

Abstract
This review article expands on the previous one (S. Selvaraju and Z. El Rassi, Electrophoresis 2012, 33, 74-88) by reviewing pertinent literature in the period extending from early 2011 to present. As the previous review article, the present one is concerned with proteomic sample preparation (e.g., depletion of high abundance proteins, reduction of the protein dynamic concentration range, enrichment of a particular sub-proteome), and the subsequent chromatographic and/or electrophoretic pre-fractionation prior to peptide separation and identification by LC-MS/MS. This review article is distinguished from its second version published in Electrophoresis 2012, 33, 74-88 by expanding on capturing/enriching sub-phosphoproteomes by immobilized metal affinity chromatography and metal oxide affinity chromatography. Seventy-seven papers published in the period extending from mid 2011 to the present have been reviewed. By no means this review article is exhaustive, given the fact that its aim is to give a concise treatment of the latest developments in the field.
Keywords: Affinity chromatography, Immunodepletion, Proteinfractionation, Proteomics, ProteoMiner
1 Introduction
Proteins are important indicators of physiological or pathological states and contribute to the early diagnosis of diseases, which may provide a basis for identifying the underlying mechanism of disease development [1]. Complexity and intensity variability make proteomic analysis of biological fluids a difficult task. Not only the very large number of the protein constituents in the samples, but also the large dynamic concentration range of proteinsnecessitates elaborate sample preparation approaches. Thus, various proteomic sample treatment approaches have been developed in recent years for reducing the complexity of biological samples including enrichment of specific subproteome (e.g., glycoproteome and phosphoproteome), depletion of high abundance proteins (HAPs), and narrowing down the dynamic concentration range of proteins. Following these sample treatments, proteomic samples are further reduced in complexity by chromatographic and electrophoretic fractionations prior to subjecting them to LC-MS/MS for protein identification.
To expand and supplement the previous review article by Selvaraju and El Rassi which covered liquid phase separation systems for proteomics analysis from the period of 2008- early 2011 [2], the current review article is aimed at updating the previously discussed topics and also to involve an additional topic, namely the enrichment of phosphoproteome over the period extending from the mid 2011 to present. This review article is divided into five major parts, including (i) depletion methods for removal of HAPs, (ii) protein equalizer technology to narrow down the dynamic protein concentration range, (iii) chromatographic and electrophoretic fractionation to reduce complexity of proteomic samples prior to LC-MS/MS analysis, (iv) enrichment of phosphoproteome by immobilized metal-ion affinity chromatography (IMAC) and metal oxide affinity chromatography (MOAC), and (v) capturing subglycoproteome by lectin affinity chromatography (LAC) to focus on specific subproteome that may provide protein information of certain diseases. Typically, steps (i) or (ii) are performed first on the sample in proteomic analysis followed by steps (iii), (iv) or (v).
2 Methodologies used to reduce the complexity of proteomic samples
2.1 Depletion methods
The depletion methods in proteomics are used primarily to reduce the complexity of proteomic samples by depleting HAPs (i.e., albumin, immunoglobulin G (IgG), immunoglobulin A (IgA), immunoglobulin M (IgM), transferrin, haptoglobin and α-2-macroglobulin) and at the same time to narrow the differences of protein concentrations in a given proteomic sample [3-10]. The reduction of complexity can be performed by classical methods such as organic solvent solubilization and precipitation methods and by immuno depletion methods [3, 4].
2.1.1 Solvent solubilization and precipitation methods
Many protein biomarkers in biological samples are present at low concentrations, which are always masked by HAPs. The depletion of these HAPs allows the identification of low abundance proteins (LAPs) in proteomic analysis as well as the discovery of new biomarkers for diseases. The organic precipitation methods have been developed to reduce sample complexity by depletion of these HAPs in plasma using organic solvents such as acetonitrile, acetone, and trichloroacetic acid (TCA) [3, 4, 11-13].
Mostovenko et al. [3] introduced semi-selective precipitation with acetonitrile at different pH (e.g., 5.0, 7.0 and 9.0) by adding acetic acid and ammonium hydroxide directly to three identical aliquots of 50 μL each. The acetonitrile was mixed with the sample in 1:1 (v/v) ratio and then incubated for 10 min in an ultrasonic bath at room temperature. Thereafter, the samples were centrifuged at 16,100 g at 4 °C for 10 min. The supernatants containing the extracted LAPs were then separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the isolated proteins were identified by LC-MS/MS. Up to 90% of albumin and other high abundance proteins were removed by adding an equal volume of acetonitrile to the blood plasma samples adjusted to pH 5.0. This method provided good reproducibility and high throughput and was suggested as a well-suited approach for the fractionation of plasma in large clinical studies.
Fattahi et al. described a TCA precipitation for the depletion of albumin in serum[11]. Albumin-TCA complex is soluble in organic solvents such as acetone. In this study, a solution of 10% TCA in acetone was added to 15 mL of serum sample, which was incubated at 20 °C for at least 90 min and then centrifuged at 4 °C for 30 min. The albumin-TCA complex remained in the supernatant phase, while the other proteins (i.e., LAPs) that precipitated out were used for further proteomic analysis.
In another study, the combination of a chemical sequential depletion method based on two protein precipitations with acetonitrile and dithiothreitol (DTT) have been reported for reducing HAPs in osteoarthritis diseased serum [4]. The two chemical depletions were combined in a sequential way in order to efficiently reduce the dynamic range of proteins in the sample. The sera were first incubated with 49.5 mM DTT for 1 h at room temperature and then the samples were centrifuged. The supernatant was further treated with 57% (v/v) ACN, vortexed and then sonicated for 10 min twice. After centrifugation, the supernatant was subjected to two-dimensional differential gel electrophoresis (2D-DIGE) followed by MALDI-TOF for protein identification.
Fischer et al. introduced a precipitation method to identify candidate proteomic biomarkers in the ankylosing spondylitis serum. After removing the IgGs using Protein G agarose beads, non-albumin proteins were removed from the serum with sodium chloride/ethanol precipitation [12]. The depleted serum was subjected to nano LC-MS/MS analysis for protein identification. A total of 316 proteins were identified with 22 proteins showing significant up or down regulations.
In addition to human biological samples, the precipitation method using organic solvent has been applied for protein extraction in plant such as crop tissues using TCA/acetone precipitation with phenol extraction [14] and A. paniculata leaves using chloroform-trichloroacetic acid-acetone with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer [15].
2.1.2 Immunoaffinity depletion methods
Immuno affinity depletion methods involve capturing one or more of the HAPs from biological samples using immobilized antibodies. This is the most widely used approach to decrease the complexity of the biological samples before proteomic analysis [16, 17].
In an interesting report, Yang et al. reported molecularly imprinted polymers known as template-induced artificial antibodies that have the abilities to recognize and selectively bind the target molecule. In this study, artificial albumin and IgG antibodies were developed by using two epitopes of human serum albumin and IgG as templates [10]. Acrylic acid, acrylamide, and N-acryl tyramine were used as monomers, N,N′-ehtylene bisacrylamide served as a crosslinker and cellulosic fibers were the supporting matrix. The epitope-imprint polymers (EIPs) were grafted onto the cellulosic fibers. The adsorption capacity of these artificial antibodies were found to be 15.2 mg, 10 mg and 15 μL per gram for human serum albumin, IgG and human serum, respectively. The dissociation constant of these artificial antibodies toward the human serum albumin and IgG was 1 μM and 0.6 μM, respectively. This shows the efficiency of the EIPs with high affinity toward human serum albumin (HSA) and IgG. The biomimetic properties of these artificial antibodies, coupled with their economical and rapid production, high specificity and their reusability, make them attractive for proteomic analysis and it could substitute monoclonal antibodies and protein A/G.
Selvaraju and El Rassi developed an integrated multi-column platform for the online depletion/capturing/fractionation of proteins from human serum samples (see Fig. 1). In this platform affinity immuno-subtraction column for HSA was combined in tandem with microbial protein-based affinity subtraction columns (e.g., protein A and protein G′) for IgG. In this regards, anti-HSA antibody and protein A and protein G′ were immobilized on the surface of glycerylmethacrylate/pentaerythritol triacrylate monolith. The affinity monoliths thus obtained were used for the simultaneous depletion of albumin and IgG, IgA and IgM from disease-free and breast cancer sera. This was followed by enrichment/concentration of glycoproteins using two lectin columns followed by the fractionation of the captured glycoproteins on an RPC column. The protein fractions were submitted for protein identification by LC-MS/MS. The depletion columns provided eight-fold reduction in albumin binding to the lectin columns. Also, 15 different Ig chains were identified in one lectin fraction in the presence of depletion columns whereas without the depletion column 25 different Ig chains were identified [18].
Figure 1.
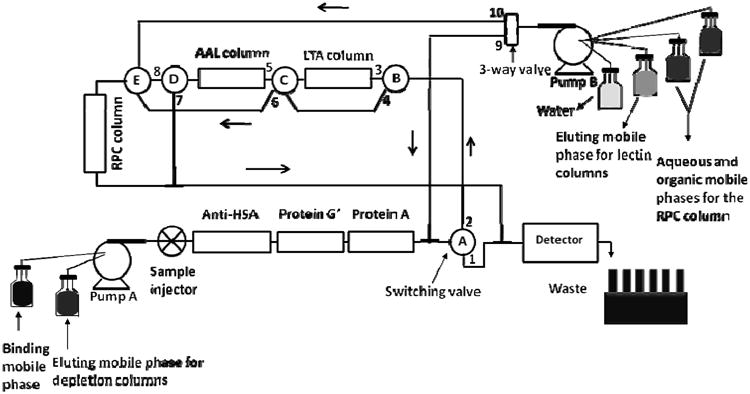
Schematic representation of the platform for the simultaneous depletion of albumin and Igs, and the enrichment of fucosylated glycoproteins, and the subsequent RPC fractionation of the captured proteins. When the switching valve (SV) A, B, C and D were in 2, 3, 5 and 7 positions, respectively, the 3-fold diluted serum was injected onto the depletion and the lectin columns, followed by washing with the binding mobile phase (BMP) using pump A. Then, the eluting mobile phase for the depletion columns was passed by changing the SV-A position to 1, thus by-passing the lectin and the RPC columns. The depletion columns were re-equilibrated again with the BMP, after which the SV-A was changed back to position 2. Then, the LTA column was eluted using pump B, while the 3-way valve was in position 9, SV-B in position 3 and SV-C in position 6, thus by-passing the AAL column and passing through the RPC column. This was followed by washing, eluting and re-equilibrating the RPC column using the mobile phase from pump B, while the 3-way valve is in position 10. Then, the AAL column was eluted by changing the 3-way valve position back to 9, SV-B position to 4, SV-C position to 5 and SV-D position to 8. This was again followed by washing, eluting and re-equilibrating the RPC column by keeping the 3-way-valve in position 10.(Reproduced with permission from Ref. [18])
Many commercial depletion kits and spin columns are finding use in Proteomics. For instance, Holewinski et al. compared five different commercially available affinity capture reagents for the depletion of HSA and IgG from serum and cerebrospinal fluid (CSF) [5]. Isolated serum and CSF albuminomes from ProteaPrep spin columns were analyzed by LC-MS/MS.128 serum proteins were identified including 45 proteins not previously reported and 94 CSF albuminome proteins were identified, 17 of which are unique to CSF.
