

Abstract
The graft-versus-leukemia (GVL) effect following allogeneic hematopoietic stem cell transplantation (allo-HCT) is critical for its curative potential. Hwever, GVL is tightly linked to graft-versus-host disease (GVHD). Among hematological malignancies, acute lymphoblastic leukemia (ALL) is the most resistant to GVL, although the reasons for this remain poorly understood. Clinical studies have identified alterations in Ikaros (Ik) transcription factor as the major marker associated with poor outcomes in ALL. We have shown that the absence of Ik in professional host-derived hematopoietic antigen-presenting cells (APCs) exacerbates GVHD. However, whether Ik expression plays a role in resistance to GVL is not known. In this study we used multiple clinically relevant murine models of allo-HCT to explore whether Ik expression in hematopoietic APCs and/or leukemic cells is critical for increasing resistance to GVL and thus inducing relapse. We found that Ik deficiency in host APCs failed to enhance GVL despite increased GVHD severity. Mechanistic studies with bone marrow (BM) chimeras and tetramer analyses demonstrated reduced tumor-specific immunodominant (gag+) antigen responses in the [B6Ik−/−→B6] group. Loss of GVL was observed when both the leukemia cells and the host APCs were deficient in Ik. We found that calreticulin (CRT) expression in host antigen-presenting dendritic cells (DCs) of Ik−/− animals was significantly lower than in wild-type animals. Rescuing CRT expression in Ik−/− DCs improved leukemic-specific cytotoxic T cell function. Together, our data demonstrate that the absence of Ikaros in host hematopoietic cells promotes resistance to GVL despite increasing GVHD and thus provides a potential mechanism for the poor outcome of Ik−/− ALL patients.
Keywords: antigen-presenting cells, bone marrow transplantation, graft-versus-leukemia, Ikaros
Abbreviations: 51Cr, Chromium-51; ALL, acute lymphoblastic leukemia; Allo-HCT, allogeneic hematopoietic stem cell transplantation; APC, allophycocyanin; APCs, antigen-presenting cells; BC, blast crisis; BLI, bioluminescence imaging; BM, bone marrow; BMDCs, bone marrow derived dendritic cells; BMT, bone marrow transplantation; CML, chronic myeloid leukemia; CRT, calreticulin; CTL, cytotoxic T cell; DCs, dendritic cells; FACS, Fluorescence-activated cell sorting; FBS, fatal bovine serum; FITC, fluorescein isothiocyanate; GVHD, graft-versus-host-disease; GVL, graft-versus-leukemia; HCT, hematopoietic stem cell transplantation; ICAM-1, intracellular adhesion molecule 1; Ik DN, Ikaros dominant negative; Ik, Ikaros; luc+, luciferase+; mAbs, monoclonal antibodies; MACS, magnetic- activated cell sorting; MBL-2, moloney-murine sarcoma virus-induced MBL-2 lymphoma cells; mCRT, murine calreticulin; MHC, major histocompatibility complex; MiHAs, multiple minor histocompatibility antigens; MLR, mixed lymphocyte reaction; PBS, phosphate buffered saline; PE, phycoerythrin; SIRP-α, signal regulatory protein α; TCD-BM, T cell depleted bone marrow; Tregs, regulatory T cells; TSA, tumor specific antigen; UCUCA, University Committee on Use and Care of Animals; WT, wild-type
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HCT) is the only curative therapy for many hematologic malignancies. The critical factor for success of allo-HCT depends upon the degree of the graft-versus-leukemia (GVL) effect. Primary disease relapse is one of the major complications and a leading cause of death following allo-HCT.1,2 Although GVL is important for curative therapy, GVL is also tightly linked with graft-versus-host disease (GVHD), which is another major complication of allo-HCT.3 Thus, attempts to increase GVL are associated with exacerbating GVHD and uncoupling the beneficial benefit of GVL from the toxic effects of GVHD is imperative to improve curative therapy. Among hematologic malignancies acute lymphoblastic leukemia (ALL) is most resistant to GVL,4 the reasons for which remain poorly understood. We have previously demonstrated that professional host-derived hematopoietic antigen-presenting cells (APCs) are necessary for the induction of robust GVL.5,6 However, the cell autonomous molecular mechanisms of APCs that are critical for mediating GVL are poorly understood.
We have recently shown that Ikaros (Ik) deficiency in host hematopoietic APCs exacerbates GVHD.7 Ik is a transcription factor known to be important for the development of lymphoid cells8,9 and for the leukemogenesis of certain hematologic malignancies, such as ALL.10 Clinical data have demonstrated that the prognosis of ALL patients with Ik deficiency or mutation is extremely poor.11,12 Our recent study revealed that Ik deficiency in host hematopoietic APCs exacerbates acute GVHD in a Notch-dependent manner.7 Therefore, the Ik–Notch axis in host APCs is an important pathway for acute GVHD. Paradoxically, despite increasing GVHD in experimental allogeneic hematopoietic stem cell transplantation (allo-HCT), clinical data suggest that Ik deficiency or mutation in ALL patients is associated with a high risk for relapse, i.e., decreased GVL. Whether Ik plays an important role in mediating this GVL resistance by its expression in the host hematopoietic cells is not known. Here, we investigated the impact of Ik in host hematopoietic APCs and leukemic cells on GVL. We show that deficiency of Ik in host hematopoietic APCs did not increase GVL despite increased GVHD severity. Mechanistic studies demonstrated that the loss of GVL was more pronounced when both the leukemia cells and the host hematopoietic-derived APCs were deficient in Ik. Absence of Ik in host-derived APCs also decreased T-cell responses to tumor-specific antigen (TSA). Calreticulin (CRT) is expressed on the surface of APCs13 and is associated with phagocytic function14 and antigen presentation via MHC class I molecules.15 Therefore, CRT expression on APCs plays an important role in regulating immune responses. We found that CRT expression in dendritic cells (DCs) of Ik−/− animals is significantly lower than that in WT mice and that forced expression of CRT in Ik−/− DCs enhanced tumor-specific cytotoxic T cell (CTL) function. Together, our data demonstrate that the absence of Ikaros in host hematopoietic cells increased GVHD but decreased GVL and thus provide a potential mechanism for the increased relapse in Ik−/− ALL after clinical allo-HCT.
