

Abstract
Conventional treatment for cancer routinely includes surgical resection and some combination of chemotherapy and radiation. These approaches are frequently accompanied by unintended and highly toxic collateral damage to healthy tissues, which are offset by only marginal prognostic improvements in patients with advanced cancers. This unfortunate balance has driven the development of novel therapies that aim to target tumors both safely and efficiently. Over the past decade, mounting evidence has supported the therapeutic utility of T-cell-centered cancer immunotherapy, which, in its various iterations, has been shown capable of eliciting highly precise and robust antitumor responses both in animal models and human trials. The identification of tumor-specific targets has further fueled a growing interest in T-cell therapies given their potential to circumvent the non-specific nature of traditional treatments. Of the several strategies geared toward achieving T-cell recognition of tumor, bispecific antibodies (bsAbs) represent a novel class of biologics that have garnered enthusiasm in recent years due to their versatility, specificity, safety, cost, and ease of production. Bispecific T-cell Engagers (BiTEs) are a subclass of bsAbs that are specific for CD3 on one arm and a tumor antigen on the second. As such, BiTEs function by recruiting and activating polyclonal populations of T-cells at tumor sites, and do so without the need for co-stimulation or conventional MHC recognition. Blinatumomab, a well-characterized BiTE, has emerged as a promising recombinant bscCD19×CD3 construct that has demonstrated remarkable antitumor activity in patients with B-cell malignancies. This clinical success has resulted in the rapid extension of BiTE technology against a greater repertoire of tumor antigens and the recent US Food and Drug Administration's (FDA) accelerated approval of blinatumomab for the treatment of a rare form of acute lymphoblastic leukemia (ALL). In this review, we dissect the role of T-cell therapeutics in the new era of cancer immunotherapy, appraise the value of CAR T-cells in the context of solid tumors, and discuss why the BiTE platform may rescue several of the apparent deficits and shortcomings of competing immunotherapies to support its widespread clinical application.
Keywords: bispecific antibodies, immunotherapy, malignancies, T lymphocytes
Abbreviations: BsAb, bispecific antibody; BiTE, bispecific T-cell engager; OS, overall survival; GBM, glioblastoma; ACT, adoptive cell therapy; TREG, regulatory T-cells; TIL, tumor infiltrating lymphocytes; TCR, T-cell receptor; TAA, tumor associated antigens; TSA, tumor specific antigens; CAR, chimeric antigen receptors; ScFv, single chain variable fragment; MAb, monoclonal antibody; MHC, major histocompatibility complex; AICD, activation induced cell death; ALL, acute lymphoblastic leukemia; CML, chronic myeloid leukemia; APC, antigen presenting cell; CHO, chinese hamster ovary; VV, vaccinia virus
Introduction
Cancer remains among the most devastating causes of death worldwide, with nearly 600,000 deaths projected to occur this year in the United States alone.1 Despite aggressive clinical intervention, systemic progression and overall disease burden have hindered improvements in prognosis for most patients suffering with solid tumors in advanced stages. This is perhaps best exemplified by glial tumors residing in the “immunologically distinct” brain, where the overall survival (OS) for patients suffering with glioblastoma (GBM), the most common primary malignant brain tumor, remains often less than 15 mo. These therapeutic shortcomings are also often compounded by the debilitating side effects of conventional therapy on patient quality of life, owing to the inherent non-specific nature of available treatment regimens. These factors have driven the development of novel strategies that can target cancers both specifically and efficiently, while eliminating tumors with minimal collateral toxicity.
The immune system presents an ideal such platform by virtue of its physiologic surveillance role, which normally proceeds in a highly precise and robust manner. Paul Ehrlich was among the first to propose the intimate connection between host immunity and neoplastic disease a century ago; it is well accepted today that the immune system plays dual roles in modulating the development of neoplastic cells through a “Darwinian selection” of sorts, as described by Schreiber and colleagues in the cancer “immunoediting” hypothesis.2 The mechanisms underlying the tumor-sculpting actions of host immunity have been described extensively elsewhere.3-5 That cancer results, at least in part, from the failure of the immune system to eliminate certain neoplastic cells - variants that are “fit” to survive-has fueled preclinical and clinical investment toward redirecting host immunity against them. Immunotherapy, then, aims to harness and redirect the immune response against cells that have replicated in the face of a frequently failed immunosurveillance task. A variety of platforms - both passive and active-have been explored for this purpose; in this review, we will discuss and compare several strategies that have recently entered or evolved in the clinical arena. Specifically, we will review adoptive-T-cell therapy and the use of bsAbs, which have emerged as a highly versatile and promising class of novel biologics. We will discuss the utility of bsAbs in facilitating T-cell mediated tumor destruction, and how a growing class of bsAbs, termed BiTEs, is particularly promising in terms of safety, cost, and ease of production.
The case for T-cell therapy
Adoptive transfer
An effective immune response against cancer requires a threshold presence of activated T-cells that recognize tumor, often specifically CD8+ cells, which mediate tumor killing through expression of granzymes and perforin. CD4+ helper T-cells are also thought to be important players in potentiating CD8+ responses, and both populations are present in high numbers in blood and organs. As such, one niche of immunotherapy aims to augment the immune response by providing or modulating T-cell responses, and this can be accomplished by direct ex vivo manipulation of autologous lymphocytes, or by passively recruiting T-cells non-specifically to the site of tumor.
Adoptive cell therapy (ACT) is one highly attractive method wherein tumor-reactive lymphocytes are identified or produced, expanded ex vivo, and transferred back into cancer-bearing patients. Since its introduction in the late 1980s, ACT has evolved dramatically to mediate the durable responses seen in patients with leukemia, metastatic melanoma, and other solid cancers this decade. In 2002, non-myeloablative lymphodepletion was introduced as a compulsory pre-conditioning regimen that drastically improved ACT response rates,6-8 in part by ablating immunosuppressive regulatory T-cells (TREG) and cytokine sinks. Additionally, the immunologic space created by an absence of host lymphocytes prior to infusion is believed to support the homeostatic cytokine-driven expansion of infused T-cells, up to a thousand-fold in some instances.6,7,9
Tumor infiltrating lymphocytes
In its earliest days, ACT often employed tumor-infiltrating lymphocytes (TILs), typically a mixed bag of CD4+ helper and CD8+ cytotoxic T-cells that had trafficked to and were isolated from tumor. Despite their tumor reactivity, they were frequently exposed to immunosuppressive cytokines within the tumor microenvironment that rendered them functionally inert. Early format ACT aimed to isolate and expand these TILs to clinically relevant numbers prior to clonal repopulation in patients. Although TILs have been shown to mediate tumor regression and durable responses in patients with melanoma,10-12 TIL-ACT has not yet reached widespread application due to the technical difficulty associated with identifying, isolating, and expanding tumor-reactive lymphocytes.10 This strategy, however, has remained as an important proof-of-principle, demonstrating the therapeutic potential of tumor-directed lymphocytes in a clinical setting. The question, then, has evolved into one surrounding the source of tumor-reactive lymphocytes; if we cannot sufficiently isolate and grow them, can we instead manufacture them?