The commercial depletion kits were used by Yu at al. who studied the depletion of albumin and IgG for the quantification of transferrin in human serum [19]. Briefly, serum was depleted using the ProteoPrep® Blue Albumin and IgG Depletion Kit according to the manufacture's protocol followed by LC-MS/MS. The validated calibration range was from 500-5000 ng/mL. The inter- and intra-day precisions were less than 4.9% and 9.9%, respectively, and the bias of quality control samples was less than 5.4%. The results demonstrated that this depletion method coupled with LC-MS/MS could measure differences in the level of transferrin in human serum and also applied to the quantitative analysis of transferrin in clinical samples.
In another attempt of using commercial depletion kits, Huillet et al. [20] reported the depletion of six most HAPs from serum using the human Multiple Affinity Removal Spin (MARS) cartridge. This technology enables the removal of albumin, IgG, antitrypsin, IgA, transferrin and haptoglobin in a single step. Also, the same MARS cartridge was used for the depletion of HAPs from serum samples by Scholl et al. [21], and the LAPs fraction was concentrated by TCA/acetone precipitation in order to evaluate the performance of affinity removal column by 2D-DIGE followed by protein identification using LC-MS/MS.
Recently, Tu et al. also reported the depletion of 14 HAPs (albumin, IgG, IgA, IgM, complement C3, antitrypsin, haptoglobin, transferrin, fibrinogen, α-2-macroglobulin, α-1-acid glycoprotein, apolipoprotein AI, apolipoprotein AII, and transthyretin) from serum samples by Agilent Human 14 MARS [22].
Chicken IgY responses against mixtures of six HAPs from plasma including α-1-antitrypsin, albumin, transferrin, haptoglobin, IgG and IgA have been studied by Tan et al [23]. The investigation was performed across a range of abundances from 1:1 for all six proteins to where one protein predominated above the other five by ≤ 1000 fold. This study demonstrated that the ability to mount an IgY response to complex human plasma protein antigens is conclusively driven by a combination of individual antigen immunogenicity and the relative abundance of components within any mixture.
Janecki et al. [6] developed a multi-column system for the depletion of abundant proteins. This involved the transformation of a conventional HPLC system to a low backpressure liquid chromatography set-up for automated serum/plasma depletion and fractionation. The system was configured to use two immunoaffinity columns. All samples were serially depleted through these two columns, first for depletion of the 14 most abundant proteins by Seppro™ IgY14 LC10, and then for next 200-300 proteins using Seppro™ SuperMix LC5 column. Up to nine samples could be depleted reproducibly within 24 h period.
The commercially available Seppro™ IgY14 LC10 column was also used for the depletion of 14 HAPs including albumin, α-1-antitrypsin, IgM, haptoglobin, fibrinogen, α-1-acid glycoprotein, HDL, LDL, IgG, IgA, transferrin, α-2-macroglobulin, and complement C3. The operation of the IgY column was optimized with buffers for sample loading, washing, eluting and column regeneration. Under the optimized conditions, the specificity and efficiency of the IgY based column allowed the quantitative removal of the 14 HAPs, which represent approximately 95% of the total protein mass in human plasma [24].
2.2 Protein equalizer technology
Protein equalizer technology or combinatorial peptide ligand library (CPLL) is an established technology for reducing protein concentration differences in biological samples. The CPLL comprises a diverse library of peptide ligands coupled to spherical porous beads. The peptide ligand libraries are peptides made of different mixtures of amino acids with different lengths, where four is the minimum and six is probably the best [25]. The CPLL beads are able to “equalize” the protein population when in contact with complex proteomic samples, by sharply reducing the concentration of the most abundant components, while simultaneously enhancing the concentration of the most dilute species.
The CPLL technology was combined with other fractionations such as tandem iminodiacetic acid (IDA)-metal chelate columns (e.g., IDA-Zn2+, IDA-Ni2+ and IDA-Cu2+) in series with an RPC column [26]. After the equalization step, the treated proteins from human serum were fractionated on a series of IDA-metal columns arranged in the order: IDA-Zn2+ →IDA-Ni2+→IDA-Cu2+ followed by the RPC column. Aliquots taken from the four collected fractions from the four tandem columns were subsequently fractionated by two-dimensional electrophoresis (2-DE), and also some part of the four collected fractions were typically digested and analyzed by LC-MS/MS. The number of identified proteins in treated serum (i.e., ProteoMiner™ treated serum) without any further fractionation was found to be 66 proteins while 82 proteins were identified with post IMAC/RPC fractionation. Moreover, the equalizer technology was compared to the immuno-subtraction approach. ProteoMiner™ and immuno-subtraction treatments of biological fluids can be viewed as complementary approaches for facilitating the comprehensive profiling of proteomics samples, while the ProteoMiner™ technology is superior in terms of the overall number of captured proteins.
The immobilization of a single chain variable fragment (scFv) displaying M13 phage library on magnetic microspheres for use as a protein equalizer for the treatment of human serum has been recently reported [27]. Firstly, the diluted human serum was loaded on 100 mg scFv displaying M13 phage library functionalized magnetic microspheres (scFv@M13@MM). After incubation, the supernatant was collected and the captured proteins on scFv@M13@MM were eluted with 2 M NaCl, 50 mM glycine-hydrochloric acid, and 20% (v/v) ACN with 0.5% (v/v) trifluoroacetic acid in sequence. Finally, scFv@M13@MM were saturated with thrombin, and the supernatant was collected. All collected fractions were first analyzed by SDS-PAGE with silver staining. The difference of protein concentration was reduced in the treated human serum sample compared with untreated sample. The eluates were also analyzed by online 2D-strong cation exchange (SCX)-RPC-MS/MS. It was found that 10 HAPs could be removed efficiently with the protein equalizer, and the number of identified proteins from the protein equalizer treated human serum could be improved 100% compared with that from the untreated sample. This demonstrates that the developed protein equalizer is of high potential for improving the human serum proteome analysis, which could lead to the discovery of candidate biomarkers or drug targets.
González-lglesias et al. [28] carried out a comparative differential proteomic analysis of blood serum from patients with Primary Open-Angle Glaucoma (POAG) and Pseudoexfoliation Glaucoma (PEXG), and healthy controls. The ProteoMiner™ technology was used to increase the relative concentration of the proteins within the medium and low dynamic concentration ranges. 2D-DIGE was used to identify changes in the concentrations of proteins between equalized serum samples from POAG, PAXG, and controls serum. The proteins were also identified using MALDI-TOF/TOF, and nano LC-MS-MS. This approach identified a panel of 35 serum proteins that were found in altered concentrations in patients with glaucoma relative to healthy controls. The top-17-ranked proteins of this panel were determined by enzyme-linked immunosorbent assays (ELISAs) to be present in glaucoma sera in higher concentrations than in serum from the healthy control group. These data offered new perspectives in the discovery of glaucoma biomarkers in the serum of POAG and PEXG patients.
Balfoussia et al. [29] reported the use of the ProteoSeek Albumin/IgG removal kit and the ProteoMiner enrichment kit for sample preparation in order to detect medium- and low-abundance proteins, whose expression may be masked due to high-abundance proteins. The athletes' plasma was analyzed by 2-DE and the differentially expressed proteins (DEPs) were identified by LC-MS/MS. Overall, 52 proteins were differentially expressed between the starting point, the finishing line and two days after the end of the race. Of these, 30 proteins were involved in inflammation, while the rest concerned anti-oxidation, anti-coagulation and iron and vitamin D transport. These results indicate that prolonged physical stress affects circulating stress-related proteins, which might be employed as biomarkers of stress-related diseases.
The protein equalizer technology was also reported for adjusting the concentration of Male New Zealand Obese (NZO) mice plasma sample [30]. The treated plasma sample was then analyzed by LC-targeted selected reaction monitoring (SRM)-MS. The results demonstrated that apolipoprotein E, mannose-binding lectin 2, and parotid secretory protein are present at significantly different quantities in equalized plasma of diabetic NZO mice compared to non-diabetic controls using AQUA peptides. In addition, the identified phenotype-specific protein signatures in plasma of NZO mice corresponded to type 2 diabetes-associated changes observed in humans.
In another study, ProteoMiner was used to “equalize” cow's milk proteins in order to investigate cow's milk allergens in human colostrum of term and preterm newborns' mothers, and other minor protein components by proteomics techniques [31]. It was found that the detection of the intact bovine α-S1-casein in human colostrum, then bovine α-1-casein could be considered the cow's milk allergen that is readily secreted in human milk and could be a cause of sensitization to cow's milk in exclusively breastfed predisposed infants. And also, the detection at very low concentrations of proteins previously not described in human milk (e.g., galectin-7, the different isoforms of the 14-3-3 protein and the serum amyloid P-component), probably involved in the regulation of the normal cell growth, in the pro-apoptotic function and in the regulation of tissue homeostasis.
The urchin Paracentrotus lividus has been characterized via previous capture and enhancement of LAPs with CPLL [32]. The treated sea urchin coelomic fluid was then analyzed using SDS-PAGE followed by LC-MS/MS for proteins identification. Whereas in the control only 26 unique gene products could be identified, 82 species could be detected after CPLL treatment.
Hexapeptide ligands were evaluated in glycan analysis [33]. A repeatable on-bead glycan release strategy was developed, and glycans were analyzed using capillary sieving electrophoresis on a DNA analyzer. Binding of proteins to the hexapeptide library occurred via the protein backbone. At neutral pH, no discrimination between protein glycoforms was observed. Interestingly, glycan profiles of plasma with and without hexapeptide library enrichment revealed very similar patterns, despite the vast changes in protein concentrations in the samples. The most significant differences in glycosylation profiles were ascribed to a reduction in immunoglobulin-derived glycans. These results suggested that specific and sensitive biomarkers are hard to access on the full plasma level using protein enrichment in combination with glycan analysis.
Malaud et al. [34] reduced the dynamic range of proteins by CPLL treatment of humancarotid artery atherosclerotic plaques. After the enrichment step, the abundance of major proteins was decreased, revealing different protein profiles as assessed by both SDS-PAGE and 2-DE comparative analyses and then identification of protein using LC-MS/MS. Novel low-abundance proteins were detected correlating very well with biological alterations related to atherosclerosis (heat shock protein 27 (HSP27) isoforms, aldehyde dehydrogenase, moesin, protein kinase C delta-binding protein, and inter-α trypsin inhibitor family heavy chain-related protein (ITIH4)). The detection of different isoforms of a low-abundance protein such as HSP27 species was actually improved after enrichment of tissue protein extracts.
