

Summary
In mammalian cells, glycogen synthase kinase-3 (GSK-3) comprises of two homologues, GSK-3α and β, encoded by independent genes, which share similar kinase domains but differ substantially in their termini. Here, we describe the generation of an allelic series of mouse embryonic stem cell (ESC) lines with 0–4 functional GSK-3 alleles and have examined GSK-3 isoform function in Wnt/β-catenin signaling. No compensatory upregulation in GSK-3 protein levels or activity was detected in cells lacking either GSK-3α or GSK-3β and Wnt/β–catenin signaling was normal. Only in cells lacking three or all four of the alleles was a gene dosage effect on β-catenin/TCF-mediated transcription observed. Indeed, GSK-3α/β double-knockout ESCs displayed hyperactivated Wnt/β–catenin signaling and were severely compromised in their ability to differentiate, but could be rescued to normality by re-expression of functional GSK-3. The rheostatic regulation of GSK-3 highlights the importance of considering the contributions of both homologues when studying GSK-3 functions in mammalian systems.
Introduction
Glycogen synthase kinase-3 (GSK-3) is a widely expressed and highly conserved serine/threonine protein kinase that is encoded by two genes in mammals, generating two related protein homologues termed GSK-3α and β (Frame and Cohen, 2001; Jope and Johnson, 2004). GSK-3α and β have almost identical central kinase domains, but differ substantially in their termini. GSK-3α, at 52 kDa, is five kDa larger than GSK-3β due to an amino-terminal glycine-rich extension of unknown function (Woodgett, 1990). GSK-3 was first isolated via its activity towards glycogen synthase, the rate-limiting enzyme of glycogen synthesis (Embi et al., 1980). However, it has since been shown to play far broader roles in many cellular processes (Doble and Woodgett, 2003). The protein kinase is unusual in that it is typically highly active in resting cells but is regulated by inhibition in response to cellular signals. Such signals include hormone and growth factor activation of receptor-tyrosine kinases that couple to the phosphatidylinositol 3′ kinase pathway, leading to the activation of protein kinase B (PKB/Akt) (Mitsiades et al., 2004). PKB phosphorylates and inactivates GSK-3α and β and this mechanism contributes to insulin-induced stimulation of glycogen deposition by interfering with the negative regulation of glycogen synthase by GSK-3 (Frame and Cohen, 2001).
GSK-3 is also a key component of the Wnt signaling pathway (Doble and Woodgett, 2003). In resting cells, a fraction of GSK-3 is associated with the Axin scaffolding protein which also binds β–catenin and adenomatous polyposis coli (APC) (Salahshor and Woodgett, 2005). GSK-3 mediated phosphorylation of three residues on β–catenin targets it for ubiquitination and subsequent proteosome-mediated degradation (Amit et al., 2002; Liu et al., 2002). Under such conditions, cytoplasmic levels of β–catenin are kept at low levels. Following engagement of the Frizzled and LRP5/6 co-receptors by certain members of the family of Wnt ligands, GSK-3 phosphorylates LRP5/6 which results in recruitment of the Axin complex to this co-receptor and leads to decreased phosphorylation of the GSK-3 sites on β-catenin, allowing the molecule to escape degradation (Zeng et al., 2005). The resultant accumulated cytoplasmic β–catenin translocates to the nucleus where, in association with members of the LEF/TCF family of DNA binding proteins, it induces transcription of a series of target genes that, in a cell context-dependent manner, promote cell fate and behavioural changes (van Noort and Clevers, 2002). Several target genes for LEF/TCF have been identified, including Axin-2, which is thought to function as a negative feedback regulator of Wnt signaling (Jho et al., 2002; Leung et al., 2002; Lustig et al., 2002).
Since the precise role of GSK-3α and β in this and other cellular processes is poorly understood, we have taken a gene targeting approach to investigate the functions of these molecules. We have previously characterized mice lacking GSK-3β (Hoeflich et al. 2000). These mice die late in gestation due to liver apoptosis. The late onset of the phenotype suggested that in mouse development at least, GSK-3α was largely capable of compensating for the absence of GSK-3β.
Most inhibitors of GSK-3 cannot discriminate between GSK-3α or β based on their ATP-competitive mode of action (Meijer et al., 2004). Thus, they are only useful in providing a way to assess the function of total cellular GSK-3 activity. Moreover, GSK-3 inhibitors are prone to non-specific “off-target” effects that become more pronounced as the working concentration of inhibitor increases (Bain et al., 2003). A particular GSK-3 inhibitor, 6-bromoindirubin-3′-oxime (BIO) has been reported to maintain the pluripotency of mouse and human ES cells through the activation Wnt-β–catenin signaling (Sato et al., 2004). Whether BIO’s effects were due solely to its effects on GSK-3 could not be determined.
Here, we report the generation of embryonic stem cells engineered to lack various combinations of the GSK-3 alleles. These cells have allowed a rigorous analysis of the effect of gene dosage on the properties of the stem cells and the importance of the two genes in Wnt signaling and cellular differentiation.
Results
Wnt Signaling in GSK-3 Knockout Embryonic Stem Cell Lines
Using sequential steps of gene-targeting and G418-induced gene conversion followed by Cre recombinase-mediated site-specific gene deletion, we generated five lines of mouse embryonic stem cells with graded levels of GSK-3 expression to examine the extent of functional redundancy between the two GSK-3 homologues (Fig. 1).
Figure 1.

Strategy used to create compound knockouts of GSK-3α and GSK-3β in ES cells. (A) Targeting strategy for GSK-3α. After homologous recombination with the targeting vector, exon 2 was replaced with a LoxP-flanked (floxxed) exon 2 and FRT-flanked (flrted) neomycin resistance cassette. B = BamHI; A = ApaI. (B) Targeting strategy for GSK-3β. After homologous recombination, exon 2 was replaced with a neomycin resistance cassette and a single FRT site. B = BglII. (C) Overview of the steps required to generate the various ESC lines used in this study (bold type). (D) Multiplex PCR analysis used to screen ESC clones for G418-mediated conversion from GSK-3α(+/flx) to GSK-3α (flx/flx) genotype (step 1–2 in panel C). (E) PCR analysis used to screen ESC clones for G418-mediated conversion from GSK-3α (flx/flx);GSK-3β(+/−) to GSK-3α(flx/flx);GSK-3β(−/−) genotype (step 4b-5 in panel C). White arrowhead indicates a non-specific band. In A-E, ovals represent FRT recombination sites, triangles represent LoxP recombination sites and asterisks represent hybridization sites for Southern blot probes. (F) Southern blots confirming proper targeting of GSK-3 loci. (G) Wild-type, GSK-3α(flx/flx), GSK-3α (−/−) and GSK-3β(−/−) ES cells all display the same level of cytosolic and membrane-associated (pellet) β–catenin. Two different exposures of the same β-catenin immunoblots are presented. (H) Quantitation of GSK-3 in WT and knockout cell lines. Three lysates, each from a different plate of cells, were analyzed per cell type. Error bars = SEM. * p <0.05 (ANOVA with Tukey’s post-hoc test). (I) Consequence of reduced GSK-3 gene dosage on kinase activity. Each bar represents the average from 2 independent kinase assay experiments with each assay performed in triplicate. Error bars = SEM. Inset – immunoblot of the GSK-3 band profiles in the eluates used in the kinase assays (normalized to total protein content).
All of the cell lines generated were viable with growth rates equivalent to WT cells. Immunoblotting with an antibody that recognized both GSK-3α and β, confirmed that the targetting strategies were effective in knocking out expression of either protein and that the conditionally targetted cells (GSK-3α(flx/flx)) expressed wild-type levels of GSK-3α and β (Fig. 1G). Quantitation of the relative amounts of GSK-3α and β in total cell lysates of WT ESCs (Fig. 1H and Supplementary Figure 1) revealed that there was no significant difference between densitometric density of the higher MW band (GSK-3α) containing 52% of the total complement of GSK-3 and the lower band containing 48% (ANOVA with Tukey’s post-hoc test). Deletion of a single GSK-3 isoform did not result in a compensatory increase of the remaining gene product and, as expected, there were no GSK-3 bands detectable in the DKO cell lysates using the GSK-3α/β antibody.
