

Abstract
The ATP-binding cassette transporters P-glycoprotein (ABCB1, MDR1) and multidrug resistance protein 4 (MRP4) efflux irinotecan and its active metabolite SN-38 in vitro, and thus may contribute to system clearance of these compounds. Mdr1a/b−/−, Mrp4−/−, and wild-type mice were administered 20 or 40 mg/kg irinotecan, and plasma samples were collected for 6 hours. Irinotecan and SN-38 lactone and carboxylate were quantitated and data were analyzed with nonlinear mixed-effects modeling. Mdr1a/b genotype was a significant covariate for the clearance of both irinotecan lactone and SN-38 lactone. Exposures to irinotecan lactone and SN-38 lactone after a 40 mg/kg dose were 1.6-fold higher in Mdr1a/b−/− mice compared to wild-type mice. Plasma concentrations of irinotecan lactone, irinotecan carboxylate, and SN-38 lactone in Mrp4−/− mice were similar to the wild-type controls. These results suggest that P-gp plays a role in irinotecan and SN-38 elimination, but Mrp4 does not affect irinotecan or SN-38 plasma pharmacokinetics.
Keywords: Irinotecan, Multidrug resistance protein 4, Pharmacokinetics, P-glycoprotein, SN-38, Transporter
Introduction
Irinotecan (7-ethyl-10-(4-[1-piperidino]-1-piperidino)-carbonyloxy-camptothecin, CPT-11, Camptosar™), a synthetic derivative of the plant alkaloid camptothecin, is an anti-cancer prodrug activated by carboxylesterase to the active metabolite SN-38 (7-ethyl-10-hydroxycamptothecin) [1]. In vitro studies suggest SN-38 is approximately 1000 times more potent than irinotecan as an inhibitor of the topoisomerase I enzyme [2]. SN-38 undergoes glucuronic acid conjugation to form SN-38 glucuronide by uridine diphosphate glucuronosyltransferase. A diagram of irinotecan metabolic pathways has been recently published {Innocenti, 2009 #7680}. Both irinotecan and SN-38 have two structural forms, the active closed-ring lactone form, which shows in vitro and in vivo antitumor activity, and the inactive open-ring carboxylate, formed by pH-dependent hydrolysis [1].
ATP-binding cassette (ABC) transporters mediate the efflux of various anticancer agents from cells. Irinotecan and SN-38 are both substrates for P-glycoprotein (P-gp, MDR1, ABCB1) and Multi Drug Resistance Protein 4 (MRP4, ABCC4) [3-9]. Results of in vitro studies suggested that P-gp can actively transport both lactone and carboxylate forms of irinotecan and SN-38 [10], and in vivo studies have shown that deletion of P-gp in mice reduces biliary excretion of irinotecan by half [11]. In vivo studies using the P-gp inhibitors cyclosporine A and verapamil show that these compounds enhance irinotecan bioavailability and decrease biliary excretion of both irinotecan and SN-38 [3, 12, 13]. However, these inhibitors are not selective and inhibit other transporters. MRP4 expression correlated with poor clinical outcome in neuroblastoma and conferred resistance to irinotecan in vitro [8, 14]. Although this evidence strongly suggests irinotecan and SN-38 are P-gp and MRP4 substrates, it has not been reported whether P-gp or MRP4 affect the systemic disposition of irinotecan or SN-38. Thus, the objectives of this study were to assess the effect of P-gp and MRP4 on the pharmacokinetics of lactone and carboxylate forms of irinotecan and SN-38 using transporter-deficient mouse models.
Materials and Methods
Chemicals
Analytical grade, purified irinotecan hydrochloride (>98% purity) and SN-38 (>98% purity) were obtained from Pharmacia and Upjohn (Kalamazoo, MI). HPLC grade acetonitrile and methanol were purchased from Burdick and Jackson (Muskegon, MI). Ammonium acetate, HPLC grade glacial acetic acid, and dimethyl sulfoxide (DMSO) were acquired from Sigma-Aldrich, Inc (St. Louis, MO). HPLC grade water was obtained from a Milli-Q UV Plus filtration system.
