells. Results summarized in Figure 1 show that the phosphorylation of these signaling molecules was not altered by JAK inhibitor treatment in any cell line or primary tumor cells tested. Similar results were obtained when different tumor cell lines were transduced with specific shRNAs to silence JAK1 or JAK2 gene expression (Fig. S1A). A representative example (Fig. S1B) shows that the constitutive activation of STAT3(pS727), STAT5(pY694), ERK1/2(pT202/pY204) and AKT(pS473) in KM12BM cells was not affected by JAK1 or JAK2 silencing.
Impact of NK cell: tumor cell interaction on JAK1 and JAK2 signaling
JAK1 and JAK2 are associated with the IFNγ receptor14 and NK cells are known to secrete IFNγ and other cytokines after activation by target cells.15,16 We therefore hypothesized that JAK1 and JAK2 were specifically activated by cytokines released when NK effector cells became engaged with tumor cell targets. To test this hypothesis, tumor cells were stimulated with supernatant from NK cells and examined for evidence of STAT, AKT and ERK activation. As shown in Figure 2, incubation with NK supernatant induced the selective phosphorylation of STAT1(pY701) in all of the tumor cell lines and primary cells tested. Activation of STAT1(pY701) did not occur when cells were treated with a JAK inhibitor (Fig. 2) or when either JAK1 or JAK2 were silenced with specific shRNAs (Fig. S2). NK supernatant did not affect phosphorylation of any of the other STATs, AKT or ERK in these cells. In data not shown, similar results were obtained when the same experiments were repeated using recombinant IFNγ instead of NK supernatant.
Figure 2.

STAT1(pY701)activation after treatment with activated NK supernatant. Different hematopoietic tumor lines and primary tumor cells were treated with activated NK supernatant from primary NK cells stimulated with tumor cells and subsequently analyzed for expression of pSTATs, pAKT and pERK by flow cytometry. Levels of phosphorylation were measured before and after treatment with NK supernatant in cells pre-treated with 40 nm JAK inhibitor1. Bar graphs indicate mean fluorescence intensity (MFI) expression determined by flow cytometry.
STAT1-KO tumor cells are more susceptible to NK cell activity
To further examine the involvement of STAT1 in modulating susceptibility to NK cell activity, we transduced three different tumor cell lines with two independent STAT1-shRNAs (STAT1-1 and STAT1-2) and two irrelevant controls (shCTRL-1 and shCTRL-2). The target sequences of the specific shRNAs and controls are detailed in Supplemental Table 1. Each cell line was examined for total STAT1 protein expression by flow cytometry (Fig. 3A) and western blot (Fig. 3B). STAT1-KO target cells were then tested for susceptibility to human purified CD3−CD56+ NK cells pre-stimulated with 100 Units IL-2. As functional markers of NK cell activity, we measured secretion of IFNγ (Fig. 3C), the level of CD107a/b degranulation17 (Fig. 3D) and NK-cell-induced apoptosis using AnnexinV/7AAD (Fig. 3E). Reduced expression of STAT1 in all three cell lines with both shRNAs significantly increased IFNγ secretion by purified NK cells from different healthy donors when tested against KM12BM-STAT1-KO (p = 0.034 and 0.014). When tested against STAT1-1-shRNA and STAT1-2-shRNA in K562 and IM9, IFNγ secretion from primary NK cells increased by 28.6%, 25.7% and 58.8%, 73.1%, respectively, but this increase was not statistically significant (Fig. 3C). Using CD107a/b degranulation as a measure of NK cell activation, STAT1 knockdown in each tumor cell line was found to significantly increase degranulation in purified NK cells from different donors (Fig. 3D). As shown in Figure 3E, STAT1-KO target cells were also more susceptible to NK-induced apoptosis when incubated with primary purified NK cells that had been stimulated with IL-2.
Figure 3.

Effects of STAT1 knockdown in hematopoietic tumor cell lines. K562, KM12BM and IM-9 were stably transduced with two independent shRNAs targeting STAT1 and two irrelevant shRNAs as control. (A) The level of total STAT1 expression was evaluated by flow cytometry and (B) western blot analysis. Controls and STAT1-KO cell lines were incubated with primary purified NK effector cells pre-stimulated with 100 Units/mL IL-2 at 1:1 E/T ratio as described in Methods. (C) Levels of IFNγ secretion in culture supernatants, (D) percent NK cells expressing CD107a/b and (E) Killing of STAT1-KO and control cell lines mediated by fresh purified NK. Bars represent the specific lysis of target cells (mean ± SEM) of four different experiments (∗p < 0.05, ∗∗p < 0.01 compared to the isotype control with the highest reactivity).