Results
Ikaros deficiency in host hematopoietic cells does not increase GVL despite increasing GVHD
We recently reported that Ik deficiency in host hematopoietic APCs exacerbates acute GVHD in a Notch-dependent manner after experimental allo-HCT.7 In light of clinical observations that deficiency or mutation of Ik in ALL increases relapse,11,12 in the present study we explored whether Ik deficiency in host hematopoietic APCs also affects GVL responses. We first generated BM chimeras to evaluate the impact of Ik deficiency only in host hematopoietic APCs, without the confounding effects of Ik deficiency in either leukemia cells or in non-hematopoietic cell-derived APCs. Wild-type (WT) B6 Ly5.2 animals were lethally irradiated with 11 Gy and infused with 5 × 106 bone marrow (BM) cells and 5 × 106 splenocytes from syngeneic B6Ly5.1 WT or B6 Ik−/− donors. The [B6→B6Ly5.2] or [Ik−/−→B6Ly5.2] animals were then used as allo-HCT recipients 4 months after primary HCT. To test GVL responses, the chimeras received 9 Gy irradiation and were injected intravenously with 0.5 × 106 CD8+ T cells together with 5 × 106 BM cells from either syngeneic B6 or major histocompatibility (MHC) antigen-matched but multiple minor histocompatibility antigens (MiHA)-mismatched allogeneic C3H.SW donors. The model that we used for the experiments is a well-established donor CD8+ T-cell mediated murine GVHD model akin to most commonly performed clinical allogeneic bone marrow transplants (BMTs).5,16 Furthermore, syngeneic moloney-murine sarcoma virus-induced lymphoma (MBL-2) cells were injected concurrently with HCT. The tumors that we used express MHC class I but not class II and thus are susceptible only to CD8−mediated antitumor cytotoxicity. Therefore, in these experiments we analyzed only CD8+ T-cell responses that cause both GVHD and GVL in these models. Consistent with previous observations, Ik deficiency in host APCs exacerbated GVHD (Fig. 1A and as shown in our previous report7). However, despite increasing GVHD, there was no statistical difference in relapse mortality of the WT [B6→B6Ly5.2] and [Ik−/−→B6Ly5.2] animals that received allogeneic T cells with MBL-2 tumor cells (Fig. 1B). The lack of increase in GVL was also observed at a lower dose (5 × 103 MBL-2 cells/mouse, Fig. 1C). To further confirm the effect on GVL, MBL-2 cells were transduced with lentivirus that contained a GFP and luciferase (luc+) construct to facilitate in vivo monitoring of the tumor burden by bioluminescence imaging (BLI) after allo-HCT. As shown in Fig. 1D, although the syngeneic WT [B6→B6Ly5.2] animals showed somewhat less tumor signal because of spinal cord infiltration, we confirmed that both allogeneic WT [B6→B6Ly5.2] and [Ik−/−→B6Ly5.2] animals showed similar tumor growth kinetics and succumbed to the tumor burden even though the [Ik−/−→B6Ly5.2] animals showed a greater severity of GVHD. Some syngeneic WT [B6→B6Ly5.2] animals showed hind-limb paralysis. To further rule out tumor and model artifacts, we used a different tumor model system and found a similar relapse mortality in other GVL with EL-4 tumor cells (1 × 104 cells/mouse; Fig. 1E).
Figure 1.

Ikaros deficiency in host APCs does not increase GVL responses regardless of enhanced GVHD in experimental HCT. WT B6 Ly5.2 animals were lethally irradiated with 11 Gy and infused with 5 × 106 bone marrow (BM) cells and 5 × 106 splenocytes from syngeneic Ly5.1 WT B6 or Ik−/− B6 donors. Four months later these [B6→B6Ly5.2] or [Ik−/−→B6Ly5.2] chimeras received 9 Gy irradiation and 1 × 106 CD90+ T cells together with 5 × 106 BM cells from either syngeneic B6 or allogeneic MHC-matched or multiple miHA-mismatched C3H.sw donors concurrently with syngeneic MBL-2 tumor at the same time as allo-HCT. (A) Overall survival data of GVHD study. (•) B6→[B6→B6Ly5.2], (▴) B6→[Ik−/−→B6Ly5.2], (Ë) C3H.sw→[B6→B6Ly5.2], (–)C3H.sw→[Ik−/−→B6Ly5.2]. Data shown are one representative dataset (n=3–5/each group) of 5 independent experiments. (B, C) Tumor mortality data for MBL-2 at 10,000 cells/mouse (n=10–18/group) (B) and 5,000 cells/mouse (n=4–10/group). (C) (•) B6→[B6→B6Ly5.2], (▴) B6→[Ik−/−→B6Ly5.2], (Ë) C3H.sw→[B6→B6Ly5.2], (–)C3H.sw→[Ik−/−→B6Ly5.2]. Data are combined from 2 or 3 independent experiments. (D) Tumor growth was monitored using bioluminescence imaging (BLI) after allo-HCT (n=2–5). Representative data from 3 independent experiments are shown. (E) Tumor mortality data of the same model using a different syngeneic tumor, EL-4 (10,000 cells/mouse, n=3–12/group). (•) B6→[B6→B6Ly5.2], (Ë) C3H.sw→[B6→B6Ly5.2], (–) C3H.sw→[Ik−/−→B6Ly5.2]. Data are combined from 2 independent experiments.