Animals
Female Mdr1a/b+/+ mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and female Mdr1a/b−/− mice [15] were purchased from Taconic (Germantown, NY), both of which have a genetic background on FVB. Female Mrp4+/+ and Mrp4−/− mice, both with a genetic background on C57/B6×129 [16] were kindly donated by Dr. John Scheutz (St. Jude Children’s Research Hospital, Memphis, TN).
Determination of Irinotecan and SN-38 Lactone and Carboxylate by HPLC
Chromatographic Conditions
A Waters Nova-Pak® C18 3.9 × 150 mm column with a Waters Nova-Pak® C18 guard column (WAT044380) was used for chromatographic separation. A Shimadzu LC-10AD VP pump was used with a flow rate of 1.0 mL/min. The temperature of the Shimadzu SIL-10AD VP autoinjector was adjusted to 4°C. A Shimadzu RF-10XL fluorescence detector was set at Ex = 370 nm and Em = 530 nm. The mobile phase was composed of 19% acetonitrile and 81% 75 mM ammonium acetate with 5mM tetrabutylammonium phosphate buffer (adjusted to pH 6.0 with glacial acetic acid). The chromatogram showed good separation of the peaks with retention times of 5.8 min (irinotecan carboxylate), 7.3 min (SN-38 carboxylate), 11.6 min (irinotecan lactone), and 15.6 min (SN-38 lactone), with no interfering peaks co-eluting with the peaks of interest in blank murine plasma.
Sample Preparation
Irinotecan and SN-38 stock solutions were prepared in DMSO at concentrations of 1 mg/mL. Working stock solutions were prepared in 0.004 N borax for irinotecan and SN-38 carboxylate, 0.005 N citric acid for irinotecan lactone, and 0.005 N citric acid: methanol (50:50) for SN-38 lactone. Calibration curves and quality controls were prepared in murine plasma, harvested from Es1emice (kindly provided by Dr. Philip M. Potter, St. Jude Children’s Research Hospital), which have reduced plasma esterase activity [17]. Plasma samples were extracted by a 1:4 dilution in ice-cold methanol. The mixture was vortexed for 15 sec and centrifuged at 10,000 rpm for 2 min. 100 μL of the methanol extract was diluted with 100 μL of the mobile phase buffer (75 mM ammonium acetate with 5mM tetrabutylammonium phosphate buffer, adjusted to pH 7.0 with glacial acetic acid) and mixed well. 100 μL of each sample was injected onto the column with an autoinjector.
Assay Validation
Calibration curves were linear (R2>0.99) for irinotecan lactone (10-500 ng/mL) and carboxylate (5-500 ng/mL), and SN-38 lactone (3-250 ng/mL) and carboxylate (2-250 ng/mL). The lowest point on each curve was the lower limit of quantitation (LLOQ), which had precision <20%, an accuracy within 80 – 120%, and a signal to noise ratio ≥ 5. The inter- and intra-day precision determined with high (400 ng/mL irinotecan; 150 ng/mL SN-38) and low (30 ng/mL irinotecan; 20 ng/mL SN-38) quality control samples, were <10%. The inter- and intra-day accuracy of both forms of irinotecan and SN-38 were within 85-110%. No peaks co-eluting with analytes were seen after injection of blank plasma samples. Less than 1% conversion between lactone and carboxylate forms of analytes was observed on column. Recovery following extraction was >85% for all analytes. The methanol extracted samples were stable at −80°C for 14 days and were stable at 4°C for 6 hours after reconstitution (< 10% loss).
Plasma Pharmacokinetic Studies of Irinotecan and SN-38
Mice were injected with either 20 or 40 mg/kg irinotecan hydrochloride via the tail vein. Each mouse was randomly sampled four times out of five time points: 0.25, 0.5, 1.5, 3 and 6 h, by retro-orbital puncture or by cardiac puncture at the final time point and plasma was obtained. Each genotype group was 5 to 7 mice and 3 to 6 mice were sampled at each time point.