Interestingly, cells lacking GSK-3α appear to contain less GSK-3β than WT cells (42% vs. 48%, respectively; p<0.05, ANOVA with Tukey’s post-hoc test). We believe that this is due to co-migration of truncated forms of GSK-3α with GSK-3β that inflate the density of the “GSK-3β” band in immunoblots of WT lysates. The presence of these truncated GSK-3α bands is clearly observed in intermediate or long exposures of chemiluminescence immunoblots of lysates from GSK-3β(−/−) cells (Supplementary Fig. 1). We quantitated these lower molecular weight GSK-3α bands in lysates from GSK-3β(−/−) and determined that their density equals approximately 8% of the total band density measured in WT ESCs. The sum of the densitometric values of the GSK-3 bands in the α(−/−) and β(−/−) cells (including the lower molecular weight GSK-3α bands) was calculated to total 103%. Statistical analyses revealed no significant difference between this amount and the sum of GSK-3 band densities in WT cells (ANOVA with Tukey’s post-hoc test).
Since GSK-3 protein levels might not correlate directly with kinase activities in the knockout cells, we assayed the GSK-3 kinase activity in WT and knockout cell lines. To do this, we partially purified GSK-3 from cell lysates using ion-exchange chromatography and assayed for GSK-3 activity in the purified fractions using an in vitro kinase assay. As a consequence of our GSK-3 purification strategy, a significant proportion of GSK-3α, relative to GSK-3β, was lost based on post-purification assessment of GSK-3 content by immunoblotting (see inset in Fig. 1I). Thus, we adjusted the kinase assay values to normalize for this loss (Fig. 1I). No compensatory upregulation in kinase activity was observed in any of the knockout cell types, with the observed kinase activities mirroring that which would be expected based on the reduced GSK-3 protein content in the knockout cell lines.
GSK-3 directly phosphorylates the transcriptional regulator β–catenin, marking it for proteosomal degradation (Liu et al., 2002). Interestingly, we observed no changes in cytosolic or membrane-associated β-catenin levels in either of the single knockout lines (Fig. 1G), revealing a clear redundancy of GSK-3 isoform function with respect to β-catenin phosphorylation. Only when the total complement of GSK-3 in the ES cells was reduced to one quarter of normal levels (“3/4KO” cells), did the cytosolic β-catenin levels begin to increase above levels observed in WT lysates (Fig. 2A). By contrast, the DKO cells exhibited a massive increase in cytosolic β-catenin levels (Fig. 2A). Overexpression of β-catenin in fibroblasts and epithelial cell lines has been reported to lead to reduced cell viability (Kim et al., 2000). However, we found no evidence of decreased growth in the DKO ES cells. Our data are consistent with ApcMin/Min mouse ES cells, which are also viable despite their highly elevated β–catenin levels (Kielman et al., 2002).
Figure 2.

Consequences of reduced GSK-3 gene dosage with respect to cytosolic β-catenin levels and β-catenin TCF-transactivation activity. (A) Immunoblot analysis of cytosolic GSK-3, β-catenin and GAPDH levels. Results from three independent clones with DKO or 3/4KO genotypes are presented. (B) Quantitative RT-PCR analysis of Axin2 transcript levels in WT and GSK-3 knockout ES cells. Error bars = SEM. (C) TCF reporter assay. Each bar represents the average from 4 independent samples. Error bars = SEM. * p < 0.05; ** p < 0.001 (ANOVA with Newman-Keuls post-hoc test). (D) Immunofluorescence analysis of a WT and DKO ES cell clone stained for β-catenin and DAPI (nuclei). Bar = 10 μm.
To assess whether the observed changes in cytosolic levels of β-catenin correlated with functional changes in β-catenin activity in the nucleus, we used a luciferase-based reporter system to measure TCF-mediated transcription (Fig. 2B). TCF reporter activity was assessed after treatments with or without Wnt-3a conditioned medium for 24 hours. Untreated WT, GSK-3α(−/−) and GSK-3β(−/−) cells showed the same basal levels of TCF reporter activity and all responded to the Wnt-3a treatment to a similar extent. Interestingly, in the 3/4KO cells, the basal TCF-reporter activity in the absence of Wnt-3a was significantly higher than the Wnt-3a-stimulated levels of the WT and single-knockout cells (p < 0.05). Wnt-3a treatment further increased the TCF-reporter activity in the 3/4KO cells (p < 0.001). However, in the DKO cells, wnt signalling is completely uncoupled resulting in a dramatic increase in TCF-reporter activity.
To determine the functional consequence of GSK-3 gene dosage manipulation on endogenous target genes, we extended our findings to a known TCF target gene, axin2 (Jho et al., 2002; Leung et al., 2002; Lustig et al., 2002). We used quantitative RT-PCR to determine the levels of axin2 gene expression in the series of knockout cells. The pattern of axin2 expression was in agreement with our TCF-reporter data with WT and single knockout ES cell lines having similar low levels of axin2 expression, 3/4KO cells beginning to show accumulation of axin2 message beyond base-line levels, and DKO cells expressing ten times more axin2 transcripts than WT cells (Fig. 2C).
We interpret these data to indicate that in the 3/4KO cells, GSK-3 becomes limiting for Wnt signaling; i.e. a single GSK-3 allele generates insufficient GSK-3 protein to occupy all β-catenin destruction complexes in the cell allowing some β–catenin to escape phosphorylation-dependent degradation. In the 3/4KO cells, the fraction of destruction complexes with GSK-3 present promote β-catenin phosphorylation and degradation until disrupted by Wnt signaling. When GSK-3 is totally absent (DKO), there are no functional destruction complexes and cytosolic and nuclear β-catenin reach very high levels (Fig. 2A–B). These levels of cytosolic and nuclear β-catenin are also clearly observed through immunofluorescence visualization in DKO cells (Fig. 2D) and are similar to cells mutant for APC (Kielman et al., 2002).