Blocking IFNγ increases tumor susceptibility to NK cells
To confirm that IFNγ secreted by NK cells induced resistance in tumor cell targets, we tested the ability of anti-IFNγ antibody (D9D10)18 to block STAT1 phosphorylation in tumor cells incubated with activated NK supernatant. Different concentrations of D9D10 antibody ranging from 0.5 μg/mL to 5 μg/mL were tested to determine the efficacy and optimal concentration of D9D10 for blocking STAT1 phosphorylation. As shown in Supplemental Figure 3A, 2 μg/mL and 5 μg/mL D9D10 completely blocked STAT1 phosphorylation when compared with an anti-IgG1-isotype control. We then added D9D10 or IgG1-isotype control at these concentrations to co-cultures of tumor cells with purified NK cells and measured levels of IFNγ secretion. As shown in Supplemental Figure 3B, addition of 2 μg/mL or 5 μg/mL D9D10 blocking antibody completely neutralized IFNγ secreted by either NK-92 or primary NK cells. Similar conditions were then used to evaluate the effect of IFNγ-neutralizing antibody on specific tumor cell lysis. In independent experiments using U937, IM-9, KM12BM, MM1S and RPMI 8226 target cells, addition of 2 μg/mL or 5 μg/mL IFNγ-blocking antibody increased specific target lysis by 51.8%, 78.5%, 25.1%, 20.6%, 28.5% and 45.6%, 87.1%, 32.4%, 15.3%, 33.3% respectively compared to similar concentrations of an IgG1-isotype control. These differences were all statistically significant (Fig. 4A). Similar experiments were also performed using primary tumor cells. As shown in Figure 4B, primary MM, AML and ALL cells were significantly more susceptible to NK lysis when IFNγ, secreted by NK cells, was blocked by 2 μg/mL (p = 0.0030, p = 0.041 and p = 0.0036 respectively) or 5 μg/mL of D9D10 (p = 0.0042, p = 0.038 and p = 0.0020 respectively) compared with the same concentrations of an IgG1-isotype control.
Figure 4.

Effects of IFNγ-blocking antibody on tumor cell susceptibility to NK lysis. (A) Five hematopoietic tumor cell lines and (B) primary tumor cells were incubated with primary purified NK cells at 1:1 E/T ratio in the presence of an anti-IFNγ-blocking antibody (D9D10) or an isotype control. Bars represent the specific lysis of target cells (mean ± SEM) of four different experiments (∗p < 0.05, ∗∗p < 0.01 compared to the isotype control with the highest reactivity).
Expression of inhibitory and activating ligands on tumor cells incubated with NK supernatant or IFNγ
Tumor susceptibility to NK cell recognition is profoundly influenced by MHC class I expression as well as other inhibitory/activating ligands.19,20 IFNγ is known to modulate expression of immunomodulary molecules on tumor cells21-23 and we therefore examined whether stimulation of tumor cells with IFNγ-affected expression of ligands known to be involved in NK cell recognition. As shown in Supplementary Figure 4, we measured the effect of activated NK supernatant or IFNγ on the expression of MHC Class I, β2M, HLA-C, HLA-A2, NKG2D, NKP44, NKP46, NKP30 ligands, MICA/B, DNAM-1 ligands (CD112, CD155), 2B4 ligand (CD48), TRAIL ligands (TRAIL-R1, TRAIL-R2) and Fas ligand (CD95) in six tumor cell lines and primary MM, AML and ALL cells. The basal expression of these ligands varied among the various tumor cell lines and primary tumors but there was no significant change in expression after in vitro stimulation with activated NK supernatant or IFNγ. We also examined expression of PD-1 ligands (PD-L1, PD-L2) as well as B7H3 and B7H4. In contrast to other activating/inhibitory ligands, PD-L1 was significantly increased in every cell line and primary sample after incubation with NK supernatant or IFNγ (Fig. 5A and B). PD-L2 was increased in U937 and primary AML cells but there was no change in B7H3 or B7H4 expression after stimulation with activated NK supernatant or IFNγ. Increased expression of PD-L1 and PD-L2 was completely abrogated if a JAK inhibitor was added together with IFNγ or activated NK supernatant. Similar results were obtained using three STAT1-KO cell lines (K562, KM12BM and IM9). As shown in Figures 5C and D, IFNγ or NK supernatant induced PD-L1 expression in each cell line, but PD-L1 upregulation was blocked when STAT1 was silenced.
Figure 5.

Tumor cell expression of PD-L1 and PD-L2 after incubation with activated NK supernatant or IFNγ. (A) Expression of PD-L1 in a representative example of AML cells treated with NK supernatant (left panel) or IFNγ (right panel) and with the addition of JAK inhibitor. (B) Expression of PD-L1 in hematopoietic tumor cell lines and primary tumor cells and expression of PD-L2 in U937 and primary AML cells. Surface antigen expression (MFI) is compared in cells incubated with medium alone, NK supernatant or IFNγ with or without JAK inhibitor. (C) Expression of PD-L1 in K562 or K562-STAT1-KO cells treated with NK supernatant (left panel) or IFNγ (right panel). (D) Expression of PD-L1 in 3 STAT1-KO or sh-CTRL cell lines. PD-L1 expression (MFI) is compared in cells incubated with medium alone, NK-supernatant or IFNγ.