Pharmacokinetic Data Analysis
The population pharmacokinetic analysis was performed using NONMEM software, version VII (GloboMax LLC, Hannover, MD, USA). The Monte Carlo importance sampling EM method with INTERACTION was used. Diagnostic plots and additional statistical analyses were completed with R (R-project, version 2.8.1). Concentration-time data for knockout mice and their respective wild-type controls were modeled simultaneously. Data from the Mdr1 and Mrp4 groups were not analyzed together because they have different genetic backgrounds. We evaluated different structural models with one or two compartments for each of the parent drug and the metabolite using the ADVAN13 subroutine. The structural model was chosen based on visual inspection of weighted residual plots and the Akaike information criterion (AIC).
The estimated pharmacokinetic parameters (Fig. 1) included irinotecan lactone systemic clearance (CLIRN), irinotecan volume of distribution (V1), irinotecan carboxylate volume of distribution (V2), clearances between irinotecan lactone and carboxylate (CLLC and CLCL), SN-38 lactone apparent systemic clearance (CLSN/FE), SN-38 apparent central volume of distribution (V3/FE), intercompartmental clearance between SN-38 central and peripheral compartments (QSN), and SN-38 peripheral volume of distribution (V4). The distribution of the parameters was assumed log-normal. Thus, interindividual (IIV) variability in parameters was modeled as exponential terms. The residual error was modeled using a mixed proportional and additive error model for all analytes. The additive error was fixed at the LLOQ of the assay and the proportional error term (ε) was assumed to have a mean of zero and variance of σ.
Figure 1.
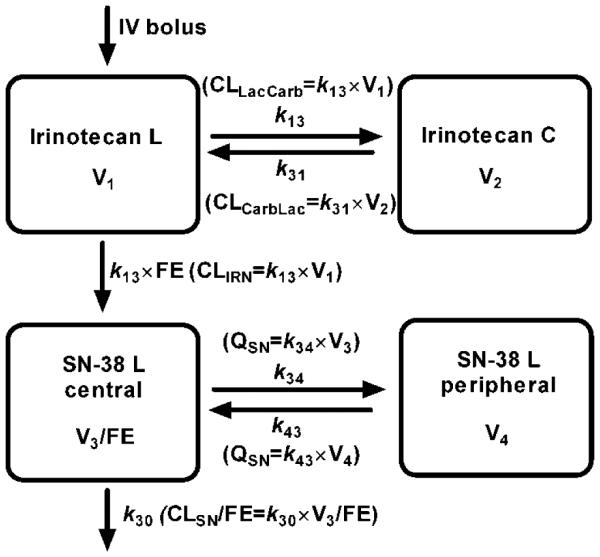
The four-compartment model for irinotecan lactone (L), irinotecan carboxylate (C), and SN-38 L. Irinotecan lactone is administered as an i.v. bolus into the central compartment. Irinotecan lactone and carboxylate are in an equilibrium described by the parameters CLLacCarb and CLCarbLac. Irinotecan lactone is converted to SN-38 lactone by the apparent clearance, CLIRN/ FE, where FE is the fraction of irinotecan lactone converted to SN-38 lactone. There is both a central and peripheral compartment for SN-38 lactone with clearance (CLSN) occurring from the central compartment.
In order to evaluate the influence of dosage and genotype in irinotecan lactone and SN-38 lactone pharmacokinetic parameters, we used the following covariate model:
| (Equation 1) |
where was the population estimate, was the population estimate in the absence of the covariate and θcov was the effect of the covariate on the model. A covariate was considered statistically significant when its addition to the model reduced the objective function value (OFV) at least 3.84 units (p < 0.05, based on the χ2 test for the difference in the log-likelihood between two hierarchical models that differ by 1 degree of freedom), and θcov was significantly different than zero (p<0.05; i.e., 1.96·SE(θp) < θp).
Results
Pharmacokinetics of Irinotecan and SN-38
SN-38 reached its maximum concentration by 15 min, indicating rapid conversion from irinotecan.The carboxylate forms of irinotecan and SN-38 were present in the plasma after injection in all strains; however, the SN-38 carboxylate plasma concentrations were mostly below the lower limit of quantitation within 1 h, and were therefore not used for compartmental modeling. A model with one compartment for each of the lactone and carboxylate forms was capable of describing the irinotecan data without any bias. The conversion from irinotecan lactone to SN-38 lactone was described by a first-order apparent rate constant. The final model was a four compartment model with central and peripheral compartments for SN-38 lactone. This model, shown in Fig. 1, did not have any significant bias in the weighted residual plots.