Validation of β–catenin as a GSK-3 Target
Phosphorylation of β-catenin by GSK-3 on residues 33, 37 and 41 has been well documented (Amit et al., 2002; Hagen et al., 2002; Liu et al., 2002; Salic et al., 2000). Still, as Frame and Cohen outlined in their 7 criteria for establishing a protein as a bona fide physiological substrate of GSK-3 (Frame and Cohen, 2001), one of the burdens of proof required for substrate validation is to show that the substrate cannot be phosphorylated on putative target residues in cells lacking GSK-3. Using commercially available phospho-specific antibodies for β-catenin, we tested whether GSK-3 was the only kinase that phosphorylated residues 33, 37 and 41 under “resting” and Wnt-3a-stimulated conditions in mouse ES cells. As expected, the antibody detected phosphorylated β-catenin species in untreated WT cells (Fig. 3A), a signal that completely disappeared upon 30 min of Wnt-3a stimulation. However, in the DKO cells, this band was completely absent in lysates from both untreated and Wnt-3a-treated, validating residues 33, 37 and 41 of β-catenin as specific GSK-3 target sites. Serine 45 (S45) of β-catenin is phosphorylated by CKIα, which primes β-catenin for subsequent phosphorylation at sites 33, 37 and 41 by GSK-3(Amit et al., 2002; Zeng et al., 2005). Phosphorylation of S45 has been suggested to be the switch which regulates β-catenin phosphorylation and subsequent degradation (Amit et al., 2002). Using an antibody that recognizes β-catenin phosphorylated on either T41, and/or S45, we found that 30 min. treatment of cells with Wnt-3a caused a decrease in phosphorylation in WT cells, but had no effect in DKO cells (Fig. 3A). The decrease seen with the T41/S45 phospho-specific antibody in WT cells is most likely due to T41 dephosphorylation alone indicating that the level of S45 phosphorylation is not regulated in response to Wnt. Axin2 serves as a negative feedback regulator of canonical Wnt signaling (Jho et al., 2002; Leung et al., 2002; Lustig et al., 2002). Long-term treatment (overnight) with Wnt3a, or tonic β-catenin signaling as observed in DKO cells would be expected to cause increases in Axin2 protein levels. This was observed: cytosolic levels of Axin2 paralleled changes in cytosolic β-catenin levels in cells with graded levels of GSK-3 in response to a 20h Wnt-3a treatment (Fig. 3B). Like Axin2, we observed similar changes with the APC protein in response to Wnt and the graded levels of GSK-3. This does not appear to be due to increased transcription of APC because we found no differences of APC transcripts by RT-PCR (data not shown). Our data are consistent with Choi et al.’s observation of increased APC accumulation without detectable changes in transcript levels upon Wnt treatment of HEK293 cells (Choi et al., 2004). Choi et al. provide evidence for the post-translational regulation of APC through ubiquitination and proteasomal degradation in a manner analogous to β-catenin regulation. Based on overexpression of kinase-dead GSK-3 and treatment of cells with the GSK-3 inhibitor lithium, these authors excluded a possible role for GSK-3 in the Wnt-mediated stabilization of APC. However, our data suggest that GSK-3 is indeed a key regulator of APC stability since DKO cells harbour APC levels that are even higher than observed with long-term Wnt treatment of WT cells (Fig. 3B).
Figure 3.

Immunoblot analysis of GSK-3 gene dosage effects on Wnt signaling intermediates. (A) Examination of β-catenin phosphorylation in WT and DKO ESCs treated 30 min. with Wnt-3a conditioned medium (CM). (B) Levels of key Wnt signaling molecules after long-term (20 hour) treatment with Wnt-3a CM.
Rescue of Wnt Signaling by Re-introduction of GSK-3
To verify that the accumulation of β-catenin in the DKO cells was due to the lack of GSK-3 and not some other event that had occurred during the multiple manipulations required to generate the DKO cells, we re-expressed WT and mutant forms of GSK-3 using stable transfection and lentiviral transduction. Re-introduction of WT GSK-3α (Fig. 4) or β (data not shown), with or without epitope or fluorescent protein tags, resulted in normalization of cytosolic and nuclear β-catenin levels as assessed by immunofluorescence microscopy (Fig. 4A, B). True WT cells (Fig. 4B) were indistinguishable from DKO cells reconstituted with GSK-3α–GFP (Fig. 4A) by stable transfection or transduced with lentivirus expressing V5-epitope-tagged WT GSK-3α. A V5-tagged S21A mutant of GSK-3α that cannot be inactivated through phosphorylation of S21 re-regulated β–catenin levels in DKO ES cells to the same extent as V5-tagged WT GSK-3α (Fig. 4B). On the other hand, a kinase-dead (K148A) GSK-3α mutant was ineffective in decreasing β-catenin staining, confirming the role of the kinase activity of GSK-3 in the regulation of β-catenin levels. Tyrosine phosphorylation of GSK-3 is required for full enzymatic activity (Y216 of GSK-3β; Y279 of GSK-3α) (Hughes et al., 1993). DKO cells transduced with lentivirus expressing V5-GSK-3α with a Y279F mutation only partially reduced cytosolic and nuclear β-catenin with respect to WT levels (Fig. 4B and supplementary Fig. 1). This finding is in agreement with the findings of Hagen et al. who reported that the Y216F mutant of GSK-3β when over-expressed in HEK293 cells was severely compromised in its ability to regulate β-catenin levels and activity in these cells (Hagen et al., 2002).
Figure 4.
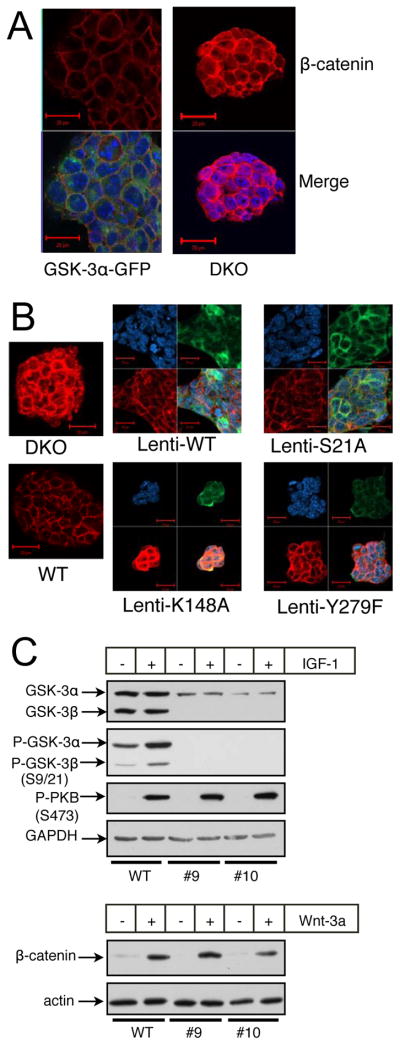
Rescue of DKO ES cells with variants of GSK-3α. (A) Immunofluorescent images of DKO ES cells and DKO ES cells reconstituted with a GSK-3α-GFP fusion protein. β-catenin staining is red, GFP staining is green and nuclear staining is blue (DAPI). (B) Immunofluorescent images of WT and DKO ES cells as well as DKO ES cells rescued with various V5-epitope-tagged mutants of GSK-3α through lentiviral transduction. V5 staining is green, β-catenin staining is red and nuclear staining is blue. All images were obtained with identical PMT, resolution and scan-rate settings. (C) DKO ES cells rescued with a GSK-3α S21A mutant still respond to Wnt-3a. #9 and #10 are independent clones of DKO ES cells reconstituted with S21A-GSK-3α. Treatments with IGF-1 were for 15 minutes, while treatments with Wnt-3a conditioned medium were overnight.
The mechanism by which GSK-3 phosphorylation of β–catenin is reduced in the destruction complex upon activation of Wnt-signaling is still unclear. Although it has been suggested that GSK-3 may be inactivated through Wnt-induced serine 9/21 phosphorylation, there are several lines of evidence that show that this is not the case (Ding et al., 2000; McManus et al., 2005). Most conclusively, mice in which both GSK-3α and β were genetically manipulated so that they expressed only S9A/S21A mutant alleles of GSK-3β and α, respectively, are viable, fertile and grossly normal (McManus et al., 2005). As expected, DKO cells stably expressing the S21A mutant of GSK-3α, while incapable of being phosphorylated at S21 by IGF-1 treatment, were fully able to respond to Wnt-3a conditioned medium as judged by an increase in cytosolic β-catenin levels similar to that seen in WT cells (Fig. 4C).
GSK-3α + β null ESCs Display Differentiation Defects
A notable feature of the DKO ES cells that we observed while generating them was that they retained an ideal undifferentiated, highly refractile, ES cell colony morphology even under conditions where WT ES cells started to differentiate. Immunofluorescence images of DKO colonies when compared to images of WT colonies revealed that the DKO cells had a much smaller cytoplasmic to nuclear ratio and were more tightly packed in their colonies than WT cells (Fig. 4A). We therefore sought to determine whether the DKO ESCs were impaired in their ability to differentiate compared to WT ESCs.