Blocking PD-L1 increases susceptibility to NK cell killing
The expression of PD-1 on T cells has been well characterized and PD-L1 is known to inhibit T cell immunity.24-27 The expression of PD-1 on NK was examined in samples from six healthy donors. Confirming results of previous studies, some level of PD-1 expression was detected on resting CD56+ NK cells in all donors28,29 and was increased after IL-2 activation while IFNγ had no effect on NK cell PD-1 expression (Fig. S5). To test whether increased PD-L1 expression on tumor target cells inhibits NK cell killing, we added 10 μg/mL anti-PD-L1 antibody to co-cultures of NK cells with various cell targets and measured tumor cell cytotoxicity. The anti-PD-L1 antibody we used was engineered to express a human IgG4 Fc domain to eliminate FcR-mediated effects and any ability to mediate ADCC. As shown in Figure 6A, blocking PD-L1 on U937, MM1S, KM12BM, RPMI, IM9, K562 primary AML, MM and ALL resulted in significantly increased lysis by purified NK cells in four out of six tumor cell lines (p = 0.037, p = 0.084, p = 0.033, p = 0.0058, p = 0.027, p = 0.063 respectively) and in the primary AML, MM and ALL targets (p = 0.024, p = 0.0098 and p = 0.027 respectively). These results were also confirmed using an NK cell line (NK-92). As shown in Figure 6B, NK-92 cell lysis of all of the tumor cell targets was significantly increased when PD-L1 was blocked. Target cell lysis was also measured when these tumor cell targets were treated with a JAK inhibitor or JAK inhibitor plus PD-L1 blocking antibody. As shown in Figures 6A and B, the JAK inhibitor also increased the level of tumor cell killing, and the extent of increased killing was generally similar to the level obtained with anti-PD-L1. However, the combination of JAK inhibitor and anti-PD-L1 did not further increase the level of tumor cell killing compared to either agent alone. Similarly, pre-incubation of K562, IM9 and U937 with IFNγ induced some level of resistance to NK cell activation or killing that was reversed by blocking PD-L1 (Fig. S6).
Figure 6.

Effect of blocking PD-L1 on NK cell lysis of tumor cells. Tumor cell lines and primary tumor cells were incubated with (A) primary purified NK cells or (B) NK-92 at 1:1 E/T ratio with or without 10 μg/mL PDL1 antibody, JAK inhibitor or JAK inhibitor + PD-L1 antibody. The percentage of AnnexinV/7AAD positive cells was calculated for gated target cells (NKG2A or CD56 negative). Bars represent specific percent target cell killing (mean ± SEM) obtained in three separate experiments (∗p < 0.05, ∗∗p < 0.01 compared to target cells without PD-L1 antibody or JAK inhibitor treatment).
Discussion
Previous studies demonstrated that reduced expression or inhibition of JAK1 or JAK2 signaling in tumor cell lines and primary leukemia cells rendered these cells more susceptible to lysis by NK cells. Silencing JAK3 or TYK2, the other members of the JAK family, had no effect. The present studies focused on the JAK1 and JAK2 signaling pathway to understand how it modulated tumor cell susceptibility to NK cell lysis. Our initial studies examined downstream signaling proteins, quantifying basal levels of phosphorylation of several STAT proteins as well as ERK and AKT by flow cytometry. In the absence of NK cells, silencing JAK1 or JAK2 or inhibition of JAK phosphorylation did not affect the constitutive activation of downstream STAT proteins, ERK or AKT in any cells we examined. These results were consistent with our previous studies showing that reduced expression of JAK1 or JAK2 did not affect tumor cell expression of activating/inhibitory NK cells ligands.
JAKs are associated with different cytokine receptors and JAK1 and JAK2 are known to be associated with the IFNγ receptor. IFNγ is one of the major cytokines secreted by NK cells and we therefore hypothesized that JAK1 and JAK2 would play a role in tumor cell resistance to NK cell activity after NK effector cells directly engage with targets and initiate NK cytokine secretion. This was examined by incubating tumor cells with NK supernatant and these experiments demonstrated rapid phosphorylation of STAT1(pY701), without activation of other STATs, ERK or AKT. STAT1(pY701) activation was blocked when JAK1 or JAK2 expression was reduced or functionally inhibited suggesting that the predominant cytokine present in the NK supernatant was IFNγ. In fact, similar results were observed when recombinant IFNγ was used instead of NK supernatant. The functional role of JAK1/JAK2 activation of STAT1 was confirmed when expression of STAT1 was reduced by stable transduction with two different STAT1-shRNAs. Tumor cells with reduced expression of STAT1 were more susceptible to NK cell activity, supporting the hypothesis that IFNγ, secreted by NK cells, played a major role in increasing resistance of tumor cells to NK lysis. This was confirmed by neutralization of IFNγ secreted by the NK cells with a specific antibody, which abrogated JAK1, JAK2 and STAT1 activation in tumor cell and increased the susceptibility of these cells to NK killing.