The volume of distribution of the carboxylate form of irinotecan was small relative to the lactone form, which is consistent with data from humans [18]. As has been previously reported in mice [19], we observed a dose-dependence of irinotecan lactone elimination. CLIRN was decreased by 33% and 41% in Mdr1a/b and Mrp4 mice, respectively, when the dosage was increased from 20 mg/kg to 40 mg/kg. Thus, irinotecan dosage was included as a covariate for CLIRN in both Mdr1a/b and Mrp4 mice. This reduced the OFV by 7.6 points (p<0.01) for the Mdr1a/b group, and by 27.5 points (p<0.01) for the Mrp4 group, and also reduced the IIV in this parameter by 28% for Mdr1a/b mice and 70% for Mrp4 mice.
Role of Mdr1a/b in Irinotecan and SN-38 Plasma Pharmacokinetics
It can be seen from the concentration-time plots that the irinotecan lactone, irinotecan carboxylate, and SN-38 lactone plasma levels were higher in the Mdr1a/b−/− mice compared to wild-type mice at both the 20 and 40 mg/kg dose levels (Fig. 2). Addition of the Mdr1a/b−/− genotype as a covariate on CLIRN reduced the OFV by 16.9 points (p<0.01) and explained 57% of IIV. Subsequent addition of the genotype as a covariate on CLSN-38/FE reduced the OFV an additional 11.2 points (p<0.01) and explained 52% of the IIV in this parameter. The Mdr1a/b−/− genotype was not a significant covariate on any of the clearance parameters related to conversion from irinotecan lactone to carboxylate (CLLacCarb or CLCarbLac). Parameter estimates of the base model and final model are presented in Table 1.
Figure 2.

Concentration-time profile of plasma irinotecan and SN-38 in Mdr1a/b−/− and wild-type mice. Plasma irinotecan lactone (A), irinotecan carboxylate (B), and SN-38 lactone (C) concentration-time data after 20 mg/kg (left column) and 40 mg/kg (right colunn) irinotecan intravenous injection. Symbols represent individual concentration-time points and the line represents the population-predicted values from the final pharmacokinetic model for Mdr1a/b−/− and wild-type mice. Each group consisted of 5-6 mice.
Table 1.
Pharmacokinetic models of irinotecan for Mdr1 and Mrp4 mice
| Parameter | Mdr1 mice |
Mrp4 mice |
||
|---|---|---|---|---|
| Base model | Final model | Base model | Final model | |
| CLIRN (L/h/kg) | 2.44 | 3.5a | 3.36 | 4.40b |
| θ Dose | - | −0.87 | − | −1.8 |
| θ Genotype | - | −1.14 | - | - |
| V1 (L/kg) | 2.23 | 2.21 | 4.44 | 4.57 |
| CLLacCarb (L/h/kg) | 2.48 | 2.53 | 3.71 | 3.16 |
| CLCarbLac (L/h/kg) | 2.07 | 2.12 | 1.96 | 1.68 |
| V2 (L/kg) | 0.74 | 0.76 | 0.52 | 0.44 |
| CLSN (L/h/kg) | 3.69 | 5.67c | 8.98 | 8.51 |
| θ Genotype | - | −3.18 | - | - |
| V3 (L/kg) | 1.21 | 1.11 | 1.48 | 1.22 |
| QSN (L/h/kg) | 4.24 | 4.1 | 7.47 | 8.04 |
| V4 (L/kg) | 63.9 | 58.7 | 108 | 118 |
| IIV CLIRN | 38.6% | 18.3% | 35.1% | 19.2% |
| IIV VIRN | 38.1% | 20.4% | 16.2% | 11.3% |
| IIV CLSN | 64.7% | 44.7% | 23.8% | 24.3% |
| ε IRN L | 15.4% | 18.3% | 23.4% | 25.2% |
| ε IRN C | 19.9% | 20.4% | 22.9% | 23.7% |
| ε SN-38 L | 1.0% | 1.0% | 0.6% | 0.2% |
| OFV | −189 | −225 | −495 | −522 |
CLIRN = Population value + (Dose × θDose) + (Genotype × θGenotype)
CLIRN = Population value + (Dose × θDose)
CLSN = Population value + (Genotype × θGenotype)
where Dose = 1 if dosage was 40 mg/kg and Genotype = 1 if mice were Mdr1a/b−/−
The model-predicted irinotecan lactone AUC0-8h for the Mdr1a/b−/− mice was 1.5-fold higher at the 20 mg/kg dosage level.and 1.8-fold higher at the 40 mg/kg dosage level compared to wild-type mice (Table 2). Similarly, the model predicted irinotecan carboxylate AUC0-8h values for the Mdr1a/b−/− mice were 1.5-fold and 1.8-fold higher. The median SN-38 lactone AUC0-8h values were 1.6-fold (20 mg/kg) and 1.5-fold (40 mg/kg) higher in Mdr1a/b−/− mice compared to wild-type mice.