Cumová et al. [35] determined an appropriate pre-fractionation method of blood plasma prior to a subsequent proteomic analysis of low-abundant fraction of proteins by 2-DE and MS to improve the resolution of 2-DE maps and protein identification. Two prefractionation methods, namely, MARS and ProteoMiner were compared preceding 2-DE analysis using 10 blood plasma samples. Based on the results of the comparative experiments, low-abundant plasma protein fractions from 18 multiple myeloma patients treated with bortezomib were analyzed. Samples that were prefractionated by ProteoMiner method yielded 2-DE maps with a significantly increased number of detected protein spots as compared to MARS.
Three different methods, including hydrogel nanoparticles, ProteoMiner(®) peptide ligand affinity beads and Sartorius Vivaspin(®) centrifugal ultrafiltration device, were compared and evaluated in order to select the best strategy for the enrichment and prefractionation of LAPs [36]. A shotgun proteomics approach was adopted, with in-solution proteolytic digestion of the whole protein mixture and determination of the resulting peptides by nano HPLC coupled with a high-resolution Orbitrap LTQ-XL mass spectrometer. The results showed that the hydrogel nanoparticles performed better in enriching the LMW protein profiles, with 115 proteins identified against 93 and 95 for ProteoMiner(®) beads and Sartorius Vivaspin(®) device, respectively.
Recently, Lichtenauer et al. have established a novel approach for proteomic biomarker identification in peritoneal dialysis effluent (PDEs) using the equalizer technology followed by 2D-DIGE [37]. The experimental approach was proven using a model system for PDE that is further referred to as artificial PDE, which was established by spiking unused peritoneal dialysis fluids with cellular proteins reflecting control conditions or cell stress. The results showed that the equalizer technology could be applied to work in this model system. Using this CPLL was not only to reduce the amount of high abundant plasma proteins but also to concentrate low abundant proteins from plasma or cellular origin while preserving changes in abundance due to any treatment or clinical condition. In addition, to near qualitative changes, quantitative assessment of spot count confirmed the greater range of individual protein spots available for protein quantification in 2D-DIGE.
In another study, LAPs from serum sample of early rheumatoid arthritis (RA) patients and healthy controls were enriched using ProteoMiner™ technology [38]. The enriched proteins were separated on SDS-PAGE, digested by trypsin, labelled with tandem mass tag reagents, and desalted by C18 stage tip column. Then, the labelled peptides were analyzed by LC-MS/MS. The DEPs were screened by intensity ratios of identified peptides. ELISAs were performed to confirm DEPs. Twenty-six proteins were found differentially expressed in RA serum by high-resolution proteomic analysis. Further study by ELISA showed a higher level of ficolin-2 in the serum of RA patients compared to healthy controls. The results of this study suggested that ficolin-2, as a newly screened biomarker by high-resolution quantitative proteomic analysis, offers the potentiality to become a diagnostic or disease evaluation tool in RA.
Very recently, ProteoMiner™ technology was evaluated as an off line step to narrow the differences of protein concentration in human serum prior to the capturing of the human fucome from disease-free and breast cancer sera by a multicolumn platform via LAC followed by the fractionation of the captured glycoproteins by RPC [39]. This platform is a slightly modified version of the one shown in Fig. 1 in the sense that the depletion columns that were used online to remove albumin and immunoglobulins have been replaced by an off line protein equalization via the CPLL technology. Two monolithic lectin columns specific to fucose, namely Aleuria aurantia lectin (AAL) and Lotus tetragonolobus agglutinin (LTA) columns were utilized to capture the fucome, which was subsequently fractionated by RPC yielding desalted fractions in volatile acetonitrile-rich mobile phase (see Figs. 2 and 3). The desalted fractions were first subjected to trypsinolysis and then to LC-MS/MS. The combined strategy consisting of the CPLL, multicolumn platform and LC-MS/MS analysis permitted the identification of the DEPs in breast cancer serum yielding 58 DEPs in both the LTA and AAL fractions with 6 DEPs common to both lectins. 17DEPs were of the low abundance type, 16 DEPs of the borderline abundance type, 4 DEPs of the medium abundance type and 15 DEPs of the high abundance type. The remaining 6 DEPs are of unknown concentration. The results showed that the combination of CPLL with the multicolumn platform facilitated the detection of DEPs by LC-MS/MS that cover a wide range of abundance spanning from very low- to borderline low-, medium-, and high-abundance proteins. One important feature of the platform shown in Fig. 1 is that it generates RPC chromatograms that show differential protein expression at a glance. In fact, upon a visual examination of the RPC chromatograms in Fig. 2 generated for the LTA captured proteins, it can be seen that in some of the fractions such as 3, 6 and 7, the peak intensity of the fractions from the cancer serum is higher as compared to those from the normal serum. In these fractions, a total of 12 proteins were found to be up regulated. Out of these 12 proteins, 6 proteins are of the low abundance proteins, 3 proteins are borderline abundance between medium and low abundance proteins, 2 proteins are medium abundance proteins, and the concentration of actin cytoplasmic 1 is unknown. Similarly, upon quick visual inspection of the RPC chromatograms in Fig. 3 corresponding to the AAL captured proteins, one can see that some of the peaks in the RPC chromatogram of the cancer serum are of higher intensity as compared to those of the RPC chromatogram of the disease-free serum. Some of the proteins that were found to be up-regulated in fractions 3, 5, 6 and 7 included some of the low abundance proteins such as C4b-binding protein alpha chain, thrombospondin-1, protein disulfide-isomerase, vitronectin and von Wille brand factor, some of the borderline abundance proteins between low and medium abundance proteins such as fibronectin, apolipoprotein C-II, apolipoprotein E and vimentin and the high abundance protein apolipoprotein A-I.
Figure 2.
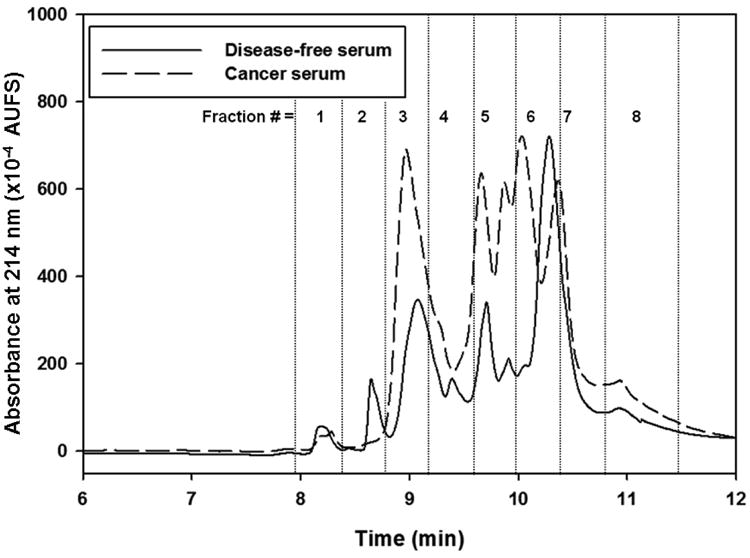
Chromatograms of RPC gradient for LTA captured band from equalized disease-free and cancer serum. The RPC gradient consisted of 0 % to 75 % of mobile phase B in 12 min. Mobile phase A consisted of H2O/ ACN (95:5 v/v) containing 0.1% TFA and mobile phase B consisted of ACN/H2O (95:5 v/v) containing 0.1% TFA. Flow rate 1 mL/min and wavelength 214 nm. (Reproduced with permission from Ref. [39])
Figure 3.
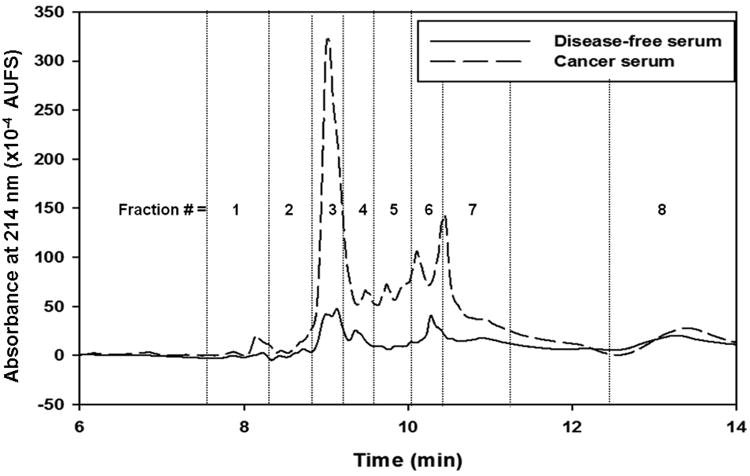
Chromatograms of RPC gradient for AAL captured band from equalized disease-free and cancer serum. Other conditions same as Fig. 2. (Reproduced with permission from Ref. [39])
2.3 Chromatographic and electrophoretic enrichment/prefractionation methods
Chromatographic and electrophoretic methods, which have been used for enrichment/prefractionation of biological samples online or offline prior to the identification of their protein components using LC-MS/MS, are the subject of this section. The progress made is highlighted and in no mean the discussion provided below is exhaustive.
2.3.1 Chromatographic enrichment/prefractionation methods
In this section, for clarity, the chromatographic methods are treated in the sequence: one-dimensional chromatographic approaches first followed by multidimensional chromatographic approaches.
Ritorto et al. [40] tested the ability of three strong anion exchanger (SAX) columns differing in their hydrophobicity to fractionate RAW 264.7 macrophage cell lysate. A strong anion exchanger column with a hyperbranched architecture quaternary ammonium ion functionality and an ultra low hydrophobicity coupled to low pH RPC has been used for the fractionation and separation of tryptic peptides from both a mixture of 6 proteins as well as mouse cell lysate. This column provided very high orthogonality and proved to be superior to an optimized high pH/low pH RPC separation approach and enabled to identify >9,000 proteins from RAW 264.7 mouse macrophage cell lysate in just one week of MS instrument time which allowed a better comparison of proteomics and transcriptomics data. This shows that hydrophilicity and the ion exchange properties of the column are important for the high orthogonality of this approach, which the authors term hydrophilic strong anion exchange chromatography.
In another report, the herbicide-resistant gene-related protein 5-enolpyruvylshikimate-3-phosphate synthase (CP4 EPSPS) of genetically modified (GM) soya was separated from high abundance proteins by using SAX chromatography and SDS-PAGE [41]. After the proteins were loaded onto the SAX column, they were eluted with eluting buffers at various NaCl concentrations. A total of 20 fractions collected were desalted and analyzed by SDS-PAGE. The gel bands at 47 kDa were excised and divided into 5 groups. Each group was combined and then subjected to in-gel digestion with trypsin. Thereafter, the tryptic peptides from the samples and reference were labelled with formaldehyde-H2 and formaldehyde-D2, respectively. The two-labelled pools were mixed and analyzed using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). The data showed a good correlation between the peak ratio of the H- and D-labelled peptides and the GM soya percentages at 0.5, 1, 3, and 5 %, with R2 of 0.99. Since the labelling reagents are readily available, and the labelling experiments as well as the detection procedures are simple, the approach was proclaimed as useful for the quantification of GM soya at a level as low as 0.5 %.