Some of the most impressive developments in ACT are thus indebted to modern advancements in DNA recombinant technology, molecular biology, and gene-engineering, which have together made possible the genetic modification of T-cells to endow them with specificity for tumor antigens, rather than depend wholly on the isolation of a tumor-reactive species. In one variation, T-cells can be engineered with a transgenic T-cell receptor (TCR) specific for a tumor-associated or -specific antigen (TAA or TSA, respectively).13,14 Results from the first-in-man trial using autologous lymphocytes engineered with MART-1-specific TCRs demonstrated safety and induced tumor regression in patients with metastatic melanoma.15,16
Chimeric antigen receptors
Similar advances have also paved way for the intricate design and production of tumor-specific synthetic receptors capable of mimicking TCR-based activation, without depending on conventional TCR signaling per se. One example of this is the advent of chimeric antigen receptors (CARs),17 which today consist of intracellular T-cell signaling domains (i.e., CD3ζ) fused to an extracellular single chain variable fragment (scFv) of a tumor-reactive monoclonal antibody (mAb).18 Since CAR specificity is derived from a mAb, these constructs can be designed to recognize any cell surface antigen and can be retrovirally integrated into T-cells to trigger in them an MHC-independent mode of activation. This MHC-independence is especially relevant, as tumors frequently downregulate MHC molecules,19-22 lose expression of MHC-I associated β2-microglobulin23 and intracellular peptide transporters,24,25 and can even alter the architecture of intracellular proteasomes, preventing adequate MHC presentation of antigens on cell surfaces. These represent mechanisms that would otherwise paralyze immunotherapies dependent on TCR-MHC complex formation (i.e., TIL-ACT and TCR gene therapy). The caveat to this MHC-independence is that targeted antigens must be on the cell surface (as with antibody therapies), limiting the number of appropriate targets available.
The first generation of CARs consisted of the CD3ζ chain fused to a tumor-associated scFv. These CARs were capable of redirecting and activating T-cells to elicit cytotoxicity and tumor cell lysis,26,27 but were limited in their activation, proliferation, and persistence in patient trials.28-30 More recently developed CARs have improved on this design to incorporate co-stimulatory signaling domains such as CD28, 4-1BB and OX40 (Fig. 1).18,31-34 These endo domains provide CARs with co-stimulatory signals that enhance T-cell activation, proliferation, survival, cytokine secretion, and prevent activation-induced cell death (AICD). Recently, these “modern” CARs have gained notable attention for their clinical efficacy against lymphoid leukemias,35-38 where CD19-directed CARs have achieved complete remissions in several patients with refractory disease. In a recent phase I/IIA study from the University of Pennsylvania, Maude and colleagues reported complete remissions in 27 out of 30 chemotherapy-resistant or -refractory CD19+ leukemia and lymphoma patients receiving CD3ζ-4-1BB second generation CD19 CARs39 - a remarkable 90% response rate in children and adults whose disease was previously considered refractory or incurable.
Figure 1.
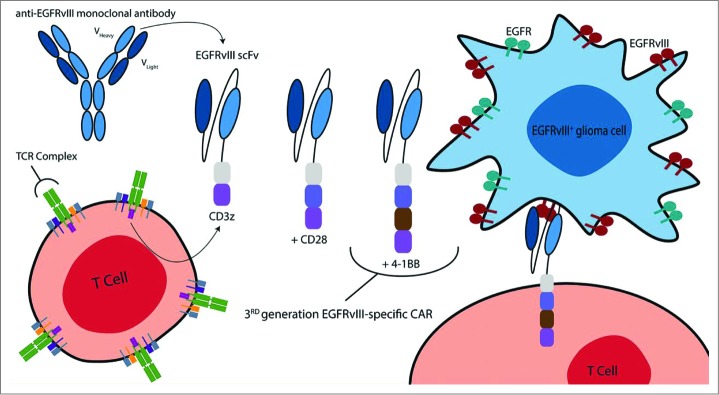
CARs directed against EGFRvIII are produced by combining the humoral specificity of an EGFRvIII-specific antibody with the intracellular signaling domains of a T-cell receptor (TCR). In general, CARs are composed of the variable heavy and light chains of a mAb fused (via a transmembrane hinge) to CD3z. More recently, CAR design has evolved to include additional costimulatory moieties – namely CD28 and/or 4–1BB – to improve OS, proliferation, and antitumor activity. The third-generation EGFRvIII-specific CAR incorporates the CD28, 4–1BB, and CD3z signaling constructs. These same CAR designs can be used to target wild-type EGFR. Reproduced from permission from reference.114
As would be expected, appropriate target selection is of critical importance with T-cell therapy, as CARs targeting shared antigens on normal tissue have led to patient death.42 The current scarcity of tumor-specific targets on the cell surface greatly complicates T-cell therapy in cases where known targets are not ubiquitously expressed by malignant cells. GBM is one such example of a heterogeneous burden where tumors vary in their expression profile of EGFRvIII, a tumor-specific mutation of the epidermal growth factor receptor. EGFRvIII-directed therapies therefore remain vulnerable to the outgrowth of tumor cells that do not express the target antigen,43 although early preclinical evidence suggests that EGFRvIII-CAR therapy may offer long-term immunologic protection, perhaps via epitope spreading.44 Similarly, one promising alternative has been the ambitious approach of eliciting a global anti-GBM immune response by improving intratumoral recruitment of dendritic cells (DCs) in conjunction with cytotoxic therapy in order to leverage the release of immunoreactive antigens from dying tumor cells. In 2009, Curtin and colleagues demonstrated this proof-of-concept by combining the intratumoral expression of a known DC chemoattractant, Fms-like tyrosine kinase 3 ligand (Flt3L), and cytotoxic agent, thymidine kinase (TK), plus ganciclovir to treat syngeneic high-grade gliomas. This study found that dying glioma cells released the TLR2 agonist high-mobility group box 1 (HMGB1), which activated local DCs migrating to sites of tumor-kill in response to Flt3L chemokine gradients.45 The authors demonstrate that the net effect of this cascade - brain tumor elimination - was dependent on HMGB1, lending greater credence to an immunotherapeutic strategy preemptively designed to elicit tumor epitope spreading.
It remains to be seen if the problems of single-antigen targeting will impede the wide scale adoption of CAR therapy, or whether preclinical evidence of immunologic memory and long-term protection will be reproduced in patients. The clinical implementation of this strategy is also met with several obstacles that complicate standardization and its “scaling up” for the masses. CAR therapy is exceedingly laborious since CAR T-cells must be produced on a patient-to-patient basis, requiring significant institutional investment in clinical infrastructure and resources that is usually only possible at major academic centers. Moreover, the capricious nature of retroviral transduction and the varied differentiation states of isolated T-cells can produce inconsistent CAR expression levels, further complicating the delivery of therapy. Retroviral integration into genomes also runs the low risk of insertional mutagenesis, causing concerns over safety. Ongoing studies, however, further substantiate T-cells as a crucial and obligatory component of immune-mediated tumor clearance.