Ikaros deficiency increases overall donor T-cell responses but not those directed against tumor-specific antigen
To determine the mechanisms underlying the comparable GVL effect despite significant differences in GVHD, we tested the expansion of total donor T cells and only T cells specific for tumor specific antigen (TSA). We used tetramer strategies of donor CD8+ T cells to detect MBL-2 TSA gag-specific T-cell expansion.6 Using a similar model, we analyzed the expansion of gag-specific CD8+ donor T cells on day 21 following allo-HCT. We found that Ik deficiency in APCs caused greater allogeneic T-cell expansion (Fig. 2A) but similar TSA responses (Fig. 2B). To further assess the functionality of the TSA-specific donor T cells, we examined the cytotoxicity of donor T cells against MBL-2 tumor. We found similar expression of granzyme B, perforin, and CD107a (Fig. 2C–E) in donor T cells 14 d after allo-BMT in both groups. However, when specifically tested for their ability to kill tumor cells using the51Cr release assay we found a reduced cytotoxicity of splenic donor T cells against MBL-2 in both groups on day 21 (Fig. 2F). These data demonstrated that Ik deficiency in host hematopoietic APCs enhances overall donor T-cell responses but does not concomitantly increase TSA-specific responses.
Figure 2.

GVL response of host antigen-presenting cells with Ikaros deficiency is equivalent to that of WT cells. Donor T cells were isolated from spleen (n = 3–4) at day 14 following allo-HCT and analyzed for (A) donor CD8+ T-cell expansion (n = 3–4), (B) donor-derived MBL-2 specific gag+CD8+ T-cell expansion (n = 3–4), and expression of (C) granzyme B, (D) perforin, and (E) CD107a on donor CD8+ T cells (n = 3–4). (F)51Cr-release assay using donor CD8+ T cells at day 14 after allo-HCT against MBL-2 tumors. One representative dataset from 3 independent experiments is shown. Data are given as mean + standard deviation.
Expression of antigen-presenting molecules on CD11c+ DCs in spleen
To evaluate the mechanism underlying the lack of increasing GVL responses despite the exacerbated GVHD between Ik and WT chimeras, we next determined the effect of Ik deficiency on the expression of antigen-presenting molecules on DCs. We found that only CD205 expression was significantly higher in Ik-deficient animals than in WT; the expression of other antigen-presenting molecules, such as signal regulatory protein (SIRP)-α, CD209, CD54/intercellular adhesion molecule 1 (ICAM1), CD47, and CD24 in the Ik-deficient animals was similar to that in WT animals (Fig. 3A, B).
Figure 3.

Expression of antigen-presenting molecules in splenic dendritic cells. The expression of antigen-presenting molecules (Sirp-α, CD209, CD205, CD54, CD27, and CD24) on CD11c+cells in the spleen was analyzed for naive Ik−/− and WT B6 animals that were not transplanted (n = 5–7/group). (A) Representative histogram. (B) Frequency of these antigen-presenting molecules on CD11c+ DCs.
We recently demonstrated that TSA cross-presentation on professional hematopoietic APCs is required for optimal GVL responses.6 Therefore, to determine whether the decreased TSA response is due to defects in cross-presentation on Ik−/− APCs, we examined the expression of CRT, which plays an important role in regulating phagocytic function and regulating antigen cross-presentation of TSA.17 We first hypothesized that CRT expression would be decreased in Ik−/− APCs. We examined the expression of CRT in spleen DCs isolated from either B6 WT or Ik−/− animals and found reduced expression of CRT in Ik−/− DCs (Fig. 4A–B). To determine whether the reduction of CRT in Ik−/− DCs was critical for the lack of CTL responses against TSA, mCRT was transduced into BM-derived DCs from Ik−/− animals and CTL function was tested with a51Cr release assay. We found that Ik−/− DCs with restored expression of CRT exhibited completely recovered CTL function (Fig. 4C). These data suggested that Ikaros in DCs might regulate CTL function in a CRT-dependent manner.
Figure 4.
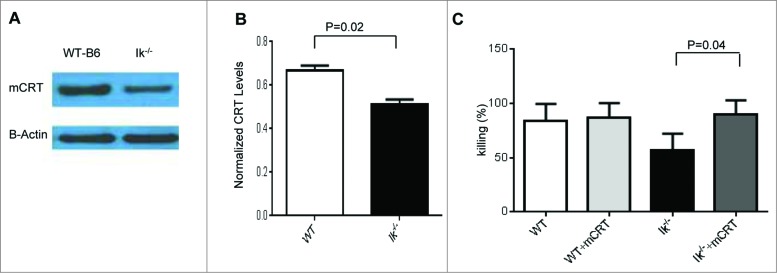
Calreticulin expression in dendritic cells from Ik−/− animals and forced expression-enhanced CTL responses. (A, B) Spleen CD11c+ DCs were isolated from either B6-WT or Ik−/− animals using CD11c-micobeads and calreticulin expression was measured by western blotting. (A) The absence of Ik decreased expression of CRT. (B) Normalized CRT level relative to actin expression. (C) BMDCs were transduced with CRT and empty vector control and used as stimulators for generating CTLs in bulk mixed-lymphocyte reaction (MLR). These generated CTLs were used as effectors against MBL-2 tumors for the51Cr release assay.
Ikaros deficiency in both hematopoietic APCs and leukemia cells contributes to lack of an optimal GVL response
Finally we determined whether absence of Ik only in the leukemic cells, and not in professional hematopoietic APCs, contributes to GVL resistance. To this end, we used the same BMT model but added p185 (Ik wild-type tumors), or Ik6 (Ik dominant negative tumors) at the time of BMT. Similar to the MBL-2 tumor model, allogeneic [Ik−/−→B6Ly5.2] animals showed greater GVHD without concomitant GVL responses compared to allogeneic WT[B6→Ly5.2] animals when they received p185 tumors (Fig. 5A). Allogeneic T cells co-transferred with p185 from allogeneic [Ik−/−→B6Ly5.2] animals on day 14 after allo-HCT demonstrated comparable cytotoxicity against p185 to that of allogeneic WT[B6→Ly5.2] animals (Fig. 5B). Conversely, allogeneic WT [B6→Ly5.2] chimeras that received Ik6 showed better GVL responses than allogeneic [Ik−/−→B6Ly5.2] animals (Fig. 5C). Allogeneic T cells co-transferred with Ik6 also effectuated greater apoptosis in T-cell cytotoxic killing assays (Fig. 5D). These data suggest that Ik deficiency in both non-leukemic and leukemic cells collectively contributes to GVL resistance (Table 1).