Sun et al. [42] reported an improved shotgun method for analyzing proteomic samples containing sodium dodecyl sulfate (SDS) by using SCX-LC for SDS removal as the first dimension of the two-dimensional LC-MS/MS workflow. Although the authors reported their work as a 2D-LC, based on the sample preparation and prefractionation part before analysis by LC-MS/MS, the work is considered in this review as a 1D-LC. The concentrations of chemical reagents and salts in the sample, the SDS concentration, and the SCX mobile phase composition, were investigated in order to optimize the performance of SDS removal. It was found that a peptide recovery rate of about 90% could be achieved while removing SDS efficiently. One key finding was that, by increasing the SDS concentration to a certain level (0.5%) in the digested peptide sample, the sample recovery rate could be increased. The peptide recovery rate of bovine serum albumin (BSA) digests was found to be 90.6 ± 1.0% (n = 3), and SDS in the SCX fractions collected was not detectable by pyrolysis GC-MS. The authors demonstrated the application of this method for 2D-LC MS/MS profiling of the MCF-7 membrane protein fraction and identified 6889 unique peptides, corresponding to 2258 unique proteins or protein groups from two replicate experiments with a false peptide discovery rate of ∼0.8%, compared to 5172 unique peptides and 1847 unique proteins identified by the RapiGest method that utilizes the acid labile surfactant sodium-3-[(2-methyl-2-undecyl-1,3-dioxolan-4-yl)-methoxyl]-1-propanesulfonate to improve membrane proteins handling by LC-MS/MS.
Similarly to 1-D fractionation, there were also some novel trends in multidimensional fractionations, and the progress made in this area is described in the following sections. Boichenko et al. [43] reported multidimensional separation of tryptic peptides from human serum proteins. The study demonstrated that peptide fractionation on SCX, weak anion exchange (WAX) in the electrostatic repulsion hydrophilic interaction chromatography (ERLIC) mode, RP C18 at pH 2.5 (low pH), fused-core fluorinated at pH 2.5, and RP C18 at pH 9.7 (high pH) stationary phases resulted in two to three times more identified proteins and three to four times more identified peptides in comparison with 1D nano Chip-LC–MS/MS quadrupole TOF analysis. The largest number of peptides and proteins was identified after prefractionation in the ERLIC mode due to the more uniform distribution of peptides among the collected fractions and on the RP column at high pH due to the high efficiency of RP separations and the complementary selectivity of both techniques to low-pH RP chromatography. A 3D separation scheme combining ERLIC, high-pH RP, and low pH nanoChip-LC–MS/MS for crude serum proteome profiling resulted in the identification of 208 proteins and 1088 peptides with the lowest reported concentration of11 ng/mL for heat shock protein 74.
Zhou et al. demonstrated an automated nanoflow 3-dimensionalLC (3D-LC)-MS/MS platform based on high-pH RP, SAX, and low-pH RP(RP-SAX-RP) separation stages for the analysis of complex proteomes [44]. The results revealed that the automated 3-D platform consisting of RP-SAX-RP outperformed RP-RP in the analysis of tryptic peptides derived from Escherichia coli and enabled the identification of proteins present at a level of 50 copies per cell in Saccharomyces cerevisiae, corresponding to an estimated detection limit of 500 amol, from 40 μg of total lysate on a low resolution 3-D ion trap mass spectrometer. A similar study was performed on A LTQ-Orbitrap yielded over 4000 unique proteins from 5 μg of total yeast lysate analyzed in a single, 101 fraction RP-SAX-RP LC-MS/MS acquisition, providing an estimated detection limit of 65 amol for proteins expressed at 50 copies per cell.
In another work, a WAX chromatography as an additional, complementary dimension to SCX and RP have been performed for the enrichment and separation of phosphopeptides [45]. Firstly, SCX was used to fractionate a human lysate digest to generate a fraction highly enriched in phosphopeptides. Thereafter, the analysis of this single fraction by RP-LC–MS with a 140 min gradient method allowed the identification of 4045 unique phosphopeptides (false discovery rate (FDR) < 1%; Mascot score > 20) using an Orbitrap Velos. Triplicate analysis (420 min total gradient time) of the same sample increased the total to just over 5000 unique phosphopeptides. The same sample was separated by WAX and analyzed 14 WAX fractions by 30 min gradient RP-LC–MS (420 min total gradient time). 7251 unique phosphopeptides were identified, an approximate increase of 40%, while maintaining the same total gradient time. Moreover, the analysis of the same 14 WAX fractions by the use of 140 min gradient LC–MS analyses resulted in the detection of over 11 000 unique phosphopeptides. These results clearly demonstrate that additional separation dimensions are still necessary for in-depth phosphoproteomics and that WAX is a suitable dimension to be combined and sandwiched between SCX and RP chromatography.
Song et al. [46] reported the use of an automated online RP-SCX-RP multidimensional separation system in a comparative phosphoproteome analysis of hepatocellular carcinoma (HCC) and normal human liver tissues. In this study, a pseudo triplex stable isotope dimethyl labelling approach was combined with the RP-SCX-RP system to improve the accuracy of large-scale quantitative phosphoproteome analyses. The phosphopeptides were labelled with light, intermediate, and heavy dimethyl labelling reagents, respectively, and were pooled together and then were subjected to Ti4+-IMAC enrichment and online RP-SCX-RP multidimensional separation. Thereafter, the samples were analyzed by LC-MS/MS. Over 1800 phosphopeptides corresponding to ∼2000 phosphorylation sites were quantified reliably in a 42 h multidimensional analysis. The pro-directed motifs, which were mainly associated with the extracellular signal-regulated kinases, were observed as being overrepresented in the regulated phosphorylation sites, and some quantification results of phosphorylation sites were validated by the other studies. Therefore, this RP-SCX-RPC combined with a pseudo triplex labelling approach was demonstrated as a promising alternative for the comprehensive quantitative phosphoproteome analysis. Another advantage of this approach is only half of the comparative sample is consumed to achieve two quantitative measurements. This is extremely important for the proteome quantification of precious samples, such as limited clinical samples.
Garbis et al. [47] developed a novel multidimensional protein identification technology (MudPIT) approach for the in-depth proteome analysis of human serum derived from patients with benign prostate hyperplasia using rational chromatographic design principles. The proposed MudPIT approach encompassed the use of three distinct yet complementary liquid chromatographic techniques. High-pressure size-exclusion chromatography (SEC) was used for the prefractionation of serum proteins followed by their dialysis exchange and solution phase trypsin proteolysis. The tryptic peptides were then subjected to offline zwitterion–ion hydrophilic interaction chromatography (ZIC-HILIC) fractionation followed by their online analysis with reversed-phase nano-ultra performance chromatography (RP-nUPLC) hyphenated to nanoelectrospray ionization-tandem mass spectrometry (nESI-MS/MS) using an ion trap mass analyzer. The SEC prefractionation was performed isocratically with two serially connected 8.0 mm i.d. × 300 mm L Shodex KW-804 SEC columns. For the SCX fractionation, a polysulfethyl A (1.0 mm i.d. × 200 mm; 5 μ particle; 300 Å pore) column was used. The proposed MudPIT approach, incorporating SEC for the protein prefractionation as segments and their dialysis exchange and trypsin proteolysis, followed by the offline first dimensional fractionation of the tryptic peptides with ZIC-HILIC fractionation and subsequent online RP-nUPLC-nESI-MS/MS analysis, resulted in the identification of 1955 proteins (p ≤ 0.05; FDR ≤ 5%) with a broad spectrum of biological and physicochemical properties including secreted, tissue-specific proteins spanning approximately 12 orders of magnitude in natural abundance levels, and encompassed in this proteome was the confident identification of 375 phosphoproteins (p ≤ 0.05; FDR ≤ 5%) of potential importance to cancer biology.
A 2D separation platform was constructed using micro SCXLC (μ-SCXLC) and RP pressurized capillary electrochromatography (RP-pCEC) for the analysis of complex samples [48]. Samples were fractionated by the first dimension μ-SCXLC with a linear solvent gradient and then injected into the second-dimension RP-pCEC for further separation. The μ-SCXLC/RP-pCEC 2D system with three separation mechanisms, namely SCX, RP chromatography and electrophoresis, provided high selectivity, high resolution and high peak capacity compared to one-dimensional chromatographic approaches. Separation effectiveness of this 2D system was demonstrated by the analysis of different kinds of complex samples, such as traditional Chinese medicine Cortex Phellodendri, BSA tryptic digest and real serum tryptic digest. A theoretical peak capacity of approximately 1200 was achieved, which proves its promising potential for the separation and analysis of complex samples.
Zhao et al. [49] developed an online 2D-LC platform for shotgun proteomics analyses of lysates of rat PC12 cell lines and Saccharomyces cerevisiae. The platform based on the online coupling of HILIC using a nonionic type of TSK gel Amide 80 at either pH 6.8 (neutral) or 2.7 (acidic) with conventional low-pH RPC. The fractions from HILIC column were collected and each fraction was further separated in the second-dimension RP column prior to mass spectrometric analysis. 2554 non-redundant proteins from duplicate analyses of a Saccharomyces cerevisiae lysate were identified, with the detected protein abundances spanning from approximately 41 to 106 copies per cell, which contained up to approximately 2092 different validated protein species with a dynamic range of concentrations of up to approximately 104. These results showed that novel fully automated online 2D HILIC–RP LC platform is a good alternative 2D-LC platform for protein identifications in complex biological samples.
Xia et al. [50] introduced an integrated multidimensional nano-flow LC platform for separation of Escherichia coli whole cell lysate. The platform allowed first the separation of proteins by a weak anion and weak cation mixed-bed micro column under a series of salt steps, then the online digestion using a trypsin immobilized enzymatic reactor, followed by the digests trapped and desalted by a C18 precolumn, and finally separated by nano-RPLC. The desalted proteins were then identified by electrospray ionization-MS/MS. In comparison with the results obtained by shotgun approach, the identified protein number was increased by 6%, with the consumed time decreased from 38 h to 14 h. And also the required sample amount was decreased to 8 μg for this current platform compared with integrate platform based on μ-HPLC.
Lam et al. [51] reported an online multidimensional LC (MDLC) for proteomics sample separation. The system contains a first-dimension RP column, which separates peptides in terms of their hydrophobicity at pH 10, and a second (last)-dimension RP column, which separates peptides in terms of their hydrophobicity at pH 2. The system can also operate in 2D mode or in 3D separation mode with the addition of a SCX separation column, which fractionates peptides based on their charge, between the two RP columns. While the 3D separation yielded 20–34 % more proteins with the extended sequence coverage relative to the performance of the 2D separation, the analytical time of 3D separation was ∼3 times longer than that when using the 2D separation. The performance of the 3D system has been benchmarked through analysis of the total lysate of Saccharomyces cerevisiae lysates at low-microgram levels and over 50 % of the yeast proteome was identified.