Navigating solid tumors
Unique physiology equips solid tumors with an assemblage of regulatory immune cell types that are key players in both the development of neoplastic tissue and the sustained resistance of established masses to cytotoxic therapy. The formation and survival of solid tumors requires not only the accumulation of aberrant mutations at the genetic level, but also a series of dynamic interactions mediated through signaling cascades between them. These events together result in the generation of abnormal vasculature, create hypoxic environments, promote tumor-beneficial modes of inflammation, and orchestrate the recruitment and retention of inhibitory immune cells that underlie the highly immunosuppressive architecture typical of solid tumors. In fact, the composition of tumor microenvironments is believed to be a critical determinant of cancer progression,46 as local structural and soluble components can be deciding factors in whether a tumor remains a benign hyperplasia or evolves into a malignant lesion.47
At onset, tumor formation has been documented to resemble “wounds that fail to heal”,48 as evidenced by cell-to-cell interactions that act in concert to promote a state of chronic inflammation. In the classical immune response to injury, resident and migratory leukocytes degrade local tissue, ingest debris, and secrete a combination of cytokines and proteinases that can drastically alter tissue architecture by activating stromal fibroblasts and vascular support cells. These processes are aimed at restoring normal tissue homeostasis by expanding the extracellular matrix (ECM) and inducing angiogenesis for new capillary formation.49,50 These pro-homeostatic mechanisms, however, are tightly regulated by immunosuppressive leukocytes that minimize potential for autoimmunity during tissue remodeling. During cancer progression, the mechanisms that typically restore these states of acute inflammation to normalcy after injury repair are absent, and instead, the chronic inflammation that persists is characterized by the accumulation of immunosuppressive cells, including TREG, regulatory B-cells,51,52 type II natural killer T-cells,53 immature macrophages, alternatively activated macrophages (M2), and TH2 CD4+ T-cells.54,55 These cellular subsets increase local concentrations of immunosuppressive cytokines TFGβ, IL-4,-5,-6,-10,-13, and -35,54,56 all of which contribute to pro-tumor immunity by promoting T-cell anergy and inhibiting cytotoxic T-cell responses.
Moreover, one critical function of these regulatory cells is their ability to simultaneously promote angiogenesis by signaling endothelial cells through the section of VEGFA, bFGF, CCL2, and ANGPT2.56 New vasculature is typically formed in disorganized fashion, owing to diminished regulation and local hypoxia. These new vessels, however, rarely rescue the deficits associated with the limitations of oxygen diffusion,57 and hypoxia has been shown to increase recruitment of TREG through the CCL28-CCR10 axis.58,59 Moreover, there is overwhelming evidence supporting the hypoxia-induced secretion of inhibitory molecules and the shedding of MHC class I chain-related proteins A and B from cell surfaces to avoid killing by immune effectors.60-62 Recent studies have also identified the hypoxia-induced upregulation of PD-L1 (B7-H1) on tumor cells as a critical mechanism of tumor immune escape, as this has been described to increase cancer cell resistance to cytolysis.63
Eradication of solid tumors therefore requires CARs to not only (i) migrate to malignant tissue, (ii) penetrate through desmoplastic stroma - which can account for up to 90% of total tumor mass48 - but to also (iii) circumvent or function in spite of the immunosuppressive and immune-evasive obstacles that otherwise protect neoplasms from surveillance. Importantly, these features have frustrated attempts at curing solid tumors since the 1950s.64 The one notable exception has been curative immunotherapy for metastatic melanoma in trials conducted at the National Cancer Institute, and the evolution of CAR therapy has been largely based on the principles established in these studies. To date, however, there remains a scarcity of promising results utilizing CARs clinically against solid cancers. An early phase I study in patients with metastatic ovarian cancer receiving α-folate receptor-directed first-generation CARs reported no evidence of tumor killing,28 and is likely explained by low persistence and the absence of radiolabeled T-cells at tumor sites, although111 labeling limits long-term T-cell tracking and carries a low resolution. Other investigators have also evaluated first- and second-generation CARs targeting PSMA, CEA, and HER2/neu, and these studies have corroborated the critical association of the persistence of transferred cells with tumor progression and clinical outcome. Based on this premise, third-generation CARs have also entered the clinical arena for other solid tumors, including GBM (NCT01454596). Whether improved persistence can overcome the obstacles of solid tumor microenvironments remain to be seen. Tremendous preclinical efforts are currently underway, however, to further engineer CAR design to include novel building blocks or intrinsic modifications that may address specifically the issues of hypoxia, TREG, and intratumoral migration.
Immunomodulatory therapeutics
A fine balance between immune stimulatory and inhibitory signals exists in the setting of cancer, modulation of which can be exploited toward a therapeutic end. Effector CD8+ T-cells require a ‘trifecta’ of TCR and costimulatory signals to achieve a sufficient activation profile for tumor killing. The TCR-CD3 complex must first bind antigen bound to MHC on an antigen presenting cell (APC) (Signal 1), followed by ligand binding of co-stimulatory or co-inhibitory receptors on T-cells that control and tune the TCR signal.65 B7 family ligand (CD80/CD86) binding to CD28, a cell-surface molecule present on approximately half of all CD8+ T-cells, produces a co-stimulatory response that promotes cytotoxicity (Signal 2). CD28 molecules respond to stimulation by eliciting an intracellular cascade of signals that together enhance cytokine secretion (Signal 3) and prevent cellular anergy. Induced anergy among tumor-reactive T-cells has been proposed as one major mechanism underlying tumor immune-evasion, due both in part to the absence of co-stimulatory ligands for CD28 and to an immunosubversive tumor microenvironment. This thought has spawned the generation of a relatively new class of therapeutic antibodies designed to mimic co-stimulation by stimulation through targets such as CD28 and 4-1BB in addition to the targeted tumor antigen, thereby perpetuating immune responses.
One concern, however, is that indiscriminate strengthening of co-stimulation carries the potential for autoimmunity and life-threatening systemic inflammatory responses. A recent phase I trial that evaluated the safety and activity of an anti-CD28 mAb, TGN1412, for example, led to severe cytokine release syndrome and multi-organ failure in several patients.66-68 A separate class of mAbs designed to instead antagonize co-inhibitory molecules, including CTLA-4 and PD-1, are currently under active investigation and warrant similar caution, as non-specific immune checkpoint blockade can remove barriers that otherwise protect hosts from autoimmunity (although antibodies to both have proven fairly well-tolerated thus far). In fact, in a recent trial for patients with metastatic melanoma, the combination of anti-CTLA-4 with a peptide vaccine was associated with the unintentional induction of colitis, dermatitis, uveitis, and hypophysitis.69 Similar symptoms were reported in other studies evaluating anti-CTLA-4 alone,70 or in combination with IL-2 administration.71 These immune-related adverse events are, however, strongly correlated with tumor regression, and are likely due to the breaking of tolerance to self-antigens. It is a continued theory that sufficiently potent anti-tumor immune responses will be accompanied, almost necessarily, by a degree of autoimmunity, as thresholds for self-tolerance are intentionally crossed. In light of the 2010 approval of anti-CTLA-4 for patients with metastatic melanoma, potential for autoimmunity in the CNS is of particular concern, given that anywhere from 10 to 50% of these patients develop CNS metastases.72,73 In a 2014 case report of a patient with stage IV melanoma m1c, treatment with low-dose anti-PD-1 for systemic disease led to non-specific CNS toxicity,74 which subsided after the drug was discontinued. Concerns over the systemic effects of wholesale immunomodulatory mAbs have therefore reinforced enthusiasm in expediting the clinical translation of antibody-based therapeutics which are tumor-specific, in order to direct biologic function exclusively to sites of tumor.
Novel biologics
BsAbs were first developed for the purpose of bridging interactions between effector mechanisms and tumor cells - similar in principle to CAR and transgenic TCR therapies - albeit through a different approach. Based in principle on the design and functionality of mAbs, construction of bsAbs sought to exploit the exquisite specificity and affinity of two separate, naturally occurring antibodies, by aligning their humoral specificities into one structure.