Figure 5.

Ikaros deficiency in both hematopoietic APCs and leukemia cells ameliorates GVL responses. WT B6Ly5.2 animals were lethally irradiated with 11 Gy and infused with 5 × 106 BM cells and 5 × 106 splenocytes from syngeneic Ly5.1 WT B6 or Ik−/− B6 donors. These animals [B6→B6Ly5.2] or [IkDN+/−→B6Ly5.2]) were used as recipients 4 months later. Mice were irradiated with 9 Gy and transplanted with 1 × 106 CD90+ T cells together with 5 × 106 BM cells from either syngeneic B6 or allogeneic MHC-matched or multiple miHA-mismatched C3H.sw donors concurrently with syngeneic P185 (Ik WT tumor) or Ik6 (Ik DN tumor). (A) Tumor mortality data for P185 200/mouse. (•) B6→[B6→B6Ly5.2] (n = 8), (Ë) C3H.sw→[B6→B6 Ly5.2] (n = 16), (–) C3H.sw→[Ik−/−→B6Ly5.2] (n = 14). Data are combined from 3 independent experiments. (B) CTL assay. Donor CD8+ T cells were isolated from spleen (n = 3–4) at day 14 following allo-HCT and used as effector T cells against P185 tumor for51Cr-release assay. One representative dataset from 3 independent experiments is shown. Data are given as mean + standard deviation. (C) Tumor mortality data of Ik6 200/mouse. (•) B6→[B6→B6Ly5.2] (n=8), (Ë) C3H.sw→[B6→B6 Ly5.2] (n = 16), (–) C3H.sw→[Ik−/−→B6Ly5.2] (n = 15). Data are combined from 3 independent experiments. (D) CTL assay. Donor CD8+ T cells were isolated from spleen (n = 3–4) at day 14 following allo-HCT and used as effector T cells against Ik6 tumor for51Cr-release assay. One representative dataset from 3 independent experiments is shown. Data are given as mean + standard deviation.
Table 1.
Summary of GVL experiments. WT and Ik-deficient host APCs showed an equivalent GVL effect for P185, Ik-WT tumor. However, Ik deficiency in both hematopoietic APCs and leukemia cells ameliorates GVL responses
| APC/Tumor | P185 (Ik WT) | Ik6 (Ik DN mutation) |
|---|---|---|
| WT APC | ++ | ++ |
| Ik−/− APC | ++ | + |
Discussion
Relapse of primary disease and GVHD are the greatest obstacles to improving the long-term outcome after allo-HCT.3 The GVL response is necessary to prevent relapse; however, GVL is tightly linked with GVHD therefore increasing GVL comes at the cost of exacerbating GVHD. Thus, separating GVL from GVHD remains the central issue in allo-HCT. Clinical data suggest that certain diseases such as ALL demonstrate greater relapse rates after allo-HCT, i.e., decreased GVL despite that fact that the patients suffer from severe GVHD. The reason for the reduced GVL in ALL could be multifactorial. Moreover, whether the decrease in GVL is due to defects in antigen presentation in addition to leukemia-intrinsic properties remains unknown. Here, we explored whether Ik deficiency in host hematopoietic cells contributes to GVL resistance. Although both host and donor APCs, specifically DCs as they are the most potent APCs, are important regulators of GVHD,16,18,19 the role of APCs in GVL is poorly understood. We have recently demonstrated that cross-presentation on professional hematopoietic APCs is critical for optimal GVL responses.5,6 Evidence suggests that professional APCs play an important role in generating effective CTLs for elimination of recipient tumor cells.20,21 Donor T cells that recognize alloantigens are critical for mediating GVHD and GVL and tumor-specific antigens also contribute to GVL. Recent data suggest that DCs may play an essential role in inducing GVL.5,6 We have shown that Batf3−/− recipients that exhibit decreased numbers of CD8α+ DCs have ameliorated GVL in a MHC-matched multiple minor mismatched BMT model.6 However, the molecular mechanisms that regulate cell-autonomous hematopoietic APC-mediated effects on GVL remain poorly understood.
Our previous data suggest that deficiency of Ik in host APCs aggravated the severity of GVHD in a Notch-dependent manner.7 Herein, we demonstrated that the absence of Ik in host APCs did not concomitantly enhance GVL, irrespective of severe GVHD, in [Ik−/−→B6 Ly5.2] animals of the MHC-matched multiple MiHA mismatched C3H.sw→B6 model. We also used a different tumor (EL-4) and confirmed that Ik-deficient host APCs demonstrated equivalent GVL responses to WT chimeras. GVT responses are usually tightly linked with GVHD severity. Thus, this uncoupling of GVHD and GVL at the level of antigen presentation is a novel observation.
Host MHC class II+ APCs play a critical role in CTL responses in mixed chimera models.22 However, the role of APC subsets and their intrinsic molecular mechanisms in GVL are poorly understood. Using Batf3−/− animals, we recently found that host CD8+ DCs play an important role in mediating optimal GVL responses. We also found that host APCs stimulated by TLR3 agonist enhanced the GVL effect without concomitant aggravation of GVHD.6 It was also recently shown that absence of the Ik–Notch pathway in host APCs increased GVHD.7 These data suggest that host type APCs are required for optimal GVL responses following allo-HCT. These observations, together with our current findings, indicate that Ik expression in host APCs is a critical regulator of GVHD and GVL. We found that Ik−/− DCs highly expressed CD205 (DEC205), which plays an important role in antigen capture and presentation23 and is predominantly expressed in CD8α+ DCs.24 CD8+CD205+ DCs also regulate T-cell immunity and homeostasis by increasing the generation of regulatory T cells (Tregs).25 Enhancement of CD205 function in DCs with tumor antigen-specific vaccine increased tumor-specific CTLs and resulted in tumor regression in some patients with solid tumors, such as melanoma.26 On the other hand, the expression of antigen-presenting related receptors other than CD205 in Ik−/− DCs was comparable to that of WT DCs. Our previous data also demonstrated that expression of co-stimulatory molecules such as CD80, CD86, CD40, CD83, and PDL1 was much lower in splenic Ik−/− DCs compared with WT DCs,7, suggesting that enhancing allo-stimulatory function of Ik−/− DCs contributes to GVHD but does not increase GVL is not in receptors including CD205, and co-stimulation molecules dependent manner. The effects of Ik deficiency are not dependent on CD205 and the co-stimulatory molecules.