IFNγ is a pleotropic cytokine that has been reported to promote tumor immunity and tumor rejection in many models. In these models, IFNγ has anti-proliferative and pro-apoptotic functions and inhibits tumor angiogenesis.30 Several studies have shown the importance of IFNγ in the efficacy of tumor vaccines and IL-12 therapies and JAK1 or JAK2 deletion has been hypothesized to be a mechanism whereby some solid tumors can evade host-protective tumor responses.31,32 In clinical trials, IFNγ therapy has been associated with melanoma regression in some patients.33,34 Nevertheless, clinical trials in patients with melanoma using IFNγ alone or in combination with IFNα failed to show improved survival compared with other therapies.35,36 Consistent with its limited clinical efficacy, other studies have found that IFNγ was associated with a more aggressive tumor phenotype as well as resistance to immune surveillance.21,37,38 In other models, IFNγ promoted protection of lymphocytic choriomeningitis virus infected mouse embryonic fibroblasts from killing by activated NK cells.39 Although the mechanisms responsible for these conflicting results are not clear, these studies suggest that similar to other cytokines such as TGF-β, TNF and IL-2, IFNγ can promote either tumor immunity or tumor resistance in different settings.40 Consistent with these findings, previous studies have shown that IFN stimulation can induce up-regulation of PD-L1 in monocytes and other cell types.41,42
One effect of IFNγ is to enhance expression of MHC Class I, thus facilitating T cell recognition.38 MHC Class I molecules also interact with inhibitory receptors on NK cells and increased expression of MHC by tumor cells is one potential mechanism whereby IFNγ can reduce NK lysis. This was carefully examined in our experiments but the expression of MHC Class I, β2M, HLA-C and HLA-A was not altered in any of our cell lines or primary cells by incubation with activated NK-supernatant or IFNγ. These findings were not expected, but previous studies have shown that various tumors do not increase MHC Class I expression after exposure to IFNγ. Dunn et al. found that this lack of response could be due to genetic disruption in JAK1/STAT1 signaling, but other studies have found no correlation between IFNγ exposure and MHC Class I expression, especially in solid tumors.32,43,44 NK cell recognition is regulated through a complex balance of inhibitory/activating ligands and receptors expressed on the surface of both NK cells and target cells.1,19 This prompted us to individually examine expression of a large number of inhibitory/activating ligands after incubation of targets with activated NK supernatant or IFNγ, but this treatment did not alter expression of any of these known NK ligands.
In contrast, expression of PD-L1 was increased in every cell line and primary cell incubated with NK-supernatant or IFNγ. PD-L2 expression was increased only in U937 cell line and primary AML cells but not in other tumors examined. Notably, increased PD-L1 expression was completely inhibited when tumor cells were pre-treated with a JAK inhibitor. The functional significance of PD-L1 expression was confirmed in experiments using a blocking PD-L1 antibody, which increased NK cell killing to an extent comparable to treatment with a JAK inhibitor. Treatment with a combination of JAK inhibitor and PD-L1-blocking antibody did not increase NK cell killing above levels observed with each agent alone suggesting that both agents mediated their effect through the same pathway.
PD-L1 and PD-L2 are ligands for PD-1, a CD28 family member that inhibits T cell activation and has been demonstrated to be an important mechanism that tumor cells use to escape T cell immunity.24,25,42,45 Our studies suggest that NK cell activation with subsequent secretion of IFNγ into the tumor microenvironment may contribute to the expression of these inhibitory ligands by tumor cells. Other molecules can also be involved in this pathway as recently shown by Chen et al., PD-L1 upregulation after IFNγ treatment could be modulated by PKD2 activation.46 Inhibition of the PD-1 pathway enhances T cell tumor immunity and clinical trials in several diseases using either anti-PD-L1 or anti-PD-1 antibodies have shown clinical efficacy.47,48 Although the role of PD-1 in the negative regulation of T cell immunity is well established, much less is known about PD-1/PD-L1 and NK reactivity. Benson et al. have previously demonstrated that anti-PD-1 enhances NK reactivity to myeloma cells, especially in combination with lenalidomide.28 Our studies suggest a much broader role for the PD-1/PD-L1 pathway as a negative regulator of NK cell antitumor activity as outlined in the model shown in Figure 7. In this model, NK cells are an important source of IFNγ in the tumor microenvironment. Tumor cells rapidly respond to IFNγ secreted by activated NK cells. IFNγ signaling in tumor cells increases expression of PD-L1, which suppresses NK cytolytic activity and further secretion of IFNγ (Fig. 7A). This negative regulatory feedback loop suppresses NK cytolytic activity and our studies identify JAK activation and PD-L1 expression as two points at which this pathway can be blocked, restoring NK-mediated tumor immunity (Fig. 7B). JAK inhibitors and anti-PD-L1 antibodies are currently available as clinical therapeutic reagents and the immunologic effects of these agents on NK may help explain some of the beneficial clinical activity of these agents. Future studies with PD-1 or PD-L1 antibodies may be designed to increase NK as well as T cell activity, further enhancing the level of tumor regression that has been observed in current clinical trials. Although JAK pathway inhibitors have not been employed as immunotherapeutic agents, our studies suggest that these agents may act synergistically with other therapies by eliminating IFNγ-induced PD-L1-mediated suppression of both T and NK cell activity.