MAbs of the IgG isotype, the most abundant isotype in blood and extracellular fluid, are composed of four peptide chains, two identical heavy chains and two identical light chains, joined by a constant fragment domain (Fc). Within the IgG structure, two identical antigen-binding fragments (Fab) can be defined; they are composed of one constant and one variable domain for each heavy and light chain, and both Fab structures can be enzymatically separated from the Fc region. The earliest bsAbs were produced by isolating antigen-binding Fab fragments from two distinct mAbs and chemically crosslinking them at their hinge residues.75 These bsAbs have also been produced by fusing two distinct hybridoma cell lines together (e.g., hybrid hybridoma or a quadroma). Quadroma-derived bsAbs are assembled by random pairing of immunoglobin heavy and light chains, and so are comprised of a heterogeneous population of pure and fusion antibodies. The bsAb of interest can be isolated chromatographically and cleaved of the Fc region afterward, which helps mitigate unintended induction of Fc-mediated effects through alternative immune mechanisms. Despite the production of functional bsAbs, these methods have been largely abandoned due to the high cost and inefficiency associated with them; quadroma technology produces a high proportion of non-functional byproducts, and both require sophisticated purification procedures, making the isolation of clinically useful amounts of material difficult or impossible.
Recent advances in recombinant technology have made novel strategies of bsAb production favorable over conventional methods by using only variable domains as starting material.76 Two formats which utilize antibody fragments have emerged as superior strategies for their efficiency, ease of production, and ability to produce the smallest bsAb constructs to date (which can improve tumor penetrance). Both formats use scFvs that are constructed by associating variable heavy (VH) and light (VL) chain domains with a stabilizing, flexible polypeptide linker. In one variation, two fusion scFvs are produced by linking the VH of one antibody with the VL of another, and both chains are held together by non-covalent forces to form a structure termed a “diabody”.77,78 In contrast to these fusion scFvs, a second format in which two scFvs are translated in tandem through a short, non-immunogenic linker has proven to be a highly successful method of bsAb production.79 In this model, the scFv of one antibody is covalently bound to the scFv of the other, and therefore offers greater stability than their diabody fusion counterparts. The linker sequence maximizes rotational flexibility for scFvs to bind two epitopes on separate cell surfaces at once. Though tandem scFvs and diabodies were first produced using Escherichia coli, this system has since been replaced with mammalian systems (e.g., Cricetulus griseu, Chinese Hamster Ovary (CHO) cell line80) that tend to produce bispecific proteins with fewer folding errors and greater efficiency,80 a breakthrough method to scale production for clinical translation.
BiTE: Bispecific Antibodies for Cancer Therapy
BiTEs are a subclass of bsAbs that consist of two scFvs originating from two separate mAbs; one scFv recognizes a tumor antigen, while the second is specified for CD3ϵ on T-cells.81,82 The combination of two linked scFvs results in a recombinant polypeptide chain of about 55-60kDa. A small fusion linker connects both variable fragments and allows them to rotate freely, facilitating optimal interaction between T-cell and target cell in an immunologic synapse (Fig. 2).83,84 BiTE-induced lytic synapses are architecturally similar in size, composition, and spatial arrangement of subdomains to lytic synapses formed by activated T-cells in a typical scenario.85 However, the T-cell-BiTE-tumor cell configuration induces greater frequency and concentration of synapse formation, allowing robust lysis of target cells.85 BiTE recruitment and activation of T-cells leads to the upregulation of typical activation markers CD69 and CD2586,87 and secretion of cytokines IFNγ, TNF-α, IL-2, IL-4, IL-6, and IL-10.87,88 Importantly, BiTE-mediated cytotoxicity does not occur by monovalent binding to CD3; both arms (i.e., to TAA as well) must be bound, linking antigen to activation and addressing concerns regarding the non-specific toxicity seen with globally activating mAbs. It is similarly important to recognize that BiTE-efficacy is not dependent on the antigen specificity of bound T-cells - it essentially confers TAA-specificity to the entire contacted T-cell populous. Target cell lysis also requires BiTE engagement of cytotoxic T-cells that express granzymes and perforin which, unlike naïve T-cells, do not require costimulatory signals to actively secrete toxins.89,90 Perforin-mediated delivery of granzyme B causes a calcium-dependent proteolytic activation of intracellular caspases which results in tumor cell death.91,92 The FasL-Fas receptor system, despite its apoptotic role in normal cells, has not been shown to be significantly involved in BiTE activity.92,93
Figure 2.
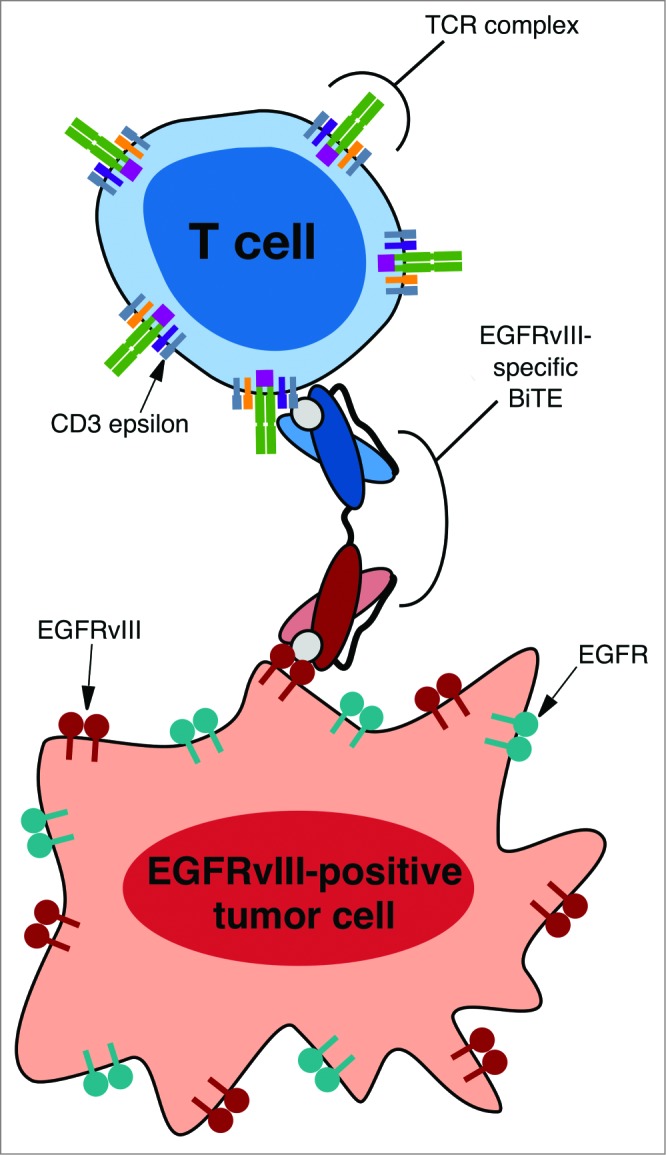
A schematic representing EGFRvIII-specific BiTE creating an immunologic synapse by binding to a tumor cell via the tumor specific antigen EGFRvIII and a T cell via CD3-epsilon. Note that the EGFRvIII binding portion does not bind to the wild-type EGFR, thus mediating tumor cell specific targeting. Reproduced with permission from reference.115
In addition to CD8+ cytotoxic T-cells, BiTEs can also bind CD4+ helper T-cells and TREG cells.94,95 Remarkably, recent data has shown that BiTE-binding of these two cell types leads to dramatic upregulation of granzyme B and perforin in response to target cells.92,96-98 Previous studies have described this perforin-granzyme axis as a means by which TREG assert their suppressive function through the killing of effector cells. Given the presence of intratumoral TREG, the finding that BiTEs carry the potential to co-opt TREG killing of lymphocytes for direct tumor cell lysis might play a dramatic role in upcoming studies evaluating BiTE efficacy.