Using the hanging drop method, we created embryoid bodies (EBs) from WT or DKO ESCs. While WT cells developed normally into EBs with transparent cystic structures reminiscent of the embryonic yolk sac, over the course of 7–8 days in differentiation medium, the DKO cells never formed these transparent cysts under identical conditions (Fig. 5A). More than 80% of the WT EBs (n > 200) contained regions of spontaneously contracting cardiomyocytes by day 8 of differentiation that were visible microscopically, while the DKO EBs never displayed these contractile cells.
Figure 5.

GSK-3 double null ES cells (DKO) display a limited potential for differentiation. (A) Embryoid bodies derived from the same number (800) of WT or DKO ES cells at various time points after the initiation of differentiation. Bar = 200 μm (B). Oct-4 immunfluorescence staining of WT and DKO EBs differentiated for 14 days in the absence of LIF. Bar = 50 μm. (C) Nanog immmunofluorescence staining of WT and DKO EBs differentiated for 12 days in the absence of LIF. Bar = 20 μm. (D) Semi-quantitative RT-PCR analysis WT and DKO EBs at day 0 (d0) and day 7 (d7) of differentiation.
The extent of the block to differentiation observed in the DKO EBs was assessed by staining EBs with markers of pluripotent mouse ES cells Nanog and Oct-4 (Chambers et al., 2003; Rosner et al., 1990). Maintained for 14 days in the absence of LIF and in 5% FBS, the DKO EBs, but not the WT EBs retained large numbers of Oct-4 positive cells (Fig. 5B). Furthermore, DKO EBs at day 12, still contained detectable levels of Nanog, while the WT EBs did not (Fig. 5C). At the transcriptional level, oct-4, nanog and rex-1, another pluripotent ES cell marker (Ben-Shushan et al., 1998), were detectable after 7 days of differentiation in DKO EBs, but transcripts of these genes were present at much lower levels in WT EBs as assessed by semi-quantitative RT-PCR analysis (Fig. 5D). Quantitative RT-PCR of oct-4, nanog and rex-1 confirmed that DKO, but not WT EBs maintained high levels of ES cell marker gene transcripts after 7 days in the absence of LIF and in reduced serum (5% FBS) conditions (supplementary Fig. 3). Surprisingly and interestingly, expression of the brachyury/transcription factor T, one of the earliest markers of mesoderm differentiation (Herrmann et al., 1990), was upregulated in the DKO cells even before withdrawal of LIF (Fig. 5D and supplementary Fig. 3). Expression of axin2 was also higher in DKO EBs compared to WT EBs at the outset, and 7 days after being cultured in differentiation medium. These data confirmed that morphologically undifferentiated DKO ES cells and “differentiated” DKO EBs both retain high tonic levels of β-catenin/TCF-mediated transcription and that our data are in agreement with previous findings that brachyury is a target of Wnt signaling (Arnold et al., 2000; Yamaguchi et al., 1999). To address whether the extremely high levels of brachyury transcript expression (40X higher than WT levels; supplementary Fig. 3) could be detected at the protein level, we examined Brachyury by immunoblotting and immunofluorescence techniques. A band running at the expected size (47 kDa) was detected by immunoblotting with a Brachyury-specific antibody in DKO lysates only (supplementary Fig. 4A). Furthermore, DKO ES cells displayed intense immunofluorescence staining for Brachyury including bright staining that co-localized with the nuclear stain DAPI, while WT cells had much weaker staining, none of which appeared to be nuclear (supplementary Fig. 4B).
Teratoma analyses confirm a DKO ESC differentiation blockade
Wild-type pluripotent embryonic stem cells, when injected into an immuno-compromised or syngeneic mouse, form tumours known as teratomas which comprise all three germ layers (i.e. endoderm, ectoderm and mesoderm) (Stevens, 1967). We tested the ability of WT and DKO ESCs to form the three germ layer lineages in teratomas generated in the hind legs of syngeneic 129Ola mice. Sections of the teratomas generated from WT ESCs had clear examples of tissues of endo-, ecto- and meso-dermal origin (Fig. 6A–D). In contrast, all teratomas generated from DKO ESCs had a grossly undifferentiated carcinomatous appearance, with the only differentiated structures observed being spicules of bone (Fig. 6E, F). Immunostaining for β-catenin had the expected localization in WT teratomas where the strongest staining was apparent at juxtaposed plasma membranes of adjacent epithelial cells where adherens junctions are located (Fig. 6G). Only weak cytoplasmic, and no nuclear, staining was observed in WT epithelial cells. In marked contrast, β-catenin staining in DKO teratomas was very strong throughout the teratoma mass, and membrane, cytoplasm and nuclear staining were observed in practically every cell of the teratomas (Fig. 6J).
Figure 6.

Histological, immunohistochemical, and PCR examination of teratomas generated from WT (GSK-3α(flx/flx)) and DKO ES cells. (A) The WT teratomas possess numerous examples of glandular epithelial structures indicative of endoderm (arrow indicates gut-like pseudostratified epithelium). (B) WT tumours also had large regions of neuronal tissue indicating differentiation into ectoderm (arrows point to neuronal nuclei). (C) Differentiation of WT ESCs into mesoderm was confirmed by observation of bone (arrow points to region of woven bone) and (D) muscle (inset shows magnification of striated skeletal muscle). (E, F) DKO teratomas contain tightly packed cells in undifferentiated tissue reminiscent of a carcinoma with scattered spicules of bone (arrows). (G) Immunohistochemical staining for β-catenin revealed normal membranous staining in epithelial cells of WT teratomas. H, I) Staining for the neuronal progenitor marker Nestin and the astrocytic marker GFAP was clearly observed in WT teratomas. (J) DKO teratomas displayed very high levels of β-catenin immunoreactivity in all cells in membrane, cytosol and nuclear compartments. (K, L) There was no detectable staining for Nestin or GFAP in DKO teratoma tissue sections. (M) Semi-quantitative RT-PCR analyses of WT vs. DKO gene expression. Bar = 100 μm.
Careful observation of numerous hematoxylin and eosin-stained tissue sections from different DKO teratomas generated in four separate animals revealed a complete absence of neuronal tissue, in contrast to WT teratoma sections which contained many examples of neuronal cell types. We sought to confirm this observation by immunostaining for the neural progenitor marker nestin and the astrocyte marker glial fibrillary acidic protein (GFAP). Nestin and GFAP staining stained strongly in regions of the teratomas generated from WT ESCs (Fig. 6H and I, respectively) while staining for these markers was completely absent in DKO teratomas (Fig. 6K and L).
The peculiar presence of bone in the largely undifferentiated teratomas generated from DKO ESCs was substantiated by RT-PCR analysis. The markers of bone development/osteoblastogenesis, osterix, runx2, osteocalcin and alkaline phosphatase (Lian et al., 2006; Salingcarnboriboon et al., 2006) were all found to be expressed at higher levels in DKO teratomas than in WT teratomas (Fig. 6M). In addition, supporting the undifferentiated nature of the DKO teratomas, expression of nanog and oct-4 was also higher in DKO teratomas than those formed from WT ESCs. Quantitative assessment of oct-4 and nanog expression revealed that DKO teratomas still expressed these genes at levels greater than 10% of those found in WT ESCs maintained in undifferentiated conditions (Fig. 6N). Wild-type teratomas had nanog and oct-4 transcript levels that were more than 10X lower than those in DKO teratomas. Together, our teratoma data strongly suggests that DKO ESCs are dramatically impaired in their ability to differentiate properly, especially into neurectodermal cell types, and that there is probably a population of ES or ES-like cells that are maintained in the DKO teratoma masses.