Figure 7.
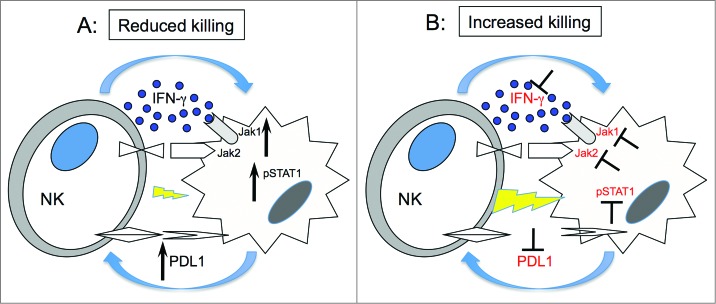
Hypothetical mechanism forIFNγ pathway modulation of tumor susceptibility to NK cells. (A) Upon engagement with target cells, NK cells secrete IFNγ, which in turn activates JAK1, JAK2 and STAT1 in tumor cells. This signal upregulates expression of PD-L1 on tumor cells resulting in increased resistance to NK cell killing. (B) Inhibition of JAK1 or JAK2 activation or IFNγ blocks upregulation of PD-L1 resulting in increased susceptibility of tumor cells to NK cell lysis.
Materials and Methods
Hematopoietic cell lines and primary tumor cells
Stable cell lines transduced with shRNAs targeting JAK1, JAK2 and STAT1 were previously described.7 Primary cells from patients with MM, AML and ALL contained at least 80% blasts or CD138+ cells. Patient samples were obtained under a protocol approved by the Institutional Review Board of the Dana-Farber/Harvard Cancer Center and informed consent was obtained from each patient. Mononuclear cells from patient bone marrow aspirates were purified by ficoll-hypaque and cryopreserved in 10% DMSO.
NK cell purification
Primary NK cells were purified from peripheral blood mononuclear cells (PBMCs) of healthy donors using MACS magnetic separation system and the NK cell isolation kit according to manufacturer's protocol (Miltenyi Biotec) and used after overnight incubation with 100 Units/mL IL-2. In all experiments, isolated CD56+CD3− NK cells were stained with anti-CD56-PE (BD PharMingen) and anti-CD3-FITC (Beckman Coulter) antibodies and the purity of NK cells was greater than 95%.
Treatment of tumor cells with activated NK supernatant or IFNγ
NK supernatant was generated by incubating NK-92, NKL49,50 or primary NK cells (1×105 cells/well) with IM9, U266 and K562 separately (1×105 cells/well) in 96 well plates for 12 h. NK supernatant was stored in aliquots at −80 C. Cell lines and primary tumor cells pre-treated with 40 nM JAK inhibitor 1 (Calbiochem) were incubated with undiluted NK supernatant or medium for 15 min, 4 h and 12 h and analyzed for STAT activation as described below. The use of this inhibitor at a concentration of 40 nM was based on previous experiments with tumor cell lines and primary tumor cells.7 JAK inhibitor 1 can also inhibit other members of the JAK family (JAK1, JAK2, JAK3, TYK2). In different experiments, cell lines or primary cells were pre-incubated with recombinant IFNγ (BD Biosciences) 10 Units/mL for the same times and analyzed for expression of phosphorylated STATs, AKT and ERK by flow cytometry.
Intracellular staining for pSTAT, pAKT and pERK
Phosphorylation of STATs, AKT and ERK was analyzed by flow cytometry using APC-conjugated antibodies (Alexa, BD Phosflow). 1×106 cells were fixed and permeabilized by incubation with formaldehyde for 10 min at RT, followed by incubation with methanol for 10 min on ice. After two washes with cold buffer, cells were incubated for 30 min at RT with anti-STAT1(pY701), STAT1(pS727), STAT3(pY705), STAT3(pS727), STAT4(pY693), STAT5(pY694), STAT6(pY641), AKT(pS473) and ERK1/2(pT202/pY204)-APC-conjugated. An APC-Alexa Fluor 647 IgG2a or IgG1-isotype control was used in every experiment. A minimum of 15,000-gated cells were acquired using a FACSCanto II (BD Biosciences) and data were analyzed using Flowjo software (Tree Star Inc.). In some experiments (Fig. S7A and B)46, expression of phosphorylated STAT protein was confirmed by western blot using rabbit anti-STAT1(pY701), pSTAT3(pS727), STAT5(pY694), total STAT1, ERK1/2, β-actin (Cell Signaling Technology) followed by a secondary horseradish peroxidase-conjugated goat anti-rabbit antibody as previously described.7
Co-cultures with NK cells
NK cell lysis of target cells was determined by flow cytometry after 12 h of co-incubation at 37°C. Cells were stained with AnnexinV-FITC/7AAD. An anti-NKG2A-PE (Beckman Coulter) or CD56-PE antibody was used to identify NK-92 or primary NK cells and the level of apoptosis was calculated for the NKG2A or CD56 negative cells. To detect NK activation, we measured IFNγ produced by NK cells using CBA-IFN-γ beads (BD Biosciences) according to manufacturer's instructions. To measure the level of NK cell degranulation, we incubated target cells and NK cells with CD107a/b-FITC (BD PharMingen), 1 h after Golgi stop was added to the culture. After a total of 4 h, cells were stained with CD56-PE-conjugated for 30 min and analyzed using the BD FACSCanto II. The percentage of CD107a/b positive cells was calculated for the CD56+ cell fraction. In all the experiments, NK cells were activated with 100 Units/mL of IL-2. Data were analyzed using Flowjo software.