2.3.2 Electrophoretic enrichment/prefractionation methods
Electrophoretic fractionation methods including 2-DE, 2D-DIGE, isoelectric focusing (IEF) and free flow electrophoresis (FFE) have been reported for separation/fractionation prior to protein identification using LC-MS/MS. For instance, Di Carli et al. [52] investigated changes in the proteome during berry withering using a 2D-DIGE quantitative approach to determine the protein dynamic changes during post-harvesting in Vitis vinifera cv. Corvina berry, which is the base grape variety for Amarone and Recioto wines. Before electrophoresis, protein samples were covalently labelled using the CyDyes DIGE Fluors (Cy2, Cy5, and Cy3). On a total of 870 detected spots, 90 were differentially expressed during berry ripening/withering and 72 proteins were identified by MS/MS analysis. The majority of these proteins were related to stress and defense activity (30%), energy and primary metabolism (25%), cytoskeleton remodelling (7%), and secondary metabolism (5%). In summary, the authors demonstrated how the protein profile associated with ripening process is modified after harvesting, thus influencing the fluctuations in the berry molecular components before vinification.
Sui et al. [53] presented a direct comparison between SCX and IEF based on immobilized pH gradient (IPG) strips (IPG-IEF) for phosphoproteomic separation. The comparison experiments were carried out using standard phosphoproteins and a real sample of HepG2 cell. SCX separation of phosphopeptides was carried out using a Hypersil SCX column and a gradient elution at increasing NH4Cl in the mobile phase. In the case of IPG-IEF separation, the protein digest was mixed with 0.5% IPG buffer, which was applied to an 18 cm linear Immobiline DryStrip at pH 3.0–10.0. After the focusing process, gel pieces were placed in Eppendorf tubes containing an extraction of peptides with 50% ACN, 0.1% TFA solution. The comparison of 18O labelling phosphopeptides' stability under IPG-IEF with that under SCX was performed by analyzing the fractions obtained from both techniques (SCX and IPG-IEF) via high mass accuracy LTQ-FTICR-MS/MS. The results demonstrated that both SCX and IPG-IEF are useful in phosphopeptides enrichment analyses on a large-scale and that SCX is superior to IPG-IEF. However, the 18O labelling phosphopeptides in SCX are less stable than those in IPG-IEF.
In another study, a 2-DE based proteomic workflow as a tool to investigate feed effects on fish by analyzing protein changes in the pyloric ceca (PC) from rainbow trout was introduced [54]. Rainbow trout divided into five groups were fed for 72 days with feeds varying in protein composition. Proteins were extracted from the PC of rainbow trout and subsequently separated using 2-DE followed by identification of proteins using MS. By using2-DE, proteins extracted from the pyloric ceca were separated, making it possible to measure the abundance of more than 440 protein spots. The expression of 41 protein spots was found to change due to differences in feed composition. Using MS, 31 of these proteins were identified, including proteins involved in digestion (trypsinogen, carboxylic ester hydrolase, and aminopeptidase). This study has clearly demonstrated that changes in the fish feed composition resulted in protein changes in the PC of rainbow trout.
Zhang et al. [55] reported a global analysis of protein expression profiles in human Esophageal squamous cell carcinoma(ESCC) tissues and adjacent normal tissues using 2-DE and MALDI-TOF/MS analysis. The results showed that a total of 104 protein spots with different expression levels were found on 2-DE, and 47 proteins were eventually identified by MALDI-TOF/MS. Among these identified proteins, 33 proteins including keratin 17, biliverdin reductase B, proteasome activator subunit 1, manganese superoxide dismutase, high-mobility group box-1, heat shock protein 70, peroxiredoxin, keratin 13, and so on were overexpressed, and 14 proteins including cystatin B, tropomyosin 2, annexin 1, transgelin, keratin 19, stratifin, and so on were down-expressed in ESCC.
Kuramitsu et al. [56] also reported the use of 2-DE and MALDI-TOF/MS for proteomics analysis of hepatitis C virus-associated 21HCCtissues to find proteins that might be involved in tumor differentiation and progression. One of the numerous spots, which was located next to three spots of glutamine synthetase showed stronger intensity in well-differentiated HCC tissues compared to non-cancerous tissues. Samples from 6 out of 21 patients showed up-regulation of this spot compared to non-cancerous tissues. After in-gel digestion, the spot was then identified by MALDITOF/MS as tubulin α-6 chain. Two-dimensional immunoblot analysis confirmed that this spot was indeed tubulin alpha, and this spot was stronger in cancerous tissues than in noncancerous tissues.
A novel approach combining FFE and DIGE was reported to identify DEPs in HT29 colorectal cells [57]. An approach analogous to 2D-DIGE where fluorescent labelling of proteins is performed prior to separation by FFE and 1-DE. The results showed potential for FFE in comparing the effects of compounds such as butyrate on cellular systems with appropriate modifications to enhance the identification of proteins detected by a combined FFE/DIGE approach. These results were verified by Western blot analysis of selected proteins. This was the first time the regulation of a number of proteins by butyrate in HT29 colorectal cells including peptidyl-prolyl cis-trans isomerase A (cyclophilin A) and profilin-1 was reported.
Wase et al. [58] demonstrated the advantages of combining robust protein fractionation using FFE and 1D-LC-MS/MS to extend protein identification deeper through the dynamic range of the model cyanobacterium, Nostoc punctiforme PCC 73102. This investigation proved that FFE is useful for the fractionation of the whole N. punctiforme cell lysate under denaturing conditions. More broadly, the technique can be used individually or as a combination with other separation technologies. Sixty-one new proteins out of 248 (all identified with ≥ 2 peptides) were successfully identified using FFE-IEF compared to all previous reports. Results demonstrated the ability of FFE to provide improved protein distributions, while also providing effective segregation of highly abundant phycobilisomes to allow access to 37 low abundance proteins (pI > 9) in the N. punctiforme proteome, which are difficult to observe using conventional methods.
In another study, FFE approach was used in Golgi enrichment and proteomic analysis of developing Pinus radiata xylem [59]. Developing xylem samples from 3-year-old pine trees were harvested and subjected to a combination of density centrifugation followed by FFE. FFE proved to be a good method for increasing both the purity of Golgi preparations from developing xylem of pine and proved to be an excellent tool in increasing the number of identified Golgi proteins from pine. A total of 30 known Golgi proteins were identified by MS including glycosyl transferases from gene families involved in glucomannan and glucuronoxylan biosynthesis.
2.4 Approaches for the enrichment of phosphoproteome
The discovery of phosphorylation as a key regulatory mechanism of cell life has initiated the study of phosphoproteome and in turn the development of rapid and sensitive analytical tools for the enrichment and identification of phosphorylated proteins or peptides in various biological samples [60]. In this section, a summary is provided for the enrichment of phosphoproteome by immobilized metal-ion affinity chromatography (IMAC) and metal oxide affinity chromatography (MOAC) prior to LC-MS/MS analysis.
2.4.1 Enrichment of phosphoproteomes by immobilized metal-ion affinity chromatography
Yue et al. [61] developed a novel strategy to perform optimized multistep IMAC enrichment from whole cell lysates followed by high-pH reversed phase fractionation (multi-IMAC-HLB; HLB means hydrophilic–lipophilic-balanced reversed-phase cartridge). Using the multi-IMAC-HLB method with 3 mg of starting material, 8969 unique phosphopeptides could be readily identified, while using the well-established SCX-IMAC method with 15 mg of starting material, 5519 unique phosphopeptides were identified. This represents an increase of 61.5% using multi-IMAC-HLB when compared to SCX-IMAC method. The increase in the numbers of identified phosphopeptides is due to the increase in the ratio of identified phosphopeptides out of all detected peptides, 70.5% with multi-IMAC-HLB method compared to 32.3% with the SCX-IMAC method. The described multi-IMAC-HLB method is a relatively easy and efficient approach to enrich phosphopeptides, especially basic and hydrophobic phosphopeptides, from less starting material than is required by other commonly used approaches.
Tsai et al. [62] designed a metal-directed IMAC for the sequential enrichment of phosphopeptides. Using Raji B cells, the sequential Ga3+-, Fe3+-IMAC method displayed a much better detection sensitivity compared to the use of a single IMAC (Fe3+, Ti4+, Ga3+, and Al3+). The low percentage of overlapping identification results (<10%) demonstrated the complementary nature between the first Ga3+-IMAC and second Fe3+-IMAC. The application of sequential Ga3+-, Fe3+-IMAC to human lung cancer tissue allowed the identification of 2560 unique phosphopeptides with only 8% overlap. In addition, the sequential Ga3+-, Fe3+-IMAC method was useful for the fractionation of basic and acidic phosphopeptides: acidophilic phosphorylation sites were predominately enriched in the first Ga3+-IMAC (72%), while Pro-directed (85%) and basophilic (79%) phosphorylation sites were enriched in the second Fe3+-IMAC.
A novel IMAC material with polydopamine coated on the surface of graphene and functionalized with titanium ions (denoted as Ti4+-G@PD) was designed and synthesized for phosphoproteome analysis [63]. The grafting layer of the polydopamine-decoration would provide enhanced hydrophilicity and biocompatibility. The high specific surface area of graphene would offer higher capacity for loading dopamine and thus enhance the amounts of immobilized titanium ions, which would result in a larger capacity of phosphopeptides binding. The newly prepared Ti4+-G@PD with enhanced hydrophilicity and biological compatibility was characterized using scanning electron microscopy, transmission electron microscopy and infrared, and its performance for selective and effective enrichment of phosphopeptide was evaluated with both standard peptide mixtures and human serum. Ti4+-G@PD exhibited the enhanced hydrophilicity and biological compatibility and showed high-performance in the selective and effective enrichment of phosphopeptides from peptide mixtures.
2.4.2 Enrichment of phosphoproteomes by metal oxide affinity chromatography
Li et al. [64] described a workflow involving a combination of titanium dioxide (TiO2) enrichment, SAX, and SCX fractionation for global phosphoproteome analysis. The workflow proposed TiO2-based high efficient enrichment with optimum peptide-to-beads ratio prior to robust IEC fractionation. With the optimum peptide-to-beads ratio, off line TiO2 enrichment provides high selectivity and large sample loading capacity compared with online TiO2 chromatography. The eluate with highly enriched phosphopeptides was then subjected to online SAX and SCX fractionation coupled to RP-LC-MS/MS analysis. The identification of phosphopeptides from SAX, SCX, and flow-through fractions showed high complementary features. Importantly, large amount of multiphosphopeptides could be recovered in SAX fractionations. In total, up to 5063 unique phosphosites were identified from 4557 unique phosphopeptides using 4-mg HeLa cell lysate as the starting material.