DKO ESC properties can be completely rescued with a WT GSK-3α transgene
We generated stable cell lines from DKO ESCs that stably express equivalent levels of WT or kinase-dead (K148A) GSK-3α with a V5-epitope tag located at the carboxy terminus. For long-term stable expression we used the human EF-1α promoter, since we found that our original “stable” lines with CMV promoter-driven GSK-3 (lines assessed in Fig. 4) lost their high expression levels within a few cell passages. Immunoblot analysis confirmed that the V5-tagged GSK-3α WT and K148A were expressed at equivalent levels in the clones we chose for further analysis (Fig. 7A). The level of transgenic GSK-3α in both the WT- and kinase -dead-rescued clones was equivalent to that produced by a single allele of GSK-3, based on comparison with endogenous GSK-3α levels in WT cells (Fig. 7A). This level of WT GSK-3α-V5 protein was sufficient to normalize cytosolic β-catenin levels to just slightly higher than the levels found in WT ESCs, while kinase-dead GSK-3α-V5 had no effect on β-catenin protein levels (Fig. 7A).
Figure 7.

The properties of DKO ESCs can be reverted to those of WT ESCs upon stable expression of WT, but not kinase-dead, GSK-3. (A) Immunoblot analyses of cytosolic lysates prepared from WT and DKO ESCs as well as DKO ESC cell lines stably expressing V5-tagged WT- or K148A-GSK-3α. (B) Semi-quantitative RT-PCR analysis of transcripts expressed in the same cell lines examined in panel A. (C–N) Immunofluorescence images of EBs maintained for 14 days in LIF-free medium. EBs derived from (C, F) V5-WT-GSK-3α-expressing DKO ESCs; (D, G) V5-K148A-GSK-3α expressing DKO ESCs; and (E, H) DKO ESCs double-stained for Oct-4 and GFAP, respectively. EBs derived from (I, L) WT ESCs; (J, M) V5-WT-GSK-3α-expressing DKO ESCs and (K, N) V5-K148A-GSK-3α-expressing DKO ESCs stained for Nestin and Nestin/DAPI (nuclear stain) merge, respectively. Bar = 100 μm in all micrographs.
To further assess the rescue of DKO ESCs by expression of WT-GSK-3α, we examined the steady-state levels of a panel of transcripts that we had observed to be differentially expressed in WT and DKO ESCs. The transcripts for axin2, brachyury, and cdx1 are all elevated in DKO ESCs when compared with WT ESCs maintained in standard ESC culture conditions with LIF supplementation (Fig. 7B). The ESC markers nanog and oct-4 are both expressed at high levels in both WT and DKO ESCs (Fig. 7B). The expression profile of DKO ESCs was changed to that of WT ESCs upon stable expression of GSK-3α-V5, but was unchanged with the kinase-dead transgene (Fig. 7B). Thus, re-expression of WT-GSK-α in DKO ESCs is sufficient to “re-program” the cells at the level of transcription.
We used WT and kinase-dead GSK-3α-expressing DKO ESCs, as well as WT and DKO ESCs to form EBs using the hanging drop method to assess their differentiative capacities. An obvious difference between EBs formed from DKO ESCs and WT-GSK-3α-V5-rescued DKO ESCs, was that while DKO ESCs never gave rise to spontaneously contracting cardiomyocytes in their EBs, some of the WT-rescued EBs did (16 of 76 EBs scored at day 7 of differentiation). As we observed with EBs derived from WT ESCs (Fig. 5B), EBs resulting from aggregation of DKO ESCs expressing WT-GSK-3α-V5, had no detectable Oct-4 protein through immunofluorescent visualization (Fig. 7C). In contrast, DKO EBs expressing kinase-dead GSK-3α-V5, still contained clusters of cells expressing Oct-4 protein (Fig. 7D) at the same frequency as DKO EBs (Fig. 7E). To confirm that the rescued DKO ESCs regained their ability to differentiate into neurectoderm upon stable expression of GSK-3α, we tested the ability of GSK-3α-V5 transgene-expressing DKO ESCs to form EBs that stained positively for GFAP and nestin. Very strong GFAP staining was observed in EBs expressing WT-GSK-3α-V5 (Fig. 7F), while only weak background staining was seen in EBs expressing kinase-dead GSK-3α-V5 or no GSK-3 (DKO) (Fig. 7G and H, respectively). Similarly, WT and WT-rescued DKO ESCs, formed EBs that stained strongly for Nestin (Figs. 7I/L and 7J/M, respectively), while kinase-dead-GSK-3α-rescued DKOs formed EBs that were devoid of Nestin staining (Figs. 7K, N). Together, our rescue data suggest that re-expression of active GSK-3 in DKO ESCs is sufficient to revert them to a WT ESC identity in all respects.
Discussion
Our data provide conclusive evidence, for the first time, that both mammalian GSK-3 homologues function identically in Wnt signaling. This redundancy in function has not been appreciated in the Wnt signaling field, where the role of GSK-3β has been overemphasized. Historically, this bias most likely arose from reports suggesting a difference in the efficacy of rescue of the zestewhite 3 (GSK-3) mutation in Drosophila by mammalian GSK-3α and β in which the β isoform appeared more effective than α (Ruel et al., 1993; Siegfried et al., 1992). However, in these studies, the amount of expression of the two homologues was not equalized. Our genetically manipulated ES cells allowed us to demonstrate that either GSK-3 homologue is necessary and sufficient to regulate Wnt-β–catenin signaling in ES cells.
Our data also clearly show that there is no compensatory upregulation of GSK-3 protein levels or kinase activity in the knockout cell lines. It should be noted that our kinase assays, which used a glycogen synthase phosphopeptide substrate known as GS-2, revealed “background” kinase activity in our DKO ES cells. It has been shown previously that while GS-2 is largely specific for phosphorylation by GSK-3, it can also be phosphorylated by other kinases (Kuma et al., 2004; Skurat and Dietrich, 2004).
We observed a clear and tonic upregulation of β-catenin/TCF-mediated signaling as assessed by observation of β-catenin levels, TCF-reporter activity and up-regulation of the TCF-responsive genes, axin2, cdx1 and brachyury. While we were surprised to see that the morphologically undifferentiated DKO ES cells expressed high levels of brachyury at both the RNA and protein levels, the existence of a population of undifferentiated colonies of mES cells that are positive for both Nanog and Oct-4 and also for Brachyury has recently been reported and termed ‘early mesoderm-specified (EM) progenitors’(Suzuki et al., 2006). The only difference between EM progenitors and ES cells is expression of Brachyury. Modulation of LIF concentrations in the culture medium can cause exchange of EM progenitor and ES cell identities. That is, EM progenitors can revert to ESCs. Thus, it is possible that the GSK-3 null, DKO cells described here that appear to be ES cells by morphological criteria, are actually more closely related to EM progenitors.
The homeobox-containing transcription factor Cdx1, has been shown to be up-regulated by Wnt-3a in embryonic stem cells and has been suggested to be a direct Wnt target gene (Lickert et al., 2000). The high levels of cdx1 gene expression in our DKO ESCs support this notion. Cdx1 is normally not expressed until embryonic day 7.5 in the mouse at which point it plays a role in the development of the posterior embryo (Lohnes, 2003).
The presence of bone in teratomas derived from DKO ESCs was somewhat surprising, since we were expecting a similar phenotype to APC(min/min) mutant ESCs which, like our DKO cells, have highly deregulated Wnt signaling due to the inability of the min mutant of APC to bind β-catenin (Kielman et al., 2002). Embryonic stem cells derived from APC(min/min) blastocysts are incapable of forming teratomas in syngeneic mice. The bone phenotype we see in DKO teratomas is most likely due to activated Wnt signaling as there is extensive evidence linking Wnt signaling to osteoblastogenesis (Lian et al., 2006). The mechanism underlying the incredible specificity of the differentiation of DKO ESCs in teratomas is unclear. The bone-specific differentiation may require extrinsic factors provided by the microenvironment of the teratomas since high levels of osteogenic transcription factors were not observed in 14 day EBs generated from DKO ESCs (data not shown).