To further explore why Ik-deficient non-leukemic hematopoietic APCs cannot enhance GVL in spite of greater GVH responses, we focused on calreticulin expression in DCs. DCs express CRT on their surface13,14,27 and this CRT expression facilitates TSA cross-presentation via MHC class I molecules to cytotoxic T cells.15,27-30 CRT plays an important role in protein folding and the maintenance of MHC class I assembly pathways,31,32 and CRT expression on the cell surface enhances cellular phagocytic uptake.14 We examined CRT expression on both WT and Ik−/− BMDCs by western blotting and found that Ik−/− BMDCs showed lower CRT expression than WT cells (Fig. 4A-B). Therefore, we next examined whether forced increase in CRT expression by gene transduction rescues CTL function in Ik−/− BMDCs and found a significant increase in cytotoxic killing by T cells primed by CRT transduced Ik−/− BMDCs, compared with WT (Fig. 4C). Therefore, the mechanism underlying the lack of concomitant increased TSA expression may be related to a lack of increasing cross-presentation capacity as a result of a deficiency on CRT expression. Future studies will determine the direct mechanism of the molecular regulation of CRT by Ik and explore the intrinsic mechanisms by which Ik in host hematopoietic-derived APCs separately regulates GVHD and GVL, as well as whether other molecular mechanisms such as Notch-dependent signaling play an important role in GVL responses.
Our observations provide novel insights into clinical observations that are particularly germane to ALL. Among all leukemias, ALL with Ik deficiency or mutation has a poor outcome and is at high risk of relapse after allo-HCT.33 Although the lack of sufficient GVL in these patients is relatively well established, available evidence suggests that these patients have a similar incidence and severity of GVHD.
Ik is not only an essential transcription factor as a tumor suppressor for leukemogenesis, such as in ALL10,34 and for blast crisis (BC) of chronic myeloid leukemia (CML),35 but Ik (Ikzf1) deficiency and mutation in leukemic cells is also associated with a poor prognosis in ALL.11,12,36,37 However, whether Ik deficiency in leukemic cells alone contributes to resistance, i.e., by increasing relapse, has not been explored. In our study using Ik DN tumor cell lines (Ik6), we determined whether the absence of Ik in leukemic cells makes them more resistant to GVL, or whether the resistance is due to primarily to its absence in the non-leukemic host hematopoietic cells. We found that when allogeneic [Ik−/−→B6Ly5.2] animals, in which Ik was deficient in the host non-leukemic hematopoietic cells, were co-transplanted with Ik6, the recipients showed greater tumor relapse and diminished cytotoxic responses against the tumor targets, compared to allogeneic WT[B6→B6Ly5.2] animals. These data suggest that Ik deficiency in leukemic cells and in non-leukemic hematopoietic APCs collectively contributes to GVL resistance.
In conclusion, we demonstrate that the absence of Ik in both host hematopoietic APCs and leukemic cells enhances GVL resistance despite an increased severity of GVHD. Therefore, strategies that overcome the impact of Ik deficiency by targeting both hematopoietic-derived APCs and hematopoietic leukemic cells are needed.
Materials and Methods
Mice
C57BL/6 (B6, H-2b, CD45.2+), C3H.sw (H-2b), BALB/c (H-2d), and B6 Ly5.2 (H-2b, CD45.1+) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and National Cancer Institute (Frederick, MD). Ik−/− mice were provided by Dr. Winandy (Boston University, Boston, MA) and backcrossed to B6 for μ(Boston 8 generations.38 BMT recipient mice were housed and maintained as in Supplemental methods. All animals were cared for under regulations reviewed and approved by the University Committee on Use and Care of Animals, based on University Laboratory Animal Medicine guidelines.
Generation of bone marrow chimeras
Bone marrow (BM) chimeras ([B6→B6 Ly5.2] and [Ik−/−→B6 Ly5.2]) were generated as described previously.5,7 Briefly, B6 Ly5.2 WT animals were subjected to 11-Gy total-body irradiation (TBI;137Cs Source) on day −1 and then injected intravenously with 5 × 106 BM cells and 5 × 106 whole spleen cells from WT B6 or the Ik−/− donor mice on day 0. Donor hematopoietic chimerism was confirmed using the CD45.2 monoclonal antibody 3–4 months after BMT (donor type >95.0%).
Bone marrow transplantation
BMT was performed as described previously.5,7,18 Briefly, splenic T cells from donors were enriched while the BM was depleted of T cells by autoMACS (Miltenyi Biotec, Bergisch Gladbach, Germany). [B6→B6Ly5.2] and [Ik−/−→B6Ly5.2] animals received 9 Gy TBI (137Cs source) on day −1 and were injected intravenously with 0.5 × 106 CD8+ T cells and 5 × 106 T-cell depleted BM (TCD-BM) from either syngeneic B6 or allogeneic C3H.sw donors on day 0. Survival was monitored daily and recipients' body weight and GVHD clinical scores39 were measured weekly.