Target cell expression of NK-activating/inhibitory ligands
Cells were pre-incubated with medium, activated NK-supernatant or IFNγ with or without JAK inhibitor for 12 h. Cells were then stained using anti-MHC Class I (clone W6/32), anti-HLA-A2 (clone BB7.2), anti-HLA-C (Santa Cruz Biotechnology), human NKp30-Fc, NKp44-Fc, NKp46-Fc, NKG2D-Fc, CD155 (R&D Systems) followed by RPE-conjugated goat anti-mouse-IgG, goat-anti-human-IgG or donkey-anti-goat-IgG (Jackson ImunoResearch). For direct staining, cells were incubated with PE-conjugated anti-TRAIL-R1, TRAIL-R2, CD95, CD112 or FITC-conjugated CD48 (Beckman Coulter) PE-conjugated anti-MICA/B (BD PharMingen), PE-conjugated PD-L1, PD-L2, B7H3 and B7H4 (BioLegend). Primary purified NK cells were also tested for PD-1 expression after 24 h incubation with medium, IL-2 (100 U/mL) or IFNγ (10 U/mL) using APC-conjugated anti-PD-1 antibody (clone EH12.2 H7) (BioLegend).
Functional blocking experiments
The anti-IFNγ antibody D9D10 (AbDSerotec) or IgG-irrelevant controls were used to block IFNγ in co-cultures of NK cells and tumor cells. After 12 h co-incubation with 2 μg/mL or 5 μg/mL of D9D10 or irrelevant-IgG, cells were stained with CD56-PE and AnnexinV-FITC/7AAD. Apoptotic cells were measured in the gated CD56 negative population. In all experiments, an aliquot of supernatant was analyzed using CBA-IFNγ beads to detect the level of IFNγ in the presence of D9D10 or control. For PD-L1 blocking experiments, we used a humanized anti-PD-L1 mAb with human IgG4 to eliminate FcR-mediated effects and S228P mutated to allow efficient dimerization (clone 29F.2A3).45,51 NK-92 or primary NK cells were incubated with cell lines or primary cells with or without 10 μg/mL anti-PD-L1 mAb for 12 h. 40 nM of JAK inhibitor 1 was also added to the culture and the level of killing was detected using AnnexinV-FITC/7AAD as described above. In some experiments, target cells were pre-treated with 10 Units/mL IFNγ and subsequently incubated with primary purified NK cells with or without anti-PD-L1 mAb. The level of tumor cell killing after incubation with NK cells was measured using CBA-IFNγ beads and AnnexinV/7AAD as described above.
Statistics
Statistical significance was determined using a Two-tailed Student's t test for all 2-sample comparisons and a p-value less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
Gordon J. Freeman has patents and receives patent royalties on the PD-1 pathway from Bristol-Myers-Squibb/Medarex, Roche/Genentech, Merck, EMD-Serono, Boehringer-Ingelheim, Amplimmune, and CoStim. Gordon J. Freeman is a scientific founder and scientific advisory board member of CoStim Pharmaceuticals. The other authors have no additional conflicts of interests.
Funding
This work was supported by NIH Grants R21 AI088521 (R.B.), CA078378, CA142106, CA183560 (J.R.), CA163125, P50 CA101942 (G.F.), RO1 CA142647 (S.H.H.). International Myeloma Foundation (IMF), Multiple Myeloma Research Foundation (MMRF) and Claudia Adams Barr Research Program (R.B.).
Supplementary Material
Supplemental data for this article can be accessed on the pub-lisher's website.