The differentiation block that we observe in the DKO ES cells is consistent with the report of Sato et al. which suggested that Wnt signaling or GSK-3 inhibition could maintain pluripotency of mouse and human ES cells (Sato et al., 2004). Their data, in part, focussed on the effects of the GSK-3 inhibitor BIO. This inhibitor, as with any small molecule inhibitor, is subject to potential “off target” effects. Our data are the first to confirm that genetic inactivation of GSK-3 is sufficient to severely compromise ES cell differentiation. Our data are also consistent with the findings of Kielman et al. who found that mES cells containing different mutants of APC, with increasing levels of β-catenin and TCF-activity, became more restricted in their developmental capacity (Kielman et al., 2002).
The mechanism through which GSK-3 regulates ES cell differentiation is at least in part due to its function in Wnt signaling, although we cannot rule out its role in other pathways. For example, the PI-3K pathway, which is known to negatively regulate GSK-3 through serine phosphorylation of its amino terminal domain, has been shown recently to play a role in the maintenance of mouse and primate ES cell pluripotency (Paling et al., 2004; Watanabe et al., 2006). Also, GSK-3 phosphorylation of Myc on threonine 58 has been implicated in the LIF/STAT3-mediated regulation of mouse ES cell pluripotency, since overexpression of a T58A mutant of Myc in mouse ES cells promotes their self-renewal and pluripotency in the absence of exogenous LIF (Cartwright et al., 2005). Indeed, we have found that our DKO ES cells do not have detectable levels of Myc phosphorylated on T58 (data not shown). Still, only a subpopulation of cells within “differentiated” DKO EBs express the pluriopotency markers Nanog and Oct-4. This suggests that even in the presence of stabilized β–catenin and Myc, other factors, perhaps related to cell polarity or local environment are required for maintenance of the true pluripotent state.
Conclusions
Taken together, our findings provide important new insights into the mode of substrate regulation by GSK-3. GSK-3 serves as a regulatory component in numerous signaling pathways. One pertinent question is understanding the rationale for the existence of two GSK-3 isoforms encoded by distinct genes. From our studies focussing on the canonical Wnt pathway, it appears that there is a very high level of functional redundancy with respect to GSK-3 homologues. We anticipate that this redundancy in function will exist in many, if not all, of the pathways that require GSK-3. We demonstrate that GSK-3α and β are equally capable of maintaining low levels of β–catenin and that only upon inactivation of 3 of the 4 alleles, or complete loss of all 4, is there any discernable impact on Wnt signaling proteins and β–catenin levels. This is of clinical relevance in conditions where elevated GSK-3 activity is deleterious, such as Alzheimer’s disease and Type II diabetes. Our data suggest that there may be a therapeutic window and dose for GSK-3 inhibitors in treating diseases without elevating β–catenin levels and thus risk of oncogenic events. Our findings with respect to embryonic stem cell differentiation suggest that pathways regulated by GSK-3, at least in part the Wnt pathway, are critically involved in the maintenance of embryonic stem cell identity. The GSK-3-less, DKO cells should serve as in important tool for the dissection of LIF-independent pathways responsible for the maintenance of ES cell identity and self-renewal in mouse and human ES cells.
Experimental Procedures
Plasmids
A clone containing the full-length cDNA for murine GSK-3α was purchased from Open Biosystems (IMAGE clone 4011973). QuikChange (Stratagene) was used to generate the following mutants of GSK-3α, each with a single amino acid substitution) S21A, K148A, and Y279F. A mammalian expression plasmid containing a single FRT recombination site was created by splicing the CMV promoter, multiple cloning site, polyA signal and puromycin resistance cassette from pPUR (Clontech) into the BspEI and Bst11071 sites of pcDNA5/FRT (Invitrogen) to yield pcDNA5/FRT_puro. WT and mutant versions of GSK-3α were subcloned into the KpnI and ApaI sites of this vector. Where necessary, appropriate restriction enzyme sites were added to the termini of PCR primers prior to ligations. To generate GFP-GSK-3α fusions, an NdeI /KpnI fragment containing part of the CMV promoter and the EGFP coding sequence from pEGFP-C1 (Clontech) was cloned into NdeI/KpnI-cleaved pcDNA5/FRT_puro_GSK-3α. Stable cell lines expressing V5-tagged GSK-3 fusion proteins were produced by cloning PCR- amplified GSK-3 variants into pEF5/FRT/V5 directional TOPO following manufacturer’s instructions (Invitrogen).
For in vitro Cre- and FLPe-mediated excisions, pIC-Cre (Gu et al., 1993) and pCAGGS-FLPe (GeneBridges, Germany) were used, respectively. The TCF reporter plasmids super8XTOPFlash and super8XFOPFlash (Veeman et al., 2003) (obtained from Randall Moon. Univ of Washington) were used in conjunction with pRL-CMV (Promega).
Cell Culture / Embryoid Body Assay
All media and supplements were obtained from Gibco (Invitrogen) unless otherwise noted. ES cells (E14K) were maintained in Knockout DMEM supplemented with 15% FBS (Hyclone), 1X non-essential amino acids, 1X GlutaMax-I, 1X 2-mercaptoethanol and 1000 units / ml ESGRO® (Chemicon). Differentiation medium for the production of embyroid bodies was ESC maintenance medium with the serum content reduced to 5% and no ESGRO® supplementation. Embryoid bodies were propagated in hanging drops on 10 cm2 bacterial dishes (800 single ES cells / 30 μl drop initially) for 3 days and were then transferred to ultra low binding 96-well dishes (Corning) and cultured with differentiation medium that was replenished every two days.
L-cells stably expressing Wnt-3a and control WT L-cells were obtained from the ATCC and conditioned medium was prepared according to the protocol provided with the cells.
Semi-Quantitative RT-PCR
Total RNA was isolated using Qiagen’s RNeasy kit. Reverse transcription was carried out using SuperScript II (Invitrogen). The template for each PCR reaction was the cDNA obtained from 50 ng of total RNA. The number of PCR cycles for β-actin, GAPDH and Eomes was 25, while all other targets were examined after 30 cycles of amplification. Primer sequences are listed in supplementary data, table 1.
Quantitative RT-PCR
RNA isolation and reverse transcription were carried out as described for semi-quantitative RT-PCR. The template for each PCR reaction was the cDNA obtained from 25 ng of total RNA in a 25 μl reaction volume. Primers were designed using the online PrimerQuest software provided by IDT, the company that synthesized the primers for us. Platinum® SYBR® green qPCR SuperMix-UDG (Invitrogen) was used according to the manufacturer’s recommendations for the Stratagene Mx3000P real-time PCR machine, which was used for all real-time experiments. The software package, MxPro (Stratagene), was used to calculate the relative differences in gene expression levels, using the delta-delta Ct method, with β-actin used as the housekeeping gene for normalization of samples. The sequences of the primers used for qPCR analyses are provided in supplementary data, table 2.
Cytosolic Lysate Preparation
Cytosolic lysates were prepared by rinsing cells 3X with PBS, then scraping them into a minimal volume of ice-cold hypotonic lysis buffer (50 mM Tris pH 7.4, 1 mM EDTA) containing a cocktail of protease and phosphatase inhibitors (Complete protease inhibitor tablet (Roche), phosphatase inhibitor cocktail 1 (P2850 Sigma); 1 mM Na-orthovanadate, 10 mM NaF, 10 mM β-glycerophosphate). Cells were pelleted by centrifuging for 1 hour at maximum speed in a benchtop microfuge at 4°C. Cytoplasm expelled from the cells was retrieved by collecting the supernatant, taking care not to aspirate any insoluble material located in the pellet or floating on the surface of the lysate. Insoluble proteins in the pellets from hypotonic lysates were extracted with 2% SDS, 50 mM Tris pH 6.8, containing protease and phosphatase inhibitors.