Induction of leukemia and lymphoma
Tumors (MBL-2 or EL-4, H-2b) were introduced during BMT at 2 different doses as described previously.5,6 MBL-2 is a Moloney-murine leukemia virus-induced T-cell lymphoma40,41 whereas EL-4 is a chemically-induced T-cell lymphoma.42 Both are of B6 origin (H-2b) and are extensively used as models of acute leukemia and lymphoma. P185 BCR-ABL1 Arf null (WT for IKZF1) and the dominant negative isoform Ik6 were kindly provided by Dr. Mullighan (St. Jude Children's Research Hospital, Memphis, TN). To observe GVL responses, we used a lower dose of tumor cells (MBL-2 0.5–1 × 104/mouse) because we had already established that with injection of this dose the animals cannot reject the tumor and injection results in tumor infiltration and engraftment of the liver and spleen with characteristic nodule formation or hind-limb paralysis caused by spinal cord infiltration.5,6,43,44 The mice that showed less tumor signal among [B6→[B6→B6Ly5.2]] animals demonstrated severe hind-limb paralysis induced by the tumor and were euthanized by the criteria established by the UCUCA protocol. Moreover, we additionally confirmed the cause of death in the allogeneic animals, regardless of BLI, by postmortem examination for tumor and histopathology. We attributed death to tumor if tumor was present at necropsy. Death was attributed to GVHD only if no tumor was evident by flow cytometry or autopsy and there was histologic evidence of GVHD. Mice surviving beyond the observation period of BMT were sacrificed for histologic evaluation to determine leukemia- and lymphoma-free survival.
Luciferase+ MBL-2 cell line
MBL-2 cells were transduced with a third-generation lentivirus coexpressing GFP and firefly luciferase (Luc) as previously described.6,45
Bioluminescence imaging
Bioluminescence imaging was performed with a cryogenically cooled CCD camera (IVIS, Caliper Life Sciences). Acquisition and analysis of images were performed as previously described.45 All animals were imaged 10 min after intraperitoneal (i.p) injection with 100 μL of 40 mg/mL firefly D-luciferin (Biosynth International Inc.). Animals were imaged for 30 s to 5 min, depending on the signal strength. All animals were maintained under isoflurane anesthesia in a 37°C environment.
FACS analysis
FACS analyses was performed as described previously.6,7 Briefly, to analyze chimerism and donor T-cell expansion, splenocytes from transplanted mice were resuspended in FACS wash buffer (2% FBS in PBS) and stained with conjugated monoclonal antibodies (mAbs) as follows: fluorescein isothiocyanate (FITC)-conjugated mAbs to mouse CD45.2 and CD229.1 (Biolegend); phycoerythrin (PE)-conjugated mAbs to CD24, CD47, CD54, CD107a, and CD205 (Biolegend), granzyme B, CD172a (signal regulatory protein α (SIRP-α) and CD209 (eBioscience); allophycocyanin (APC)-conjugated mAbs to CD4, CD229.1, and perforin; and PerCPcy5.5-conjugated mAb to CD8 (Biolegend). Cells were stained and fixed with 1× BD FACS™ Lysing Solution (BD Bioscience), and analyzed using BD Accuri™C6 cytometer (BD Bioscience).
Analysis of MBL-2–specific T-cell responses
Donor MBL-2 specific CD8+ T cells were analyzed on day 21 after BMT with the immune dominant PE-conjugated peptide tetramer CCLCLTVFL epitope, a gag-encoded protein of the Friend/Mononey/Rauscher (FMR) retrovirus that is recognized in the context of H-2b.40,41 An H-2Db restricted influenza peptide (DbPA, SSLENFRAYV) was used as negative control. Tetramers were made by the NIH tetramer core facility (Atlanta, GA). Briefly, splenocytes from transplanted mice were resuspended in FACS wash buffer (2% FBS in PBS), stained with conjugated monoclonal antibodies, and incubated with PE-conjugated gag specific tetramer. Cells were washed twice with FACS wash buffer, fixed with 1× BD FACS™ Lysing Solution (BD Biosciences), and analyzed using BD Accuri™C6 cytometer (BD Bioscience).
51Cr release assay
Splenic CD8+ T cells (4 × 106/ml) were isolated from transplanted mice 21 d after allo-BMT, purified with a CD8+ T-cell isolation kit (Miltenyi Biotec), and used as effector cells. MBL-2, P185 BCR-ABL1 Arf null, and Ik6 tumor cells were labeled by incubating 2 × 106 cells with 2 MBq of Na251CrO4 (PerkinElmer Life) for 2 h at 37 °C in a 5% CO2 atmosphere and were used as target cells. After washing, 4 × 103 labeled targets were resuspended and added to triplicate wells at varying effector-to-target ratios and then incubated for 4 h. Maximal and minimum release was determined by the addition of Triton-X (MP Biomedicals) or media alone to targets, respectively. Supernatants were transferred to a Luma plate (PerkinElmer) after 4 h and 51Cr activity was determined using an autogamma counter (Packard).
Calreticulin expression and overexpression transfection to DCs
Spleens were harvested from either B6-WT or Ik−/− animals and incubated with collagenase D (1 mg/mL, Roche Diagnostics, Indianapolis, IN) in a 37°C/5% CO2 incubator for 45 min. Cells were mashed and purified with CD11c microbeads (Miltenyi Biotec) and auto MACS (Miltenyi Biotec). Calreticulin expression was detected by western blotting.
For overexpression transfection into DCs, we used the plasmid pMSV-mCRT (a gift from Dr. Malini Raghavan, Department of Microbiology and Immunology, the University of Michigan Medical School), which contains full-length mouse wild-type calreticulin cDNA. The pMSV-mCRT plasmid was transformed into Oneshot Top 10 competent cells (Life Technologies) and transfectants were selected by ampicillin. Positive strains were expanded and purified using an Endofree Plasmid Purification kit (Qiagen). Purified pMSV-mCRT plasmids and empty vector control were transfected into DCs from WT-B6 or Ik−/− animals in a 6-well plate (at 80% confluency) using X-treme GENE HP DNA Transfection Reagent (Roche) according to the manufacturer's instruction. After incubation at 37°C for 48 h, the transfected cells were collected for assays.