BiTEs are also being investigated in combinatorial formats. In a recent study by Yu and colleagues,99 an oncolytic vaccinia virus (VV) encoding secretory EphA2-specific BiTEs was shown to enhance T-cell mediated tumor killing of human A549 lung cancer cells. Importantly, specifying VV for secretory BiTEs did not interfere with either viral replication or the antitumor activity of oncolytic VV. Instead, the secretion of BiTEs synergized with the antitumor efficacy of VV by recruiting T-cells to the site of tumor. The use of VV as both an oncolytic agent and delivery mechanism represents a novel application of two exciting therapies, which may also further enhance the safety profile of the BiTE platform.
Clinical Translation
The BiTE format has been evaluated against an impressive variety of tumor-associated targets. These include CD19, CD20, EpCAM, EGFR, MUC-1, CEA, and HER2. Blinatumomab (MT103), a CD19/CD3 targeted BiTE, became the first BiTE ever tested in man in 2001 and has shown safety and promising efficacy in treating ALL and B non-Hodgkin's lymphoma (NHL) in phase I/II trials. Importantly, the phase II data from ALL patients supported the notion that BiTEs are efficacious against both advanced and minimal residual disease.100,101 In a separate trial by Schlegel and colleagues, nine post-transplant relapsed pediatric patients suffering with ALL were treated with blinatumomab at infusion dosages of 5 to 15 µg/m2/day,102 and remarkably, six patients responded with complete remission after one or two cycles. The remaining three patients did not respond to treatment. Early preclinical studies by Wong et al. also demonstrate promising results for patients with chronic lymphocytic leukemia - an incurable B-cell malignancy. When peripheral blood mononuclear cells from 28 patients were tested with blinatumomab against leukemic cells, cytotoxicity and tumor cell death was observed at low T cell:tumor cell ratios.103 Blinatumomab was recently approved by the FDA for Philadelphia chromosome-negative precursor B-cell ALL under the FDA's accelerated approval program after receiving ‘breakthrough therapy designation’ earlier this year.
The EpCAM-targeting MT110 is the second BiTE to have entered a phase I clinical trial for solid tumors including gastric, colorectal, ovarian, breast, prostate, and small cell lung cancer. Minimal doses (1 µg/day) of MT110 were found to be well-tolerated, and dose escalation is currently under evaluation. BiTEs under active clinical investigation in trials registered with www.clinicaltrials.gov are summarized in Table 1.
Table 1.
Summary of patient trials investigating Bispecific T-cell Engagers (BiTEs). EpCAM = epithelial cell adhesion molecule; CEA = carcinoembryonic antigen; PSMA = prostate specific membrane antigen
| BiTE® | Target antigen | Disease | Phase | Status | ClinicalTrials.gov ID |
|---|---|---|---|---|---|
| Blinatumomab (MT103 / AMG 103) | CD19 | Relapsed NHL | I | Completed | NCT00274742 |
| AMG 110 / MT110 | EpCAM | Lung cancer (adenocarcinoma and small cell), gastric cancer or adenocarcinoma of the gastro-esophageal junction, colorectal cancer, breast cancer, hormone-refractory prostate cancer, and ovarian cancer | I | Active, not recruiting | NCT00635596 |
| AMG 211 / MEDI-565 | CEA | Gastrointestinal Adenocarcinomas | I | Active and recruiting | NCT01284231 |
| AMG 212 / BAY2010112 | PSMA | Prostate cancer | I | Active and recruiting | NCT01723475 |
| Blinatumomab | CD19 | Pediatric and adolescent patients with relapsed/refractory B-ALL | I / II | Active and recruiting | NCT01471782 |
| Blinatumomab | CD19 | Relapsed/Refractory B-ALL | II | Active, not Recruiting | NCT01466179 |
| Blinatumomab | Philadelphia Positive/BCR-ABL Positive ALL | II | Active and recruiting | NCT02000427 | |
| Blinatumomab | CD19 | Relapsed/Refractory B-ALL | II | Active, not recruiting | NCT01209286 |
| Blinatumomab | CD19 | Minimal residual Disease of B-ALL | II | Active, not recruiting | NCT01207388 |
| Blinatumomab | CD19 | Minimal residual disease of B-ALL | II | Active, not recruiting | NCT00560794 |
| Blinatumomab | CD19 | Relapsed/Refractory B-ALL | II | Active, not recruiting | NCT01209286 |
| Blinatumomab | CD19 | Relapsed/Refractory diffuse large B-cell lymphoma | II | Active, not recruiting | NCT01741792 |
| Blinatumomab | CD19 | Relapsed/Refractory ALL | III | Active and recruiting | NCT02013167 |
| Blinatumomab | CD19 | Newly diagnosed BCR-ABL-negative B-ALL in adults | III | Active and recruiting | NCT02003222 |
Expert Opinion
BiTEs are novel biologics that are beginning to demonstrate great promise in tumor therapy. The growing interest and enthusiasm surrounding this platform is owed in part to the widespread clinical and commercial success of mAbs over the past two decades. As of 2012, 40 mAbs have received FDA-clearance for clinical use against several cancers, including lymphoma,104 breast cancer,105 and colorectal cancer,106 among others. Recent advances in DNA recombinant and hybridoma technologies have helped streamline the bench-to-bedside translation of mAbs, and their low cost of manufacturing has helped them become the highest selling class of biologics in industry, peaking to nearly $25 billion in US sales in 2012.107 BiTEs therefore represent an extension of a class of drugs that is cheaply made, “off-the-shelf”, and within a market that has already proven lucrative.
These factors may ultimately determine whether BiTEs enjoy greater commercial success than competing immunotherapies. The pharmaceutical industry, for example, has had relatively limited experience with ACT, and there will be significant obstacles in delivering this platform to clinics and community hospitals that do not have the resources of large, well-funded academic centers to support the infrastructure necessary for cell banks and production of clinical-grade retrovirus for CAR T-cell production. BiTEs also carry many of the same advantages of CARs, namely MHC-independence and tumor-specificity, with several added benefits. CAR T-cells, for example, are prone to tumor immune-subterfuge, in a similar fashion to inert TILs found within tumors. CARs can therefore be overwhelmed by immunosuppressive cytokines or intratumoral TREG inhibition, impeding their antitumor activity. It remains to be seen if novel modifications to the CAR design will overcome these factors. By contrast, BiTEs have been shown to not only re-activate T-cells within tumor microenvironments,108,109 but can also transform the immunosuppression of TREG into tumor-directed cytotoxicity. To our knowledge, there is currently no evidence to support a similar conversion of function in CAR+TREG. ACT-based therapies also depend on the trafficking, localization, and in situ expansion of adoptively transferred T-cells near tumor, which can vary dramatically based on the patient and type of cancer. BiTEs do not depend on the trafficking patterns of infused lymphocytes, but may instead recruit and activate T-cells polyclonally, conferring upon all contacted T-cells specificity for and activation by tumor. These latter two factors may prove particularly important; given their small size, BiTEs may be able to penetrate deep into solid masses to either activate local TILs or co-opt TREG that are within the vicinity of tumor cells expressing the BiTE-directed antigen. This is in comparison to CARs, which depend completely on the successful migration of single-cells deep into tumor for clonal expansion upon antigen engagement. Like CARs, BiTEs also carry the potential to confer hosts with immunologic protection against tumor antigens by coupling DC recruitment with BiTE-mediated tumor killing. In theory, activated DCs from the CNS could stimulate an endogenous host response through antigen cross-presentation in the draining lymph nodes, which has been shown to confer primed T-cells with a tropism for homing to the CNS.110 Furthermore, BiTEs do not run the safety risk of insertional mutagenesis associated with gene-therapy, and the short half-life of BiTEs allows a relatively quick cessation of any unintended severe adverse events resulting from therapy. Regardless of the specific therapeutic platform, however, it remains clear that the identification of cell surface tumor-specific targets represents the most important barrier to the clinical application of either therapy.