An offline phosphopeptide enrichment procedure using TiO2 columns was recently described by Yu and Veenstra [65]. In this study, 0.5 mg of peptides from a HeLa cell lysate was loaded onto the TiO2 column in an acidic environment. The column was then washed with aqueous, organic, and NH4Glu buffers at acidic conditions. Phosphopeptides were eluted with a high recovery rate using an ammonia solution at high pH (∼11.7) This offline approach is flexible for both analytical and preparative scale isolation of phosphoproteomes by using different size of TiO2 columns. A large-scale enrichment of phosphopeptides from 1 to 2 mg of digested proteome sample is suitable for current multidimensional LC-MS/MS.
Hoehenwarter et al. [66] introduced a novel approach combining tandem MOAC with LC-MS/MS to identify and quantify site-specific phosphorylation of in vivo mitogen-activated protein kinase (MPK) substrate candidates in Arabidopsis thaliana. This approach represents a two-step chromatography combining phosphoprotein enrichment using Al(OH)3-based MOAC, tryptic digest of enriched phosphoproteins, and TiO2-based MOAC to enrich phosphopeptides from complex protein samples. The approach effectively targets the phosphate moiety of phosphoproteins and phosphopeptides and, thus allows probing of the phosphoproteome to an unprecedented depth. Application of tandem MOAC disclosed the identity of numerous novel phosphorylation sites and potential in vivo targets of ArabidopsisMPKs, particularly of MPK3 and MPK6, in just a single experiment.
Yan et al. [67] developed a facile route for the preparation of a novel MOAC by grafting titania nanoparticles on polydopamine (PD)-coated graphene (denoted as G@PD@TiO2). In the first step, self-assemble polymerization of dopamine on graphene was performed in basic solution at room temperature, which not only offered the coupling linker between titania and graphene but also improved the hydrophilicity and biological compatibility of the nanohybrids. Thereafter, the titania nanoparticles were grafted on the surface of the PD-coated graphene via a simple hydrothermal treatment. The prepared nanohybrids have well-defined 2D morphology, nanoscaled thickness, hydrophilic property, and high surface area. The nanohybrids were investigated in the enrichment of phosphopeptides from real biological samples (human serum and mouse brain). A total number of 556 phosphorylation sites were identified from the digest of mouse brain proteins, showing great potential in the practical application.
2.5 Capturing/enrichment of subglycoproteomes by lectin affinity chromatography
The glycosylation of proteins, which involves the covalent linkages between carbohydrates and proteins, is one of the post-translational modifications that are regulated in various biological processes. Aberrant protein glycosylation is implicated in many diseases (e.g., cancers). Therefore, the analyses of glycoproteomics expression patterns are of great value in clinical study.
2.5.1 Single or serial lectin affinity chromatography
Single lectin affinity chromatography (LAC) refers to analyzing a given sample on each lectin column separately regardless of the number of lectin columns involved, while serial LAC refers to using LAC in a sequential or serial fashion where the pass through fraction from one lectin column is collected and further analyzed on a second lectin column and so forth.
A dual-LAC and LC-MS/MS platform was introduced for the enrichment and identification of glycoprotein biomarkers in urine samples [68]. An experiment was carried out via a dual-LAC including concanavalin A (ConA) and wheat germ agglutinin (WGA) to isolate a wide range of N-linked glycoproteins from urine samples. It incorporated an LC/MS-MS–based label-free shotgun method to identify glycoproteins that were differentially expressed according to disease status. From a primary sample set obtained from 54 cancer patients and 46 controls, a total of 265 distinct glycoproteins were identified with high confidence, and changes in glycoprotein abundance between groups were quantified by a label-free spectral counting method. One of the most discriminatory novel candidate biomarkers, α-1-antitrypsin, was validated in an independent sample cohort by using ELISA.
Bell et al. reported the use of subcellular fractionation in combination with WGA affinity based membrane for the enrichment of the M. tuberculosis proteins [69]. Glycoproteins and other membrane proteins were enriched based on affinity purification in an aqueous two-phase system consisting of the polymers polyethylene glycol and dextran. The M. tuberculosis fractions were digested and then analyzed by MS. It was found that 1051 M. tuberculosis protein groups including 183 transmembrane proteins have been identified by LC-MS/MS. Due to the many mycobacterial antigens and lipoglycoproteins identified, the authors stated that many of these newly discovered proteins could represent potential candidates mediating host pathogen interactions. Furthermore, the results achieved in this work could be used in the design of new antigens for vaccines.
A study involving the utilization of Con A, Ricinus communis agglutinin (RCA-I) and Ulex europaeus agglutinin (UEA-I) based LAC for the enrichment of salivary glycoproteins has been reported by Gonzalez-Begne et al. [70]. Ten milligrams of dry-weight lyophilized submandibular/sublingual (SM/SL) saliva dissolved in 5 mL of buffers was applied to each lectin column containing 1 mL of settled agarose-lectin beads covalently linked to Con A, RCA-I or UEA-I. The columns were then incubated with the salivary mixture overnight at 4 °C. Elution of bound salivary glycoproteins was carried out as mentioned by the manufacturer's instructions. The fractions corresponding to the two elution steps (from neutral and acidic pHs) were pooled together for each Con A, RCA-I and UEA-I lectin experiment, dialyzed against 50 mM ammonium bicarbonate to remove salts and carbohydrates and then freeze-dried. A total of 262 N- and O-linked glycoproteins were identified by MudPIT. Only 38 were previously described in SM/SL saliva glycoproteome salivas from the human salivary N-linked glycoproteome, while 224 were unique. The authors stated that this approach is robust and selective enough to achieve a purification efficiency of more than 90% in spite of the broad specificities and nonglycosylated co-purification problems associated with the use of lectin fractionation. Moreover, the study revealed 125 glycoproteins not previously identified in the SM/SL human saliva proteome. The strategy identified specific glycoprotein biomarkers with potential utility in noninvasive bladder cancer detection.
Selvaraju and El Rassi [18] introduced a fully integrated platform for capturing/fractionating human serum fucome from disease-free and breast cancer sera (see Fig. 1). The platform comprised multicolumn operated by HPLC pumps and switching valves for the simultaneous depletion of HAPs (i.e., HSA and Ig's) via affinity-based subtraction using two anti-HSA columns, and Protein G′ and Protein A columns as previously discussed above in the depletion section 2.1.2. The capturing of fucosylated glycoproteins was performed via LAC followed by the fractionation of the captured glycoproteins by RPC. Two lectin columns specific to fucose, namely AAL and LTA were utilized. The fractionated fucome by RPC were subjected to trypsinolysis for LC-MS/MS analysis. The platform allowed the “cascading” of the serum sample from column-to-column in the liquid phase with no sample manipulation between the various steps. The platform allowed the convenient comparison of the fucosylated proteins in breast cancer serum to those in disease-free serum yielding a broad panel of 35 DEPs from the combined LTA and AAL captured proteins and a narrower panel of 8 DEPs that were commonly differentially expressed in both LTA and AAL captured proteins, which are considered as more representative of the altered human serum fucome in breast cancer due the complementary specificity of both lectins. Selvaraju and El Rassi also reported the use of the same AAL and LTA columns for capturing the fucome [39] by performing CPLL treatment of the serum sample prior to fucome capture by serial LAC and the details were discussed above in the Proteo Miner technology section. Briefly, the combined offline CPLL treatment of the human serum with the multicolumn platform for the capturing and fractionation of the human serum fucome permitted the identification of the differentially expressed proteins (DEPs) in breast cancer serum yielding 58 DEPs in both the LTA and AAL fractions with 6 DEPs common to both lectins. 17 DEPs were of the low abundance type, 16 DEPs of the borderline abundance type, 4 DEPs of the medium abundance type and 15 DEPs of the high abundance type. The remaining 6 DEPs are of unknown concentration (see Figs 2 and 3 for fucome fractionation by RPC).
The separation and enrichment of water soluble glycoproteins in barley malt using a monolithic ConA affinity HPLC column have been reported [71]. The high capacity monolithic ConA column allowed to enrich minor glycoproteins present in such heterogeneous malt samples and thus make them easier to analyze by MS methods. Also, it provides higher reproducibility, efficiency, and faster performance in comparison with the previously used manually filled columns. The ConA captured glycoproteins from barley malt were separated on SDS-PAGE and then chymotryptic digested and identified using MALDI-TOF/TOF MS. This ConA affinity HPLC column allowed the identification of several putative glycoproteins present in barley malt.
In another study, a new silica microparticle with macrosized internal pores for affinity chromatography was developed by Mann et al. [72]. The particles have a high surface area for a macroporous material, ∼200 m2/g, making them suitable for large biomolecular separations. These microparticles have been utilized to fabricate lectin affinity chromatography columns for microscale enrichment of glycoproteins. Firstly, the microparticles were functionalized with two lectins, ConA or AAL and binding properties were tested with standard glycoproteins. These two lectin microcolumns shown excellent binding capacities for microaffinity enrichment. In addition, the lectin microcolumns were utilized for enrichment of glycoproteins from 1 μL volumes of blood serum sample. The enriched serum fractions were then subjected to analysis with MS. The results demonstrated that the new particles offer excellent sensitivity for microscale analyses of precious biological sample materials.
Jung and Cho [73] examined the utility of serial affinity columns in determining whether individual glycan structures appear alone or together with other glycans in specific proteins. Four lectin columns namely, agarose-bound Lycopersicon esculentum lectin (LEL), agarose-bound Helix pomatia agglutinin (HPA), anti-Lewis x antibody (anti-LexAb) and anti-sialyl Lewis x antibody (anti-sLexAb) were utilized. These four different types of affinity columns were examined in two series: the LEL → HPA → anti-LexAb → anti-sLexAb series and the anti-sLexAb → anti-LexAb → HPA → LEL series. Patterns in protein capture from these two series were very different. It is concluded that Serial LAC can be a valuable tool in recognizing diversity in protein glycosylation, especially when the order of columns in the Serial LAC series is varied. It is further concluded that two clear types of diversity were recognized in this study. One is the independent occurrence of different affinity-targetable glycan features in the same glycoprotein. The second is the case in which multiple targetable glycan features were co-resident in the same glycoprotein.
2.5.2 Multilectin affinity chromatography
Multi-LAC (M-LAC) refers to using a mixture of immobilized lectins having complementary specificities in a given column, i.e., mixed bed column. After loading the sample onto the column, the M-LAC column is eluted sequentially using specific displacer (i.e., haptenic sugar) for each lectin. Lee et al. reported a native M-LAC column, combining three lectins, namely, Con A, jacalin, and WGA for the fractionation of cellular glycoproteins from MCF-7 breast cancer lysate [74]. Several conditions for optimum recovery of total proteins and glycoproteins such as low pH and saccharide elution buffers, and the inclusion of detergents in the binding and elution mobile phases were evaluated. Optimum recovery was observed with overnight incubation of cell lysate with lectins at 4°C, and inclusion of detergent in the binding and saccharide elution mobile phases. Total protein and bound recoveries were 80 and 9%, respectively. After LC-MS/MS analysis, the proteins such as mucin 6, bile salt-activated lipase, and pyruvate kinase lysozyme M1/M2 with significant differential expression, which have strong association with pancreatic cancer were identified.