Statistical analysis
The Mann-Whitney U test was used for statistical analysis of in vitro data, and the Wilcoxon rank test was used to analyze survival data. A p value <0.05 was considered statistically significant.
Acknowledgments
The authors thank Dr. Susan Winandy (Boston University) for providing Ikaros KO mice, Dr. Charles G Mullighan (St Jude Children's Research Hospital) for providing P185 and Ik6 tumors, and Dr. Malini Raghavan (University of Michigan Medical School) for providing plasmid of pMSV-mCRT.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health AI-075284, HL-090775, and CA-173878 (PR).
References
- 1.Gooley TA, Chien JW, Pergam SA, Hingorani S, Sorror ML, Boeckh M, Martin PJ, Sandmaier BM, Marr KA, Appelbaum FR, et al.. Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med 2010; 363:2091-101; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storb R, Gyurkocza B, Storer BE, Sorror ML, Blume K, Niederwieser D, Chauncey TR, Pulsipher MA, Petersen FB, Sahebi F, et al.. Graft-versus-host disease and graft-versus-tumor effects after allogeneic hematopoietic cell transplantation. J Clin Oncol 2013; 31:1530-8; PMID:; http://dx.doi.org/ 10.1200/JCO.2012.45.0247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol 2012; 12:443-58; PMID:; http://dx.doi.org/ 10.1038/nri3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khaled SK, Thomas SH, Forman SJ. Allogeneic hematopoietic cell transplantation for acute lymphoblastic leukemia in adults. Curr Opin Oncol 2012; 24:182-90; PMID:; http://dx.doi.org/ 10.1097/CCO.0b013e32834f5c41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med 2005; 11:1244-9; PMID:; http://dx.doi.org/ 10.1038/nm1309 [DOI] [PubMed] [Google Scholar]
- 6.Toubai T, Sun Y, Luker G, Liu J, Luker KE, Tawara I, Evers R, Liu C, Mathewson N, Malter C, et al.. Host-derived CD8+ dendritic cells are required for induction of optimal graft-versus-tumor responses after experimental allogeneic bone marrow transplantation. Blood 2013; 121:4231-41; PMID:; http://dx.doi.org/ 10.1182/blood-2012-05-432872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toubai T, Sun Y, Tawara I, Friedman A, Liu C, Evers R, Nieves E, Malter C, Chockley P, Maillard I, et al.. Ikaros-Notch axis in host hematopoietic cells regulates experimental graft-versus-host disease. Blood 2011; 118:192-204; PMID:; http://dx.doi.org/ 10.1182/blood-2010-12-324616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science 1992; 258:808-12; PMID:; http://dx.doi.org/ 10.1126/science.1439790 [DOI] [PubMed] [Google Scholar]
- 9.Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, Sharpe A. The Ikaros gene is required for the development of all lymphoid lineages. Cell 1994; 79:143-56; PMID:; http://dx.doi.org/ 10.1016/0092-8674(94)90407-3 [DOI] [PubMed] [Google Scholar]
- 10.Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell 1995; 83:289-99; PMID:; http://dx.doi.org/ 10.1016/0092-8674(95)90170-1 [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, et al.. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360:470-80; PMID:; http://dx.doi.org/ 10.1056/NEJMoa0808253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, Soverini S, Vitale A, Chiaretti S, Cimino G, et al.. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol 2009; 27:5202-7; PMID:; http://dx.doi.org/ 10.1200/JCO.2008.21.6408 [DOI] [PubMed] [Google Scholar]
- 13.Vegh Z, Goyarts EC, Rozengarten K, Mazumder A, Ghebrehiwet B. Maturation-dependent expression of C1q-binding proteins on the cell surface of human monocyte-derived dendritic cells. Int Immunopharmacol 2003; 3:345-57; PMID:; http://dx.doi.org/ 10.1016/S1567-5769(02)00234-5 [DOI] [PubMed] [Google Scholar]
- 14.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123:321-34; PMID:; http://dx.doi.org/ 10.1016/j.cell.2005.08.032 [DOI] [PubMed] [Google Scholar]
- 15.Gao B, Adhikari R, Howarth M, Nakamura K, Gold MC, Hill AB, Knee R, Michalak M, Elliott T. Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity 2002; 16:99-109; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(01)00260-6 [DOI] [PubMed] [Google Scholar]
- 16.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 1999; 285:412-5; PMID:; http://dx.doi.org/ 10.1126/science.285.5426.412 [DOI] [PubMed] [Google Scholar]
- 17.Raghavan M, Wijeyesakere SJ, Peters LR, Del Cid N. Calreticulin in the immune system: ins and outs. Trends Immunol 2013; 34:13-21; PMID:; http://dx.doi.org/ 10.1016/j.it.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, Ferrara JL. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med 2002; 8:575-81; PMID:; http://dx.doi.org/ 10.1038/nm0602-575 [DOI] [PubMed] [Google Scholar]
- 19.Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD. Donor APCs are required for maximal GVHD but not for GVL. Nat Med 2004; 10:987-92; PMID:; http://dx.doi.org/ 10.1038/nm1089 [DOI] [PubMed] [Google Scholar]
- 20.Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol 2014; 27:26-32; PMID:; http://dx.doi.org/ 10.1016/j.coi.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 21.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:; http://dx.doi.org/ 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakraverty R, Eom HS, Sachs J, Buchli J, Cotter P, Hsu R, Zhao G, Sykes M. Host MHC class II+ antigen-presenting cells and CD4 cells are required for CD8-mediated graft-versus-leukemia responses following delayed donor leukocyte infusions. Blood 2006; 108:2106-13; PMID:; http://dx.doi.org/ 10.1182/blood-2006-03-007427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, Nussenzweig MC, Piperno AG, Steinman RM. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci U S A 2007; 104:1289-94; PMID:; http://dx.doi.org/ 10.1073/pnas.0610383104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moron G, Rueda P, Casal I, Leclerc C. CD8alpha- CD11b+ dendritic cells present exogenous virus-like particles to CD8+ T cells and subsequently express CD8alpha and CD205 molecules. J Exp Med 2002; 195:1233-45; PMID:; http://dx.doi.org/ 10.1084/jem.20011930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, Nussenzweig MC, Steinman RM. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol 2008; 181:6923-33; PMID:; http://dx.doi.org/ 10.4049/jimmunol.181.10.6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, Chuang E, Sanborn RE, Lutzky J, Powderly J, et al.. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med 2014; 6:232ra51; PMID:; http://dx.doi.org/ 10.1126/scitranslmed.3008068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng G, Aldridge ME, Tian X, Seiler D, Zhang X, Jin Y, Rao J, Li W, Chen D, Langford MP, et al.. Dendritic cell surface calreticulin is a receptor for NY-ESO-1: direct interactions between tumor-associated antigen and the innate immune system. J Immunol 2006; 177:3582-9; PMID:; http://dx.doi.org/ 10.4049/jimmunol.177.6.3582 [DOI] [PubMed] [Google Scholar]
- 28.Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med 1999; 189:797-802; PMID:; http://dx.doi.org/ 10.1084/jem.189.5.797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair S, Wearsch PA, Mitchell DA, Wassenberg JJ, Gilboa E, Nicchitta CV. Calreticulin displays in vivo peptide-binding activity and can elicit CTL responses against bound peptides. J Immunol 1999; 162:6426-32; PMID: [PubMed] [Google Scholar]
- 30.Bak SP, Amiel E, Walters JJ, Berwin B. Calreticulin requires an ancillary adjuvant for the induction of efficient cytotoxic T cell responses. Mol Immunol 2008; 45:1414-23; PMID:; http://dx.doi.org/ 10.1016/j.molimm.2007.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Cid N, Jeffery E, Rizvi SM, Stamper E, Peters LR, Brown WC, Provoda C, Raghavan M. Modes of calreticulin recruitment to the major histocompatibility complex class I assembly pathway. J Biol Chem 2010; 285:4520-35; PMID:; http://dx.doi.org/ 10.1074/jbc.M110.166330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howe C, Garstka M, Al-Balushi M, Ghanem E, Antoniou AN, Fritzsche S, Jankevicius G, Kontouli N, Schneeweiss C, Williams A, et al.. Calreticulin-dependent recycling in the early secretory pathway mediates optimal peptide loading of MHC class I molecules. EMBO J 2009; 28:3730-44; PMID:; http://dx.doi.org/ 10.1038/emboj.2009.296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ, Cavé H, Trka J, Saha V, Schrappe M, et al.. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014; 123:1691-8; PMID:; http://dx.doi.org/ 10.1182/blood-2013-06-509794 [DOI] [PubMed] [Google Scholar]
- 34.Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, et al.. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat Genet 2011; 43:673-8; PMID:; http://dx.doi.org/ 10.1038/ng.858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama H, Ishimaru F, Avitahl N, Sezaki N, Fujii N, Nakase K, Ninomiya Y, Harashima A, Minowada J, Tsuchiyama J, et al.. Decreases in Ikaros activity correlate with blast crisis in patients with chronic myelogenous leukemia. Cancer Res 1999; 59:3931-4; PMID: [PubMed] [Google Scholar]
- 36.Kuiper RP, Waanders E, van der Velden VH, van Reijmersdal SV, Venkatachalam R, Scheijen B, Sonneveld E, van Dongen JJ, Veerman AJ, van Leeuwen FN, et al.. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010; 24:1258-64; PMID:; http://dx.doi.org/ 10.1038/leu.2010.87 [DOI] [PubMed] [Google Scholar]
- 37.Waanders E, van der Velden VH, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V, Veerman AJ, van Kessel AG, Hoogerbrugge PM, Kuiper RP, et al.. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 2011; 25:254-8; PMID:; http://dx.doi.org/ 10.1038/leu.2010.275 [DOI] [PubMed] [Google Scholar]
- 38.Wang JH, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, Georgopoulos K. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity 1996; 5:537-49; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(00)80269-1 [DOI] [PubMed] [Google Scholar]
- 39.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr, Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 1996; 88:3230-9; PMID: [PubMed] [Google Scholar]
- 40.Chen W, Qin H, Chesebro B, Cheever MA. Identification of a gag-encoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J Virol 1996; 70:7773-82; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Facchinetti A, Dalla Santa S, Mezzalira S, Rosato A, Biasi G. A large number of T lymphocytes recognize Moloney-murine leukemia virus-induced antigens, but a few mediate long-lasting tumor immunosurveillance. J Immunol 2005; 174:5398-406; PMID:; http://dx.doi.org/ 10.4049/jimmunol.174.9.5398 [DOI] [PubMed] [Google Scholar]
- 42.van Hall T, van Bergen J, van Veelen PA, Kraakman M, Heukamp LC, Koning F, Melief CJ, Ossendorp F, Offringa R. Identification of a novel tumor-specific CTL epitope presented by RMA, EL-4, and MBL-2 lymphomas reveals their common origin. J Immunol 2000; 165:869-77; PMID:; http://dx.doi.org/ 10.4049/jimmunol.165.2.869 [DOI] [PubMed] [Google Scholar]
- 43.Teshima T, Hill GR, Pan L, Brinson YS, van den Brink MR, Cooke KR, Ferrara JL. IL-11 separates graft-versus-leukemia effects from graft-versus-host disease after bone marrow transplantation. J Clin Invest 1999; 104:317-25; PMID:; http://dx.doi.org/ 10.1172/JCI7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, Rossignol DP, Ferrara JL. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest 2001; 107:1581-9; PMID:; http://dx.doi.org/ 10.1172/JCI12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duran-Struuck R, Tawara I, Lowler K, Clouthier SG, Weisiger E, Rogers C, Luker G, Kumanogoh A, Liu C, Ferrara JL, et al.. A novel role for the semaphorin Sema4D in the induction of allo-responses. Biol Blood Marrow Transplant 2007; 13:1294-303; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]