Importantly, BiTEs have also been shown to elicit antitumor activity against tumors residing in the brain, overcoming previous concerns regarding the treatment of tumors in immunologically “distinct” areas such as the CNS.87,111,112 BiTEs recognizing EGFRvIII have been studied extensively in preclinical models of GBM.87,111,113 These studies have demonstrated localization of BiTEs to brain tumors resulting in prolonged survival of tumor-bearing mice with up to a 75% complete cure rate.87 Other potential surface targets for the treatment of GBM by BiTE therapy include HER2 and IL-13R. While the clinical translation of BiTEs in this setting has yet to be realized, the technology represents a new boon for patients where the need for improved therapy is great.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:; http://dx.doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 3.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol 2014; 27C:16--25; PMID: ; http://dx.doi.org/ 10.1016/j.coi.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004; 22:329-60; PMID:; http://dx.doi.org/ 10.1146/annurev.immunol.22.012703.104803 [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002; 3:991-8; PMID:; http://dx.doi.org/ 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 6.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM et al.. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850-4; PMID:; http://dx.doi.org/ 10.1126/science.1076514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012; 4:127ps8; PMID:; http://dx.doi.org/ 10.1126/scitranslmed.3003634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, Herndon JE 2nd, Bigner DD et al.. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PloS One 2013; 8:e59082; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0059082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer-what clinicians need to know. Nat Rev Clin Oncol 2011; 8:577-85; PMID:; http://dx.doi.org/ 10.1038/nrclinonc.2011.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8:299-308; PMID:; http://dx.doi.org/ 10.1038/nrc2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR et al.. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550-7; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zippel DB, Besser M, Shapira R, Ben-Nun A, Goitein D, Davidson T, Treves AJ, Markel G, Schachter J, Papa MZ. Adoptive cell therapy with autologous tumor-infiltrating lymphocytes and high-dose interleukin-2 for metastatic melanoma: The surgeon's perspective. Exp Ther Med 2012; 3:898-902; PMID:; http:dx.doi.org/ 10.3892/etm.2012.498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coccoris M, de Witte MA, Schumacher TN. Prospects and limitations of T cell receptor gene therapy. Curr Gene Ther 2005; 5:583-93; PMID:; http://dx.doi.org/ 10.2174/156652305774964730 [DOI] [PubMed] [Google Scholar]
- 14.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM et al.. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36:133-51; PMID:; http://dx.doi.org/ 10.1097/CJI.0b013e3182829903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duval L, Schmidt H, Kaltoft K, Fode K, Jensen JJ, Sorensen SM, Nishimura MI, von der Maase H. Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: a phase I trial in metastatic melanoma. Clin Cancer Res 2006; 12:1229-36; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1485 [DOI] [PubMed] [Google Scholar]
- 16.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP et al.. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126-9; PMID:; http://dx.doi.org/ 10.1126/science.1129003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989; 86:10024-8; PMID:; http://dx.doi.org/ 10.1073/pnas.86.24.10024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, Temme A, Schmitz M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol 2010; 2010:956304; PMID:; http://dx.doi.org/ 10.1155/2010/956304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bubenik J. MHC class I down-regulation: tumour escape from immune surveillance? (review). Int J Oncol 2004; 25:487-91; PMID:; http://dx.doi.org/ 10.3892/ijo.25.2.487. [DOI] [PubMed] [Google Scholar]
- 20.Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today 1999; 5:178-86; PMID:; http://dx.doi.org/ 10.1016/S1357-4310(99)01451-3 [DOI] [PubMed] [Google Scholar]
- 21.Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S. Down-regulation of HLA class I antigen-processing molecules in malignant melanoma: association with disease progression. Am J Pathol 1999; 154:745-54; PMID:; http://dx.doi.org/ 10.1016/S0002-9440(10)65321-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rees RC, Mian S. Selective MHC expression in tumours modulates adaptive and innate antitumour responses. Cancer Immunol Immunother 1999; 48:374-81; PMID:; http://dx.doi.org/ 10.1007/s002620050589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabrera CM, Jimenez P, Cabrera T, Esparza C, Ruiz-Cabello F, Garrido F. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 2003; 61:211-9; PMID:; http://dx.doi.org/ 10.1034/j.1399-0039.2003.00020.x [DOI] [PubMed] [Google Scholar]
- 24.Johnsen AK, Templeton DJ, Sy M, Harding CV. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J Immunol 1999; 163:4224-31; PMID: . [PubMed] [Google Scholar]
- 25.Ritz U, Momburg F, Pilch H, Huber C, Maeurer MJ, Seliger B. Deficient expression of components of the MHC class I antigen processing machinery in human cervical carcinoma. Int J Oncol 2001; 19:1211-20; PMID:; http://dx.doi.org/ 10.3892/ijo.19.6.1211 [DOI] [PubMed] [Google Scholar]
- 26.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991; 64:891-901; PMID:; http://dx.doi.org/ 10.1016/0092-8674(91)90314-O [DOI] [PubMed] [Google Scholar]
- 27.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 1993; 90:720-4; PMID:; http://dx.doi.org/ 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L et al.. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006; 12:6106-15; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24:e20-2; PMID:; http://dx.doi.org/ 10.1200/JCO.2006.05.9964 [DOI] [PubMed] [Google Scholar]
- 30.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ et al.. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008; 112:2261-71; PMID:; http://dx.doi.org/ 10.1182/blood-2007-12-128843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 2002; 20:70-5; PMID:; http://dx.doi.org/ 10.1038/nbt0102-70 [DOI] [PubMed] [Google Scholar]
- 32.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther 2005; 12:933-41; PMID:; http://dx.doi.org/ 10.1016/j.ymthe.2005.04.016 [DOI] [PubMed] [Google Scholar]