References
- 1.Caligiuri MA. Human natural killer cells. Blood 2008; 112:461-9; PMID:; http://dx.doi.org/ 10.1182/blood-2007-09-077438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell 2010; 142:847-56; PMID:; http://dx.doi.org/ 10.1016/j.cell.2010.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science 2011; 331:44-9; PMID:; http://dx.doi.org/ 10.1126/science.1198687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495-502; PMID:; http://dx.doi.org/ 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood 1990; 76:2421-38; PMID: [PubMed] [Google Scholar]
- 6.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. AdvImmunol 2006; 90:1-50; PMID:; http://dx.doi.org/ 10.1016/S0065-2776(06)90001-7 [DOI] [PubMed] [Google Scholar]
- 7.Bellucci R, Nguyen HN, Martin A, Heinrichs S, Schinzel AC, Hahn WC, Ritz J. Tyrosine kinase pathways modulate tumor susceptibility to natural killer cells. J Clin Invest 2012; 122:2369-83; PMID:; http://dx.doi.org/ 10.1172/JCI58457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen E, Staudt LM, Green AR. Janus kinase deregulation in leukemia and lymphoma. Immunity 2012; 36:529-41; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jelinek J, Oki Y, Gharibyan V, Bueso-Ramos C, Prchal JT, Verstovsek S, Beran M, Estey E, Kantarjian HM, Issa JP. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood 2005; 106:3370-3; PMID:; http://dx.doi.org/ 10.1182/blood-2005-05-1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J et al.. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997; 278:1309-12; PMID:; http://dx.doi.org/ 10.1126/science.278.5341.1309 [DOI] [PubMed] [Google Scholar]
- 11.Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264:1415-21; PMID:; http://dx.doi.org/ 10.1126/science.8197455 [DOI] [PubMed] [Google Scholar]
- 12.Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-gamma Elicits Macrophage Autophagy via the p38 MAPK Signaling Pathway. J Immunol 2012; 189:813-8; PMID:; http://dx.doi.org/ 10.4049/jimmunol.1102041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro A, Anand-Apte B, Tanabe Y, Feldman G, Larner AC. A PI-3 kinase-dependent, Stat1-independent signaling pathway regulates interferon-stimulated monocyte adhesion. J LeukocBiol 2003; 73:540-5; PMID:; http://dx.doi.org/ 10.1189/jlb.1002508 [DOI] [PubMed] [Google Scholar]
- 14.O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013; 368:161-70; PMID:; http://dx.doi.org/ 10.1056/NEJMra1202117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farag SS, Caligiuri MA. Human natural killer cell development and biology. Blood Rev 2006; 20:123-37; PMID:; http://dx.doi.org/ 10.1016/j.blre.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 16.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol 2008; 9:503-10; PMID:; http://dx.doi.org/ 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 17.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. JImmunolMethods 2004; 294:15-22; PMID:; http://dx.doi.org/ 10.1016/j.jim.2004.08.008 [DOI] [PubMed] [Google Scholar]
- 18.Depraetere H, Depla E, Haelewyn J, De Ley M. An anti-idiotypic antibody with an internal image of human interferon-gamma and human interferon-gamma-like antiviral activity. EurJBiochem 2000; 267:2260-7; PMID:; http://dx.doi.org/ 10.1046/j.1432-1327.2000.01231.x [DOI] [PubMed] [Google Scholar]
- 19.Lanier LL. NK cell recognition. Annu Rev Immunol 2005; 23:225-74; PMID:; http://dx.doi.org/ 10.1146/annurev.immunol.23.021704.115526 [DOI] [PubMed] [Google Scholar]
- 20.Maio M, Altomonte M, Tatake R, Zeff RA, Ferrone S. Reduction in susceptibility to natural killer cell-mediated lysis of human FO-1 melanoma cells after induction of HLA class I antigen expression by transfection with B2m gene. J Clin Invest 1991; 88:282-9; PMID:; http://dx.doi.org/ 10.1172/JCI115289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beatty GL, Paterson Y. IFN-gamma can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J Immunol 2000; 165:5502-8; PMID:; http://dx.doi.org/ 10.4049/jimmunol.165.10.5502 [DOI] [PubMed] [Google Scholar]
- 22.Boraschi D, Censini S, Tagliabue A. Interferon-gamma reduces macrophage-suppressive activity by inhibiting prostaglandin E2 release and inducing interleukin 1 production. J Immunol 1984; 133:764-8; PMID: [PubMed] [Google Scholar]
- 23.Naganuma H, Sasaki A, Satoh E, Nagasaka M, Nakano S, Isoe S, Nukui H. Down-regulation of transforming growth factor-beta and interleukin-10 secretion from malignant glioma cells by cytokines and anticancer drugs. JNeurooncol 1998; 39:227-36; PMID:; http://dx.doi.org/ 10.1023/A:1005902120612 [DOI] [PubMed] [Google Scholar]
- 24.Dolen Y, Esendagli G. Myeloid leukemia cells with a B7-2(+) subpopulation provoke Th-cell responses and become immuno-suppressive through the modulation of B7 ligands. Eur J Immunol 2013; 43:747-57; PMID:; http://dx.doi.org/ 10.1002/eji.201242814 [DOI] [PubMed] [Google Scholar]