GSK-3 kinase assay
ES cells from a confluent T175 flask were harvested using 2ml of detergent-free lysis buffer (50mM Tris-HCl, 4mM EDTA, 2mM EGTA, 10mM Naβ-glycerophosphate, 25mM NaF, 1mM PMSF, 1 mM Na-orthovanadate, protease and phosphatase inhibitor cocktail, pH 7.5) and homogenised in a 1ml Dounce homogeniser with 10 strokes of pestle A and B. The lysates were centrifuged at 14000×g for 30 min and the supernatant was applied to a pre-equilibrated (25mM Tris-HCl, 2mM EDTA, 1mM EGTA, 10mM Naβ-glycerophosphate, 5% glycerol, 25mM NaF, 1mM PMSF, 1 mM Na-orthovanadate, protease and phosphatase inhibitor cocktail, pH 7.5; buffer A) CM-Sepharose fast flow resin (Amersham). The column was washed with 5 column volumes of low-salt wash buffer (buffer A + 25mM NaCl) and GSK-3 eluted using elution buffer (buffer A + 200mM NaCl). Fractions containing the highest concentrations of protein were pooled and used in a kinase assay to assess GSK-3 activity.
GSK-3 kinase assays were performed with equivalent amount of eluted protein at 30C for 15 min in a 50 ul reaction volume containing 8mM MOPS pH 7.4, 0.2mM EDTA, 15mM MgCl2, 0.1 mM EGTA, 2.5 μM protein kinase A inhibitor (PKI – Upstate Biotech) 10 mM magnesium acetate, 5 mM Na β-glycerophosphate, 25uM glycogen synthase 2 peptide (Upstate Biotech) and initiated with 2uCi/ul [γ32P]-ATP (6000Ci/mmol). 40 μl of the reaction mixture was spotted onto 2 cm2 squares of Whatman phosphocellulose P81 paper and washed 5 times with 0.75% phosphoric acid, and once with acetone and the incorporation of radioactivity in the peptide determined using a Beckman LS 6500 liquid scintillation counter. As the ratio of GSK-3α:GSK-3β was altered in the partial purification compared to whole cell lysate (in which it is 1:1), activities were corrected for the partial purification loss of GSK-3α as determined by immunoblotting of the assayed fractions. The relative activities are an average of 6 assays from 2 independent experiments with each kinase assay performed in triplicate.
Antibodies
All phospho-specific antibodies were purchased from Cell Signaling Technologies: anti-phospho-β–catenin, S33/S37/T41 (#9561: Lot1) and S45/41 (#9565); anti-phospho-GSK-3, S9/S21 (#9331); and anti-phospho-PKB, S473 (#4058). Other antibodies used were: anti-βactin, ab6276 (Abcam); anti-APC, sc-895 and (Santa Cruz); anti-brachyury, sc-17743 (Santa Cruz); anti-β–catenin, clone 14 (BD Biosciences); anti-Conductin, sc-20784 (Santa Cruz); anti-GAPDH, ab8245 (Abcam); anti-GFAP, Z0334 (DAKO); anti-GSK-3, clone 4G-1E (Upstate); anti-Nanog, ab21603 (Abcam); anti-Nestin, MAB353 (Chemicon); and anti-Oct-4, sc-9081 (Santa Cruz). Species-specific antibodies conjugated to Alexa fluors 488 and 594 were obtained from Molecular Probes.
Immunofluorescence Microscopy
Cells / EBs were fixed with 4% paraformaldehyde / PBS for 15 minutes at RT, and permeabilized with 0.1% Triton X-100 / PBS at RT for 2 minutes (cells) or 15 minutes (EBs). EBs were freeze-fractured before fixation and permeabilization by sandwiching them between positively charged slides (Fisher Scientific), freezing for 10 minutes on dry ice, and then quickly prying apart the slides to fracture the EBs (modification of: (Toumadje et al., 2003)). Primary and secondary antibodies were diluted to final working concentrations in 2% BSA/PBS. Specimens were mounted with ProLong gold anti-fade reagent containing DAPI (Molecular Probes). Images of immunofluorescent samples were obtained using a Zeiss LSM510 confocal microscope.
Immunohistological Staining of Teratomas
Sections from paraffin embedded teratomas were dewaxed and rehydrated following standard procedures. Antigen retrieval for β–catenin and Nestin was achieved using heat-induced retrieval in citrate buffer (pH 6.0), while proteolytic antigen retrieval with proteinase K (DAKO) was used for GFAP. The following primary antibody concentrations were used: anti-β–catenin, 1:250; anti-GFAP, 1:500; anti-nestin, 1:200. Kits specific for rabbit or mouse antigen detection in mouse tissues (Vector labs and DAKO, respectively) were used according to the manufacturers’ recommendations.
Transient Transfections
Single cell suspensions of ES cells were transfected with Lipofectamine 2000 (Invitrogen) using 1–2 million cells, 2 μg DNA and 4 μl Lipofectamine 2000 and plated on a single well of a gelatin-coated 6-well plate (Bugeon et al., 2000).
TCF Reporter Assay
ES cells were co-transfected with TCF reporter constructs (1.8 μg) driving firefly luciferase production and a pRL-CMV (0.2 μg) driving constitutive expression of renilla luciferase for normalization. Treatments with Wnt-3a conditioned medium were done the day after transfection with reporter constructs and lysed using 1X passive reporter lysis buffer. Firefly and Renilla reporter activities was measured using a 96 well based luminometer and detected as per manufacturer’s instructions (Promega Dual-Light system).
Lentivirus Production
WT and mutant versions of GSK-3α were cloned into the pLenti6 /V5 directional TOPO vector and lentiviruses were produced according to the instructions provided by the manufacturer of the ViraPower™ lentiviral expression system (Invitrogen).
Teratoma Production
GSK-3α(flx/flx) and DKO ES cells grown for 2 passages on gelatin-coated dishes to eliminate feeder fibroblasts, were trypsinized into single cell suspensions and resuspended in PBS to a concentration of 107 cells / ml. These cells were injected subcutaneously into the hind limbs of syngeneic 129Ola mice (Harlan) using a 25 Ga needle (200 μl). Teratomas were collected after 3 weeks. Small samples were collected for RNA isolation and the remainder of the tissue was fixed with neutral buffered formalin overnight at 4°C for subsequent paraffin embedding, sectioning and histological / immuno-histological staining.
Statistical Analyses
All statistical analyses were performed using the GraphPad Prism software package. The TCF-reporter assay data were analyzed by ANOVA with the Newman-Keuls post-hoc test. There were four replicate samples for each treatment group. The quantitation of the relative amounts of GSK-3 in the various cell lines was analyzed by ANOVA with Tukey’s post-hoc test. The immunoblot band profiles from 3 independent cell lysates per cell type were used in the analyses.
Supplementary Material
Supplementary Fig. 1. Relative levels of GSK-3α and GSK-3β protein in WT and knockout ES cell lines. Immunoblots of total cell lysates (RIPA buffer) were probed with antibodies detecting total GSK-3 and GAPDH (loading control). The total protein samples in each lane are from independent plates of cells with the denoted genotype.
Supplementary Fig. 2. Effects on cytosolic β-catenin levels in WT, DKO ES cells and DKO ES cells “rescued” with V5-tagged variants of GSK-3α. Immunoblots of cytosolic preparations were probed with antibodies detecting β-catenin, the V5-epitope, total GSK-3 and GAPDH (loading control).
Supplementary Fig. 3. DKO ES cells retain high levels of ES cell marker transcription 7 days after differentiation in medium lacking LIF. Quantitative RT-PCR data for the expression levels of the ES cell markers nanog, oct-4, and rex-1 in WT and DKO ES cells at days 0 and 7 of differentiation. Quantitative RT-PCR data is also presented for brachyury expression in undifferentiated WT and DKO ES cells. Error bars = SEM.