In another work, a lectin affinity based approach was applied to enrich and increase the detection number of secreted proteins in colorectal cancer (CRC) tumor tissues and paired normal colorectal (NC) tissues to identify novel serum biomarkers [75]. For the enrichment of glycoproteins, 100 μL of agarose-bound Con A and 100 μL of agarose-bound WGA were mixed and equilibrated with lectin-binding buffer in a micro-spin column. Nine groups of conditioned medium (CM) from CRC tumor tissues at the early stage and their paired NC tissues were collected, and the concentrated CM were incubated with mixed agarose-bound lectins (Con A and WGA) overnight at 4 °C in 250 μL of lectin-binding buffer. The captured proteins were then analyzed by 1-DE coupled to LC-MS/MS. Based on label-free spectral counting, differentially expressed secreted proteins (DESPs) between the CRC tissues and NC tissues were identified, with 68 DESPs up-regulated in CRC tissues. EFEMP2, one of the top10 up-regulated DESPs, was further validated by immunohistochemistry at tissue level and ELISA at serum level. The authors stated that the expression level of EFEMP2 was dramatically increased in CRC patients, even at an early stage of CRC. These results indicated that EFEMP2 is a promising tissue and serum biomarker for the early detection of CRC.
The application of glycoproteins enrichment via a high-performance M-LAC (HP-M-LAC) method to differentiate mucinous pancreatic cyst fluid subtypes from nonmucinous pancreatic cyst fluid subtypes has been reported [76]. HP-M-LAC is a robust and high-throughput high-performance platform that combines the depletion of two HAPs (human immunoglobulins and albumin), enrichment of glycoproteins and glycoisoforms by multiple lectins (ConA, WGA, and Jacalin), followed by 1D SDS-PAGE fractionation. The platform successfully allows the enrichment and characterization of glycoproteins that are present at different levels in mucinous and nonmucinous cyst fluid subtypes.
The determination of the relative abundance of glycoproteins in rheumatoid arthritis (RA) and spondyloarthropathies (SpAs) by LAC coupled to iTRAQ labelling and LC-MS/MS analysis has been reported [77]. Glycoproteins from the synovial fluid of RA and SpA patients were incubated overnight at 4 °C with a mixture of three agarose bound lectins, WGA, ConA and Jacalin. Thereafter, the bound glycoproteins were eluted using competitive elution, performed by a mixture of sugars (100 mM each of M-pyranoside, galactose, melibiose and N-acetyl glucoseamine in Tris buffered saline, pH 7.5). The eluates were then washed and concentrated using 3 kDa MWCO filters. The concentrated proteins were then identified by LC-MS/MS. Of the 210 proteins identified, 131 (62%) were already reported to be glycosylated. This approach allowed the identification of proteins that were previously reported to be overexpressed in RA including metalloproteinase inhibitor 1, myeloperoxidase and several S100 proteins. In addition, several novel candidates that were overexpressed in SpA including apolipoproteins C-II and C-III and the SUN domain-containing protein 3 were discovered. Novel molecules found overexpressed in RA included extracellular matrix protein 1 and lumican.
3. Concluding remarks
This review article has assembled the major advances made in the area of proteomic sample preparation and fractionation in the aim of facilitating proteomic profiling of complex biological samples. It is clear that integrated approaches for proteomic sample preparation prior to LC-MS/MS have been the center for development and improvement in order to minimize sample loss and propagation of experimental biases and consequently ensure reliable proteomic profiling by LC-MS/MS. This trend is expected to continue in the future, and most likely to see more miniaturized integrated sample preparation platforms.
Acknowledgments
The authors gratefully acknowledge the grant no. 1R15GM096286-01 from the Department of Health and Human Services at the National Institute of Health. CP acknowledges a scholarship from the Institute for the Promotion of the Teaching Science and Technology, Ministry of Education, Thailand towards a postdoctoral research fellow.
Nonstandard Abbreviations
- AAL
Aleuria aurantia lectin
- Con A
concanavalin A
- CPLL
combinatorial peptide ligand library
- CSF
cerebrospinal fluid
- DEPs
differentially expressed proteins
- DESPs
differentially expressed secreted proteins
- ELISA
enzyme-linked immunosorbent assays
- ERLIC
electrostatic repulsion hydrophilic interaction chromatography
- FDR
false discovery rate
- FFE
free flow electrophoresis
- HAPs
high abundance proteins
- HPA
Helix pomatia agglutinin
- IDA
iminodiacetic acid
- IMAC
immobilized metal-ion affinity chromatography
- LAC
lectin affinity chromatography
- LAPs
low abundance proteins
- LEL
Lycopersicon esculentum lectin
- LTA
Lotus tetragonolobus agglutinin
- MARS
multiple affinity removal spin cartridge
- M-LAC
multi- lectin affinity chromatography
- MOAC
metal oxide affinity chromatography
- MudPIT
multidimensional protein identification technology
- RCA-I
Ricinus communis agglutinin
- RPC
reversed-phase chromatography
- SAX
strong anion exchange
- SCX
strong cation exchange
- WAX
weak anion exchange
- WGA
wheat germ agglutinin
- UEA-I
Ulex europaeus agglutinin
- ZIC-HILIC
zwitterion−ion hydrophilic interaction chromatography
References
- 1.Zhang Ah, Sun H, Yan Gl, Han Y, Wang Xj. Appl Biochem Biotechnol. 2013;170:774–786. doi: 10.1007/s12010-013-0238-7. [DOI] [PubMed] [Google Scholar]
- 2.Selvaraju S, El Rassi Z. J Sep Sci. 2012;35:1785–1795. doi: 10.1002/jssc.201200230. [DOI] [PubMed] [Google Scholar]
- 3.Ekaterina M, Scott HC, Oleg K, Hans D, Deelder AM, Magnus P. J Proteomics Bioinform. 2012;5:217–221. [Google Scholar]
- 4.Fernandez-Costa C, Calamia V, Fernandez-Puente P, Capelo-Martinez JL, Ruiz-Romero C, Blanco F. Proteome Sci. 2012;10:55. doi: 10.1186/1477-5956-10-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holewinski RJ, Jin Z, Powell MJ, Maust MD, Van Eyk JE. Proteomics. 2013;13:743–750. doi: 10.1002/pmic.201200192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janecki DJ, Pomerantz SC, Beil EJ, Nemeth JF. J Chromatogr B. 2012;902:35–41. doi: 10.1016/j.jchromb.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Jmeian Y, El Rassi Z. J Proteome Res. 2007;6:947–954. doi: 10.1021/pr060660o. [DOI] [PubMed] [Google Scholar]
- 8.Mortezai N, Wagener C, Buck F. Methods. 2012;56:254–259. doi: 10.1016/j.ymeth.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Roche S, Tiers L, Provansal M, Seveno M, Piva M, Jouin P, Lehmann S. J Proteomics. 2009;72:945–951. doi: 10.1016/j.jprot.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Yang HH, Lu KH, Lin YF, Tsai SH, Chakraborty S, Zhai WJ, Tai DF. J Biomed Mater Res, A. 2013;101A:1935–1942. doi: 10.1002/jbm.a.34491. [DOI] [PubMed] [Google Scholar]
- 11.Fattahi S, Kazemipour N, Valizadeh J, Hashemi M, Ghazizade H. Zahedan J Res Med Sci. 2012;14:1–5. [Google Scholar]
- 12.Fischer R, Trudgian D, Wright C, Thomas G, Bradbury L, Brown M, Bowness P, Kessler B. Mol Cell Proteomics. 2012;11:M111 013904–11. doi: 10.1074/mcp.M111.013904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pont L, Benavente F, Barbosa J, Sanz-Nebot V. J Sep Sci. 2013;36:3896–3902. doi: 10.1002/jssc.201300838. [DOI] [PubMed] [Google Scholar]
- 14.Xiaolin W, Erhui X, Wei W, Monica S, Mauro C. Nature Protocols. 2014;9:362–374. doi: 10.1038/nprot.2014.022. [DOI] [PubMed] [Google Scholar]
- 15.Talei D, Valdiani A, MA P. Biotechnic & Histochemistry. 2013;60:521–526. [Google Scholar]
- 16.Echan L, Tang H, Ali-Khan N, Lee K, Speicher D. Proteomics. 2005;5:3292–3303. doi: 10.1002/pmic.200401228. [DOI] [PubMed] [Google Scholar]
- 17.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, Xie F, Wu S, Kim JS, Jones N, Moore RJ, Paša-Tolić L, Kagan J, Rodland KD, Liu T, Tang K, Camp DG, Smith RD, Qian WJ. Proc Nat Acad Sci. 2012;109:15395–15400. doi: 10.1073/pnas.1204366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selvaraju S, Rassi ZE. Proteomics. 2013;13:1701–1713. doi: 10.1002/pmic.201200524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Xu J, Liu Y, Chen Y. J Chromatogr B. 2012;902:10–15. doi: 10.1016/j.jchromb.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Huillet C, Adrait A, Lebert D, Picard G, Trauchessec M, Louwagie M, Dupuis A, Hittinger L, Ghaleh B, Le Corvoisier P, Jaquinod M, Garin J, Bruley C, Brun V. Mol Cell Proteomics. 2012;11:M111.008235–12. doi: 10.1074/mcp.M111.008235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholl PF, Cole RN, Ruczinski I, Gucek M, Diez R, Rennie A, Nathasingh C, Schulze K, Christian P, Yager JD, Groopman JD, West KP., Jr Placenta. 2012;33:424–432. doi: 10.1016/j.placenta.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu WJ, Liu XY, Dong H, Yu Y, Wang Y, Chen H. J Mol Neurosci. 2014;52:507–514. doi: 10.1007/s12031-013-0206-2. [DOI] [PubMed] [Google Scholar]