- 33.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18:676-84; PMID:; http://dx.doi.org/ 10.1038/sj.leu.2403302 [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol 2009; 183:5563-74; PMID: ; http://dx.doi.org/ 10.4049/jimmunol.0900447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M et al.. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra38; PMID:; http://dx.doi.org/ 10.1126/scitranslmed.3005930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S et al.. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 2013; 122:2965-73; PMID:; http://dx.doi.org/ 10.1182/blood-2013-06-506741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725-33; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1103849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z et al.. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264-70; PMID:; http://dx.doi.org/ 10.1038/nm.1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H et al.. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 2012; 23:1043-53; PMID:; http://dx.doi.org/ 10.1089/hum.2012.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843-51; PMID:; http://dx.doi.org/ 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE 2nd, McLendon RE, Mitchell DA et al.. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010; 28:4722-9; PMID:; http://dx.doi.org/ 10.1200/JCO.2010.28.6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK et al.. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 2014; 20:972-84; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C et al.. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 2009; 6:e10; PMID:; http://dx.doi.org/ 10.1371/journal.pmed.1000010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingber DE. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol 2008; 18:356-64; PMID:; http://dx.doi.org/ 10.1016/j.semcancer.2008.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989; 339:58-61; PMID:; http://dx.doi.org/ 10.1038/339058a0 [DOI] [PubMed] [Google Scholar]
- 48.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986; 315:1650-9; PMID:; http://dx.doi.org/ 10.1056/NEJM198612253152606 [DOI] [PubMed] [Google Scholar]
- 49.van Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur J Cell Biol 2003; 82:539-48; PMID:; http://dx.doi.org/ 10.1078/0171-9335-00346 [DOI] [PubMed] [Google Scholar]
- 50.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev 2011; 25:2559-72; PMID:; http://dx.doi.org/ 10.1101/gad.169029.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat Med 1998; 4:627-30; PMID:; http://dx.doi.org/ 10.1038/nm0598-627 [DOI] [PubMed] [Google Scholar]
- 52.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res 2006; 66:7741-7; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3766 [DOI] [PubMed] [Google Scholar]
- 53.Halder RC, Aguilera C, Maricic I, Kumar V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest 2007; 117:2302-12; PMID:; http://dx.doi.org/ 10.1172/JCI31602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014-22; PMID:; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev 2010; 29:309-16; PMID:; http://dx.doi.org/ 10.1007/s10555-010-9223-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol 2011; 11:702-11; PMID:; http://dx.doi.org/ 10.1038/nri3064 [DOI] [PubMed] [Google Scholar]
- 57.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011; 11:393-410; PMID:; http://dx.doi.org/ 10.1038/nrc3064 [DOI] [PubMed] [Google Scholar]
- 58.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72:2162-71; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L et al.. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011; 475:226-30; PMID:; http://dx.doi.org/ 10.1038/nature10169 [DOI] [PubMed] [Google Scholar]
- 60.Arreygue-Garcia NA, Daneri-Navarro A, del Toro-Arreola A, Cid-Arregui A, Gonzalez-Ramella O, Jave-Suarez LF, Aguilar-Lemarroy A, Troyo-Sanroman R, Bravo-Cuellar A, Delgado-Rizo V et al.. Augmented serum level of major histocompatibility complex class I-related chain A (MICA) protein and reduced NKG2D expression on NK and T cells in patients with cervical cancer and precursor lesions. BMC Cancer 2008; 8:16; PMID:; http://dx.doi.org/ 10.1186/1471-2407-8-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 2003; 3:781-90; PMID:; http://dx.doi.org/ 10.1038/nri1199 [DOI] [PubMed] [Google Scholar]
- 62.Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, Graham CH. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res 2008; 68:4746-53; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0054 [DOI] [PubMed] [Google Scholar]
- 63.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 2014; 74:665-74; PMID:; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0992 [DOI] [PubMed] [Google Scholar]
- 64.Li MC, Hertz R, Bergenstal DM. Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med 1958; 259:66-74; PMID:; http://dx.doi.org/ 10.1056/NEJM195807102590204 [DOI] [PubMed] [Google Scholar]
- 65.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol 2004; 4:336-47; PMID:; http://dx.doi.org/ 10.1038/nri1349 [DOI] [PubMed] [Google Scholar]
- 66.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer 2007; 7:95-106; PMID:; http://dx.doi.org/ 10.1038/nrc2051 [DOI] [PubMed] [Google Scholar]
- 67.Sheridan C. TeGenero fiasco prompts regulatory rethink. Nat Biotechnol 2006; 24:475-6; PMID:; http://dx.doi.org/ 10.1038/nbt0506-475 [DOI] [PubMed] [Google Scholar]
- 68.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355:1018-28; PMID:; http://dx.doi.org/ 10.1056/NEJMoa063842 [DOI] [PubMed] [Google Scholar]
- 69.Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE et al.. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 2005; 23:6043-53; PMID:; http://dx.doi.org/ 10.1200/JCO.2005.06.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fischkoff SA, Hersh E, Weber J, Powderly J, Khan K, Pavlick A, Samlowski W, O'Day S, Nichol G, Yellin M. Durable responses and long-term progression-free survival observed in a phase II study of MOX-010 alone or in combination with dacarbazine (DTIC) in metastatic melanoma. J Clin Oncol 2005; 23:716s. [Google Scholar]
- 71.Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Haworth LR, Levy C et al.. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol 2005; 12:1005-16; PMID:; http://dx.doi.org/ 10.1245/ASO.2005.03.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bafaloukos D, Gogas H. The treatment of brain metastases in melanoma patients. Cancer Treat Rev 2004; 30:515-20; PMID:; http://dx.doi.org/ 10.1016/j.ctrv.2004.05.001 [DOI] [PubMed] [Google Scholar]
- 73.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mandel JJ, Olar A, Aldape KD, Tremont-Lukats IW. Lambrolizumab induced central nervous system (CNS) toxicity. J Neurol Sci 2014; 344:229-31; PMID:; http://dx.doi.org/ 10.1016/j.jns.2014.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glennie MJ, McBride HM, Worth AT, Stevenson GT. Preparation and performance of bispecific F(ab' gamma)2 antibody containing thioether-linked Fab' gamma fragments. J Immunol 1987; 139:2367-75. [PubMed] [Google Scholar]
- 76.Kriangkum J, Xu B, Nagata LP, Fulton RE, Suresh MR. Bispecific and bifunctional single chain recombinant antibodies. Biomol Eng 2001; 18:31-40; PMID:; http://dx.doi.org/ 10.1016/S1389-0344(01)00083-1 [DOI] [PubMed] [Google Scholar]
- 77.Holliger P, Prospero T, Winter G. “Diabodies:” small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A 1993; 90:6444-8; PMID:; http://dx.doi.org/ 10.1073/pnas.90.14.6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Atwell JL, Pearce LA, Lah M, Gruen LC, Kortt AA, Hudson PJ. Design and expression of a stable bispecific scFv dimer with affinity for both glycophorin and N9 neuraminidase. Mol Immunol 1996; 33:1301-12; PMID:; http://dx.doi.org/ 10.1016/S0161-5890(96)00097-1 [DOI] [PubMed] [Google Scholar]
- 79.De Jonge J, Brissinck J, Heirman C, Demanet C, Leo O, Moser M, Thielemans K. Production and characterization of bispecific single-chain antibody fragments. Mol Immunol 1995; 32:1405-12; PMID:; http://dx.doi.org/ 10.1016/0161-5890(95)00089-5 [DOI] [PubMed] [Google Scholar]
- 80.Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A 1995; 92:7021-5; PMID:; http://dx.doi.org/ 10.1073/pnas.92.15.7021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today 2005; 10:1237-44; PMID:; http://dx.doi.org/ 10.1016/S1359-6446(05)03554-3 [DOI] [PubMed] [Google Scholar]
- 82.Choi BD, Cai M, Bigner DD, Mehta AI, Kuan CT, Sampson JH. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther 2011; 11:843-53; PMID:; http://dx.doi.org/ 10.1517/14712598.2011.572874 [DOI] [PubMed] [Google Scholar]