Supplementary Fig. 4. DKO ES cells have highly elevated levels of brachury/T protein compared to WT, single and 3/4 knockout ES cells. A. Immunoblots from WT and knockout ES cells probed with antibodies detecting brachyury/T, GSK-3 and GAPDH (loading control). B. Immunofluorescence data from WT and DKO ES cells immunostained with an antibody against the N-terminus of brachyury/T. Nuclei have been stained with DAPI. Bar = 10 μm.
Acknowledgments
We thank Dr. R. Moon for providing us with TCF reporter plasmids. We also thank Sophia George, from Dr. Andras Nagy’s lab for teaching us how to perform the hanging drop embryoid body assay. We are also indebted to Dr. Scott Pownall for his advice early on in the project with respect to gene targeting strategies and for supplying us with the core plasmid reagents for gene targeting. Funding for this work was provided by the Canadian Institutes of Health Research (J.R.W, S.P. and B.W.D), the Howard Hughes Medical Institute (J.R.W) and the Banting and Best Diabetes Centre (J.R.W. and S.P.).
References
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SJ, Stappert J, Bauer A, Kispert A, Herrmann BG, Kemler R. Brachyury is a target gene of the Wnt/beta-catenin signaling pathway. Mech Dev. 2000;91:249–258. doi: 10.1016/s0925-4773(99)00309-3. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shushan E, Thompson JR, Gudas LJ, Bergman Y. Rex-1, a gene encoding a transcription factor expressed in the early embryo, is regulated via Oct-3/4 and Oct-6 binding to an octamer site and a novel protein, Rox-1, binding to an adjacent site. Mol Cell Biol. 1998;18:1866–1878. doi: 10.1128/mcb.18.4.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugeon L, Syed N, Dallman MJ. A fast and efficient method for transiently transfecting ES cells: application to the development of systems for conditional gene expression. Transgenic Res. 2000;9:229–232. doi: 10.1023/a:1008990510849. [DOI] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Choi J, Park SY, Costantini F, Jho EH, Joo CK. Adenomatous polyposis coli is down-regulated by the ubiquitin-proteasome pathway in a process facilitated by Axin. J Biol Chem. 2004;279:49188–49198. doi: 10.1074/jbc.M404655200. [DOI] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–32481. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Hagen T, Di Daniel E, Culbert AA, Reith AD. Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J Biol Chem. 2002;277:23330–23335. doi: 10.1074/jbc.M201364200. [DOI] [PubMed] [Google Scholar]
- Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kielman MF, Rindapaa M, Gaspar C, van Poppel N, Breukel C, van Leeuwen S, Taketo MM, Roberts S, Smits R, Fodde R. Apc modulates embryonic stem-cell differentiation by controlling the dosage of beta-catenin signaling. Nat Genet. 2002;32:594–605. doi: 10.1038/ng1045. [DOI] [PubMed] [Google Scholar]
- Kim K, Pang KM, Evans M, Hay ED. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell. 2000;11:3509–3523. doi: 10.1091/mbc.11.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma Y, Campbell DG, Cuenda A. Identification of glycogen synthase as a new substrate for stress-activated protein kinase 2b/p38beta. Biochem J. 2004;379:133–139. doi: 10.1042/BJ20031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–21665. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006 doi: 10.1007/s11154-006-9001-5. [DOI] [PubMed] [Google Scholar]
- Lickert H, Domon C, Huls G, Wehrle C, Duluc I, Clevers H, Meyer BI, Freund JN, Kemler R. Wnt/(beta)-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development. 2000;127:3805–3813. doi: 10.1242/dev.127.17.3805. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Lohnes D. The Cdx1 homeodomain protein: an integrator of posterior signaling in the mouse. Bioessays. 2003;25:971–980. doi: 10.1002/bies.10340. [DOI] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. Embo J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends in pharmacological sciences. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- Paling NR, Wheadon H, Bone HK, Welham MJ. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem. 2004;279:48063–48070. doi: 10.1074/jbc.M406467200. [DOI] [PubMed] [Google Scholar]
- Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–692. doi: 10.1038/345686a0. [DOI] [PubMed] [Google Scholar]
- Ruel L, Bourouis M, Heitzler P, Pantesco V, Simpson P. Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature. 1993;362:557–560. doi: 10.1038/362557a0. [DOI] [PubMed] [Google Scholar]
- Salahshor S, Woodgett JR. The links between axin and carcinogenesis. Journal of clinical pathology. 2005;58:225–236. doi: 10.1136/jcp.2003.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–532. doi: 10.1016/s1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- Salingcarnboriboon R, Tsuji K, Komori T, Nakashima K, Ezura Y, Noda M. Runx2 is a target of mechanical unloading to alter osteoblastic activity and bone formation in vivo. Endocrinology. 2006;147:2296–2305. doi: 10.1210/en.2005-1020. [DOI] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Siegfried E, Chou TB, Perrimon N. wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell. 1992;71:1167–1179. doi: 10.1016/s0092-8674(05)80065-0. [DOI] [PubMed] [Google Scholar]
- Skurat AV, Dietrich AD. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem. 2004;279:2490–2498. doi: 10.1074/jbc.M301769200. [DOI] [PubMed] [Google Scholar]
- Stevens LC. The biology of teratomas. Advances in morphogenesis. 1967;6:1–31. doi: 10.1016/b978-1-4831-9953-5.50005-6. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Raya A, Kawakami Y, Morita M, Matsui T, Nakashima K, Gage FH, Rodriguez-Esteban C, Belmonte JC. Maintenance of embryonic stem cell pluripotency by Nanog-mediated reversal of mesoderm specification. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S114–122. doi: 10.1038/ncpcardio0442. [DOI] [PubMed] [Google Scholar]
- Toumadje A, Kusumoto K, Parton A, Mericko P, Dowell L, Ma G, Chen L, Barnes DW, Sato JD. Pluripotent differentiation in vitro of murine ES-D3 embryonic stem cells. In vitro cellular & developmental biology. 2003;39:449–453. doi: 10.1290/1543-706X(2003)039<0449:PDIVOM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- van Noort M, Clevers H. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev Biol. 2002;244:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Umehara H, Murayama K, Okabe M, Kimura T, Nakano T. Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene. 2006;25:2697–2707. doi: 10.1038/sj.onc.1209307. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/Factor A. Embo J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP. T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev. 1999;13:3185–3190. doi: 10.1101/gad.13.24.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Relative levels of GSK-3α and GSK-3β protein in WT and knockout ES cell lines. Immunoblots of total cell lysates (RIPA buffer) were probed with antibodies detecting total GSK-3 and GAPDH (loading control). The total protein samples in each lane are from independent plates of cells with the denoted genotype.
Supplementary Fig. 2. Effects on cytosolic β-catenin levels in WT, DKO ES cells and DKO ES cells “rescued” with V5-tagged variants of GSK-3α. Immunoblots of cytosolic preparations were probed with antibodies detecting β-catenin, the V5-epitope, total GSK-3 and GAPDH (loading control).
Supplementary Fig. 3. DKO ES cells retain high levels of ES cell marker transcription 7 days after differentiation in medium lacking LIF. Quantitative RT-PCR data for the expression levels of the ES cell markers nanog, oct-4, and rex-1 in WT and DKO ES cells at days 0 and 7 of differentiation. Quantitative RT-PCR data is also presented for brachyury expression in undifferentiated WT and DKO ES cells. Error bars = SEM.
Supplementary Fig. 4. DKO ES cells have highly elevated levels of brachury/T protein compared to WT, single and 3/4 knockout ES cells. A. Immunoblots from WT and knockout ES cells probed with antibodies detecting brachyury/T, GSK-3 and GAPDH (loading control). B. Immunofluorescence data from WT and DKO ES cells immunostained with an antibody against the N-terminus of brachyury/T. Nuclei have been stained with DAPI. Bar = 10 μm.