- 23.Tan SH, Kapur A, Baker MS. J Proteome Res. 2012;11:6291–6294. doi: 10.1021/pr300717b. [DOI] [PubMed] [Google Scholar]
- 24.Liu T, Hossain M, Schepmoes AA, Fillmore TL, Sokoll LJ, Kronewitter SR, Izmirlian G, Shi T, Qian WJ, Leach RJ, Thompson IM, Chan DW, Smith RD, Kagan J, Srivastava S, Rodland KD, Camp DG., Ii J Proteome. 2012;75:4747–4757. doi: 10.1016/j.jprot.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santucci L, Candiano G, Bruschi M, D'Ambrosio C, Petretto A, Scaloni A, Urbani A, Righetti PG, Ghiggeri GM. Proteomics. 2012;12:509–515. doi: 10.1002/pmic.201100404. [DOI] [PubMed] [Google Scholar]
- 26.Selvaraju S, El Rassi Z. Electrophoresis. 2011;32:674–685. doi: 10.1002/elps.201000606. [DOI] [PubMed] [Google Scholar]
- 27.Zhu G, Zhao P, Deng N, Tao D, Sun L, Liang Z, Zhang L, Zhang Y. Anal Chem. 2012;84:7633–7637. doi: 10.1021/ac3017746. [DOI] [PubMed] [Google Scholar]
- 28.González-Iglesias H, Álvarez L, García M, Escribano J, Rodríguez-Calvo PP, Fernández-Vega L, Coca-Prados M. J Proteomics. 2014;98:65–78. doi: 10.1016/j.jprot.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Balfoussia E, Skenderi K, Tsironi M, Anagnostopoulos AK, Parthimos N, Vougas K, Papassotiriou I, Tsangaris GT, Chrousos GP. J Proteomics. 2014;98:1–14. doi: 10.1016/j.jprot.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 30.von Toerne C, Kahle M, Schäfer A, Ispiryan R, Blindert M, Hrabe De Angelis M, Neschen S, Ueffing M, Hauck SM. J Proteome Res. 2013;12:1331–1343. doi: 10.1021/pr3009836. [DOI] [PubMed] [Google Scholar]
- 31.Coscia A, Orrù S, Di Nicola P, Giuliani F, Varalda A, Peila C, Fabris C, Conti A, Bertino E. J Matern Fetal Neonatal Med. 2012;25:49–51. doi: 10.3109/14767058.2012.715015. [DOI] [PubMed] [Google Scholar]
- 32.Fasoli E, D'Amato A, Righetti PG, Barbieri R, Bellavia D. Biol Bull. 2012;222:93–104. doi: 10.1086/BBLv222n2p93. [DOI] [PubMed] [Google Scholar]
- 33.Huhn C, Ruhaak LR, Wuhrer M, Deelder AM. J Proteomics. 2012;75:1515–1528. doi: 10.1016/j.jprot.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Malaud E, Piquer D, Merle D, Molina L, Guerrier L, Boschetti E, Saussine M, Marty-Ané C, Albat B, Fareh J. Electrophoresis. 2012;33:470–482. doi: 10.1002/elps.201100395. [DOI] [PubMed] [Google Scholar]
- 35.Cumová J, Jedličková L, Potěšil D, Sedo O, Stejskal K, Potáčová A, Zdráhal Z, H R. Klin Onkol. 2012;25:17–25. [PubMed] [Google Scholar]
- 36.Capriotti AL, Caruso G, Cavaliere C, Piovesana S, Samperi R, Laganà A. Anal Chim Acta. 2012;740:58–65. doi: 10.1016/j.aca.2012.06.033. [DOI] [PubMed] [Google Scholar]
- 37.Lichtenauer AM, Herzog R, Tarantino S, Aufricht C, Kratochwill K. Electrophoresis. 2014;35:1387–1394. doi: 10.1002/elps.201300499. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Y, Chen Y, Sun X, Li Y, Huang C, Deng H, Li Z. Inflammation. 2014:1–9. doi: 10.1007/s10753-014-9871-8. [DOI] [PubMed] [Google Scholar]
- 39.Selvaraju S, El Rassi Z. J Chromatogr B. 2014;951–952:135–142. doi: 10.1016/j.jchromb.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritorto MS, Cook K, Tyagi K, Pedrioli PGA, Trost M. J Proteome Res. 2013;12:2449–2457. doi: 10.1021/pr301011r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang PC, Reddy PM, Ho YP. Anal Bioanal Chem. 2014:1–8. doi: 10.1007/s00216-014-7965-7. [DOI] [PubMed] [Google Scholar]
- 42.Sun D, Wang N, Li L. J Proteome Res. 2012;11:818–828. doi: 10.1021/pr200676v. [DOI] [PubMed] [Google Scholar]
- 43.Boichenko AP, Govorukhina N, van der Zee AGJ, Bischoff R. J Sep Sci. 2013;36:3463–3470. doi: 10.1002/jssc.201300750. [DOI] [PubMed] [Google Scholar]
- 44.Zhou F, Sikorski TW, Ficarro SB, Webber JT, Marto JA. Anal Chem. 2011;83:6996–7005. doi: 10.1021/ac200639v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hennrich ML, Groenewold V, Kops GJPL, Heck AJR, Mohammed S. Anal Chem. 2011;83:7137–7143. doi: 10.1021/ac2015068. [DOI] [PubMed] [Google Scholar]
- 46.Song C, Wang F, Ye M, Cheng K, Chen R, Zhu J, Tan Y, Wang H, Figeys D, Zou H. Anal Chem. 2011;83:7755–7762. doi: 10.1021/ac201299j. [DOI] [PubMed] [Google Scholar]
- 47.Garbis SD, Roumeliotis TI, Tyritzis SI, Zorpas KM, Pavlakis K, Constantinides CA. Anal Chem. 2011;83:708–718. doi: 10.1021/ac102075d. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Wang Y, Gu X, Zhang L, Y C. J Sep Sci. 2011;34:1027–1034. doi: 10.1002/jssc.201000815. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Kong RP, Li G, Lam MP, Law CH, Lee SM, Lam HC, CI K. J Sep Sci. 2012;35:1755–1763. doi: 10.1002/jssc.201200054. [DOI] [PubMed] [Google Scholar]
- 50.Xia S, Tao D, Yuan H, Zhou Y, Liang Z, Zhang L, Z Y. J Sep Sci. 2012;35:1764–1770. doi: 10.1002/jssc.201200052. [DOI] [PubMed] [Google Scholar]
- 51.Lam MY, Law C, Quan Q, Zhao Y, Chu I. In: Shotgun Proteomics. Martins-de-Souza D, editor. Springer; New York: 2014. pp. 39–51. [Google Scholar]
- 52.Di Carli M, Zamboni A, Pè ME, Pezzotti M, Lilley KS, Benvenuto E, Desiderio A. J Proteome Res. 2011;10:429–446. doi: 10.1021/pr1005313. [DOI] [PubMed] [Google Scholar]
- 53.Sui SH, Dong JJ, Wang JL, Cai Y, Qian XH. Chin J Anal Chem. 2012;40:177–183. [Google Scholar]
- 54.Wulff T, Petersen J, Nørrelykke MR, Jessen F, Nielsen HH. J Agric Food Chem. 2012;60:8457–8464. doi: 10.1021/jf3016943. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Wang K, Zhang J, Liu SS, Dai L, Zhang JY. J Proteome Res. 2011;10:2863–2872. doi: 10.1021/pr200141c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuramitsu Y, Takashima M, Yokoyama Y, Iizuka N, Tamesa T, Akada JK, Wang Y, Toda T, Sakaida I, Okita K, Oka M, Nakamura K. Anticancer Res. 2011;31:3331–3336. [PubMed] [Google Scholar]
- 57.Fung KYC, Cursaro C, Lewanowitsch T, Brierley GV, McColl SR, Lockett T, Head R, Hoffmann P, C L. Proteomics. 2011;11:964–971. doi: 10.1002/pmic.201000429. [DOI] [PubMed] [Google Scholar]
- 58.Wase NV, Ow SY, Salim M, Nissum M, Whalley MC, P W. J Proteomics Bioinform. 2012;5:067–072. [Google Scholar]
- 59.Parsons HT, Weinberg CS, Macdonald LJ, Adams PD, Petzold CJ, Strabala TJ, Wagner A, Heazlewood JL. PLOS One. 2013;8:1–12. doi: 10.1371/journal.pone.0084669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kailasa S, Wu HF. Microchim Acta. 2014;181:853–864. [Google Scholar]
- 61.Yue XS, Hummon AB. J Proteome Res. 2013;12:4176–4186. doi: 10.1021/pr4005234. [DOI] [PubMed] [Google Scholar]
- 62.Tsai CF, Hsu CC, Hung JN, Wang YT, Choong WK, Zeng MY, Lin PY, Hong RW, Sung TY, Chen YJ. Anal Chem. 2013;86:685–693. doi: 10.1021/ac4031175. [DOI] [PubMed] [Google Scholar]
- 63.Yan Y, Zheng Z, Deng C, Li Y, Zhang X, Yang P. Anal Chem. 2013;85:8483–8487. doi: 10.1021/ac401668e. [DOI] [PubMed] [Google Scholar]
- 64.Li QR, Ning ZB, Yang XL, Wu JR, Zeng R. Electrophoresis. 2012;33:3291–3298. doi: 10.1002/elps.201200124. [DOI] [PubMed] [Google Scholar]
- 65.Yu LR, Veenstra T. In: Proteomics for Biomarker Discovery. Zhou M, Veenstra T, editors. Humana Press; 2013. pp. 93–103. [Google Scholar]
- 66.Hoehenwarter W, Thomas M, Nukarinen E, Egelhofer V, Röhrig H, Weckwerth W, Conrath U M, GJ B. Mol Cell Proteomics. 2013;12:369–380. doi: 10.1074/mcp.M112.020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan Y, Sun X, Deng C, Li Y, Zhang X. Anal Chem. 2014;86:4327–4332. doi: 10.1021/ac500047p. [DOI] [PubMed] [Google Scholar]
- 68.Yang N, Feng S, Shedden K, Xie X, Liu Y, Rosser CJ, Lubman DM, Goodison S. Clin Cancer Res. 2011;17:3349–3359. doi: 10.1158/1078-0432.CCR-10-3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bell C, Smith GT, Sweredoski MJ, Hess S. J Proteome Res. 2012;11:119–130. doi: 10.1021/pr2007939. [DOI] [PubMed] [Google Scholar]
- 70.Gonzalez-Begne M, Lu B, Liao L, Xu T, Bedi G, Melvin JE, Yates JR. J Proteome Res. 2011;10:5031–5046. doi: 10.1021/pr200505t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benkovska D, Flodrova D, Bobalova J. J Liq Chrom & Rel Technol. 2012;36:561–572. [Google Scholar]
- 72.Mann BF, Mann AKP, Skrabalak SE, Novotny MV. Anal Chem. 2013;85:1905–1912. doi: 10.1021/ac303274w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jung K, Cho W. Anal Chem. 2013;85:7125–7132. doi: 10.1021/ac400653z. [DOI] [PubMed] [Google Scholar]
- 74.Lee LY, Hincapie M, Packer N, Baker MS, Hancock WS, Fanayan S. J Sep Sci. 2012;35:2445–2452. doi: 10.1002/jssc.201200049. [DOI] [PubMed] [Google Scholar]
- 75.Yao L, Lao W, Zhang Y, Tang X, Hu X, He C, Hu X, Xu LX. J Proteome Res. 2012;11:3281–3294. doi: 10.1021/pr300020p. [DOI] [PubMed] [Google Scholar]
- 76.Gbormittah FO, Haab BB, Partyka K, Garcia-Ott C, Hancapie M, Hancock WS. J Proteome Res. 2013;13:289–299. doi: 10.1021/pr400813u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhattacharjee M, Sharma R, Goel R, Balakrishnan L, Renuse S, Advani J, Gupta S, Verma R, Pinto S, Sekhar N, Nair B, Prasad TSK, Harsha H, Jois R, Shankar S, Pandey A. Clin Proteomics. 2013;10:1–11. doi: 10.1186/1559-0275-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]