- 83.Dustin ML, Shaw AS. Costimulation: building an immunological synapse. Science 1999; 283:649-50; PMID:; http://dx.doi.org/ 10.1126/science.283.5402.649 [DOI] [PubMed] [Google Scholar]
- 84.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001; 15:751-61; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(01)00234-5 [DOI] [PubMed] [Google Scholar]
- 85.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol 2006; 43:763-71; PMID:; http://dx.doi.org/ 10.1016/j.molimm.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 86.Mack M, Gruber R, Schmidt S, Riethmuller G, Kufer P. Biologic properties of a bispecific single-chain antibody directed against 17-1A (EpCAM) and CD3: tumor cell-dependent T cell stimulation and cytotoxic activity. J Immunol 1997; 158:3965-70; PMID: . [PubMed] [Google Scholar]
- 87.Choi BD, Kuan CT, Cai M, Archer GE, Mitchell DA, Gedeon PC, Sanchez-Perez L, Pastan I, Bigner DD, Sampson JH. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci U S A 2013; 110:270-5; PMID:; http://dx.doi.org/ 10.1073/pnas.1219817110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brandl C, Haas C, d'Argouges S, Fisch T, Kufer P, Brischwein K, Prang N, Bargou R, Suzich J, Baeuerle PA, et al.. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother 2007; 56:1551-63; PMID:; http://dx.doi.org/ 10.1007/s00262-007-0298-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther 2012; 136:334-42; PMID:; http://dx.doi.org/ 10.1016/j.pharmthera.2012.07.013 [DOI] [PubMed] [Google Scholar]
- 90.Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, Kufer P, Riethmuller G, Bargou R, Baeuerle PA. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer 2002; 100:690-7; PMID:; http://dx.doi.org/ 10.1002/ijc.10557 [DOI] [PubMed] [Google Scholar]
- 91.Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol Cell Biol 1999; 19:8604-15; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haas C, Krinner E, Brischwein K, Hoffmann P, Lutterbuse R, Schlereth B, Kufer P, Baeuerle PA. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology 2009; 214:441-53; PMID:; http://dx.doi.org/ 10.1016/j.imbio.2008.11.014 [DOI] [PubMed] [Google Scholar]
- 93.Gruen M, Bommert K, Bargou RC. T-cell-mediated lysis of B cells induced by a CD19xCD3 bispecific single-chain antibody is perforin dependent and death receptor independent. Cancer Immunol Immunother 2004; 53:625-32; PMID:; http://dx.doi.org/ 10.1007/s00262-003-0496-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi BD, Gedeon PC, Sanchez-Perez L, Bigner DD, Sampson JH. Regulatory T cells are redirected to kill glioblastoma by an EGFRvIII-targeted bispecific antibody. Oncoimmunology 2013; 2:e26757; PMID:; http://dx.doi.org/ 10.4161/onci.26757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choi BD, Gedeon PC, Herndon JE 2nd, Archer GE, Reap EA, Sanchez-Perez L, Mitchell DA, Bigner DD, Sampson JH. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol Res 2013; 1:163; PMID:; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, Locher M, Hammond SA, Kiener P, Kufer P et al.. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J immunother 2007; 30:798-807; PMID:; http://dx.doi.org/ 10.1097/CJI.0b013e318156750c [DOI] [PubMed] [Google Scholar]
- 97.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, et al.. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol 2006; 43:1129-43; PMID:; http://dx.doi.org/ 10.1016/j.molimm.2005.07.034 [DOI] [PubMed] [Google Scholar]
- 98.Nah JH, Kim HJ, Lee HN, Lee MJ, Choi SS, Kim ES. Identification and biotechnological application of novel regulatory genes involved in Streptomyces polyketide overproduction through reverse engineering strategy. Biomed Res Int 2013; 2013:549737; PMID:; http://dx.doi.org/ 10.1155/2013/549737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther 2014; 22:102-11; PMID:; http://dx.doi.org/ 10.1038/mt.2013.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res 2011; 317:1255-60; PMID:; http://dx.doi.org/ 10.1016/j.yexcr.2011.03.010 [DOI] [PubMed] [Google Scholar]
- 101.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M et al.. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974-7; PMID:; http://dx.doi.org/ 10.1126/science.1158545 [DOI] [PubMed] [Google Scholar]
- 102.Schlegel P, Lang P, Zugmaier G, Ebinger M, Kreyenberg H, Witte KE, Feucht J, Pfeiffer M, Teltschik HM, Kyzirakos C et al.. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014; 99:1212-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong R, Pepper C, Brennan P, Nagorsen D, Man S, Fegan C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica 2013; 98:1930-8; PMID:; http://dx.doi.org/ 10.3324/haematol.2012.082248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK et al.. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood 1997; 90:2188-95; PMID: [PubMed] [Google Scholar]
- 105.Hudis CA. Trastuzumab-mechanism of action and use in clinical practice. N Engl J Med 2007; 357:39-51; PMID:; http://dx.doi.org/ 10.1056/NEJMra043186 [DOI] [PubMed] [Google Scholar]
- 106.Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ et al.. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040-8; PMID:; http://dx.doi.org/ 10.1056/NEJMoa071834 [DOI] [PubMed] [Google Scholar]
- 107.Aggarwal RS. What's fueling the biotech engine-2012 to 2013. Nat Biotechnol 2014; 32:32-9; PMID:; http://dx.doi.org/ 10.1038/nbt.2794 [DOI] [PubMed] [Google Scholar]
- 108.Baeuerle PA, Kufer P, Bargou R. BiTE: teaching antibodies to engage T-cells for cancer therapy. Curr Opin Mol Ther 2009; 11:22-30; PMID: [PubMed] [Google Scholar]
- 109.Topp MGN, Kufer P et al.. Treatment with anti-CD19 BiTE antibody blinatumomab (MT103/MEDI-538) is able to eliminate minimal residual disease (MRD) in patients with B-precursor acute lmphoblastic leukemia (ALL): First results of ongoing phase 2 study ASH Annual Meeting: Blood, 2008:1926. [Google Scholar]
- 110.Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, Ruegg C, Dietrich PY, Walker PR. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 2005; 22:175-84; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 111.Gedeon PC, Choi BD, Hodges TR, Mitchell DA, Bigner DD, Sampson JH. An EGFRvIII-targeted bispecific T-cell engager overcomes limitations of the standard of care for glioblastoma. Expert Rev Clin Pharmacol 2013; 6:375-86; PMID:; http://dx.doi.org/ 10.1586/17512433.2013.811806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Choi BD, Pastan I, Bigner DD, Sampson JH. A novel bispecific antibody recruits T cells to eradicate tumors in the “immunologically privileged” central nervous system. Oncoimmunology 2013; 2:e23639; PMID:; http://dx.doi.org/ 10.4161/onci.23639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, Sampson JH. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol 2009; 19:713-23; PMID:; http://dx.doi.org/ 10.1111/j.1750-3639.2009.00318.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Congdon KL, Gedeon PC, Suryadevara CM, Caruso HG, Cooper LJ, Heimberger AB, Sampson JH. Epidermal growth factor receptor and variant III targeted immunotherapy. Neuro-oncol 2014; 16:viii20-viii5; PMID:; http://dx.doi.org/ 10.1093/neuonc/nou236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gedeon PC, Riccione KA, Fecci PE, Sampson JH. Antibody-based immunotherapy for malignant glioma. Seminn Oncol 2014; 41:496-510; PMID:; http://dx.doi.org/ 10.1053/j.seminoncol.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]