- 25.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ et al.. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009; 458:206-10; PMID:; http://dx.doi.org/ 10.1038/nature07662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. NatRev Drug Discov 2013; 12:130-46; PMID:; http://dx.doi.org/ 10.1038/nrd3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012; 1:1223-5; PMID:; http://dx.doi.org/ 10.4161/onci.21335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, Zhang J, Yu J, Smith MK et al.. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010; 116:2286-94; PMID:; http://dx.doi.org/ 10.1182/blood-2010-02-271874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, Metes D. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol 2012; 42:541-50; PMID:; http://dx.doi.org/ 10.1002/eji.201141832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coughlin CM, Salhany KE, Gee MS, LaTemple DC, Kotenko S, Ma X, Gri G, Wysocka M, Kim JE, Liu L et al.. Tumor cell responses to IFNgamma affect tumorigenicity and response to IL-12 therapy and antiangiogenesis. Immunity 1998; 9:25-34; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(00)80585-3 [DOI] [PubMed] [Google Scholar]
- 31.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994; 1:447-56; PMID:; http://dx.doi.org/ 10.1016/1074-7613(94)90087-6 [DOI] [PubMed] [Google Scholar]
- 32.Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res 2005; 65:3447-53; PMID: [DOI] [PubMed] [Google Scholar]
- 33.Fisher PB, Miranda AF, Babiss LE. Measurement of the effect of interferons on cellular differentiation in murine and human melanoma cells. Methods Enzymol 1986; 119:611-8; PMID:; http://dx.doi.org/ 10.1016/0076-6879(86)19082-3 [DOI] [PubMed] [Google Scholar]
- 34.Kortylewski M, Komyod W, Kauffmann ME, Bosserhoff A, Heinrich PC, Behrmann I. Interferon-gamma-mediated growth regulation of melanoma cells: involvement of STAT1-dependent and STAT1-independent signals. JInvestDermatol 2004; 122:414-22; PMID:; http://dx.doi.org/ 10.1046/j.0022-202X.2004.22237.x [DOI] [PubMed] [Google Scholar]
- 35.Creagan ET, Ahmann DL, Long HJ, Frytak S, Sherwin SA, Chang MN. Phase II study of recombinant interferon-gamma in patients with disseminated malignant melanoma. Cancer TreatReports 1987; 71:843-4; PMID: [PubMed] [Google Scholar]
- 36.Meyskens FL Jr, Kopecky K, Samson M, Hersh E, Macdonald J, Jaffe H, Crowley J, Coltman C. Recombinant human interferon gamma: adverse effects in high-risk stage I and II cutaneous malignant melanoma. J Natl Cancer Inst 1990; 82:1071; PMID:; http://dx.doi.org/ 10.1093/jnci/82.12.1071-a [DOI] [PubMed] [Google Scholar]
- 37.Lollini PL, Bosco MC, Cavallo F, De Giovanni C, Giovarelli M, Landuzzi L, Musiani P, Modesti A, Nicoletti G, Palmieri G et al.. Inhibition of tumor growth and enhancement of metastasis after transfection of the gamma-interferon gene. Int J Cancer 1993; 55:320-9; PMID:; http://dx.doi.org/ 10.1002/ijc.2910550224 [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi K, Petersson M, Hoglund P, Kiessling R, Klein G, Karre K. Interferon gamma induces lung colonization by intravenously inoculated B16 melanoma cells in parallel with enhanced expression of class I major histocompatibility complex antigens. ProcNatlAcadSci US A 1987; 84:3405-9; PMID:; http://dx.doi.org/ 10.1073/pnas.84.10.3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bukowski JF, Welsh RM. Inability of interferon to protect virus-infected cells against lysis by natural killer (NK) cells correlates with NK cell-mediated antiviral effects in vivo. J Immunol 1985; 135:3537-41; PMID: [PubMed] [Google Scholar]
- 40.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res 2011; 17:6118-24; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999; 5:1365-9; PMID:; http://dx.doi.org/ 10.1038/70932 [DOI] [PubMed] [Google Scholar]
- 42.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al.. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027-34; PMID:; http://dx.doi.org/ 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. ProcNatlAcadSci U S A 1998; 95:7556-61; PMID:; http://dx.doi.org/ 10.1073/pnas.95.13.7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pena J, Alonso C, Solana R, Serrano R, Carracedo J, Ramirez R. Natural killer susceptibility is independent of HLA class I antigen expression on cell lines obtained from human solid tumors. Eur J Immunol 1990; 20:2445-8; PMID:; http://dx.doi.org/ 10.1002/eji.1830201113 [DOI] [PubMed] [Google Scholar]
- 45.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 2003; 170:1257-66; PMID:; http://dx.doi.org/ 10.4049/jimmunol.170.3.1257 [DOI] [PubMed] [Google Scholar]
- 46.Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y, Zhang P. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology 2012; 217:385-93; PMID:; http://dx.doi.org/ 10.1016/j.imbio.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 47.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994; 8:652-8; PMID: [PubMed] [Google Scholar]
- 50.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. ExpHematol 1996; 24:406-15; PMID: [PubMed] [Google Scholar]
- 51.Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, Sharpe AH, Dustin LB, Rice CM, Grakoui A et al.. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). ProcNatlAcadSci U S A 2013; 110:15001-6; PMID:; http://dx.doi.org/ 10.1073/pnas.1312772110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.