Table 2.
Systemic exposures of irinotecan and metabolites in Mdr1a/b mice
| Compound | Dosage | AUC0-8h (μM*h) | |
|---|---|---|---|
| Mdr1a/b +/+ | Mdr1a/b −/− | ||
| Irinotecan L | 20 mg/kg | 5.9 | 8.8 |
| 40 mg/kg | 15.8 | 27.9 | |
| Irinotecan C | 20 mg/kg | 7.0 | 10.5 |
| 40 mg/kg | 18.8 | 33.1 | |
| SN-38 L | 20 mg/kg | 2.4 | 3.8 |
| 40 mg/kg | 4.8 | 7.2 | |
Role of Mrp4 in Irinotecan and SN-38 Plasma Pharmacokinetics
Despite evidence that irinotecan and SN-38 are substrates for MRP4 in vitro, the irinotecan and SN-38 plasma concentration-time data from Mrp4−/− mice were very similar to that obtained from wild-type mice at both 20 mg/kg and 40 mg/kg dosages levels (Fig. 3). As expected from this, inclusion of the Mrp4 genotype as a covariate for CLIRN or CLSN38/FE did not improve the model fit to the data (p>0.05) or reduce the IIV in these parameters (Table 1). Thus, MRP4 deletion does not affect the plasma disposition of irinotecan lactone or SN-38 lactone in mice.
Figure 3.

Concentration-time profile of plasma irinotecan and SN-38 in Mrp4−/− and wild-type mice. Plasma irinotecan lactone (A), irinotecan carboxylate (B), and SN-38 lactone (C) concentration-time data after 20 mg/kg (left column) and 40 mg/kg (right colunn) irinotecan intravenous injection. Symbols represent individual concentration-time points, and the line represents the population-predicted values from the final pharmacokinetic model of wild-type and Mrp4−/− mice. Only one line is seen because Mrp4−/− genotype was not a significant covariate on any pharmacokinetic parameter. Each group consisted of 6-7 mice.
Discussion
Structure-based studies have shown that the intact lactone ring is essential for topoisomerase I binding and in vivo anti-tumor activity of camptothecin analogs [20]. Thus, it is important to differentiate between lactone and carboxylate forms in pharmacokinetic studies. As has been previously reported in mice [19], we observed a dose-dependence of irinotecan lactone elimination. The volume of distribution of the carboxylate form of irinotecan was small relative to the lactone form, which is consistent with data from humans [18] and may relate to more extensive tissue distribution or differential plasma protein binding of irinotecan lactone compared to the carboxylate form. Since much of the SN-38 carboxylate concentrations were below the LLOQ, our population pharmacokinetic modeling focused on the other analytes.
A clinical study of irinotecan excretion showed that 22% and 32% of an irinotecan dose were recovered unchanged in the urine and feces, respectively {Slatter, 2000 #7681}. This indicates that active transport may play an important role in irinotecan excretion. In vitro experiments strongly support that irinotecan and SN-38 are substrates for P-gp and MRP4 [3, 8, 14], suggesting that these transporters may play a role in the pharmacokinetics of irinotecan and SN-38. We found that ablation of Mdr1a/b resulted in a significant increase in the systemic exposure of irinotecan lactone and SN-38 lactone. The concentration-time profile suggests decreased systemic clearance in Mdr1a/b−/− mice compared to wild-type mice, and this was confirmed by the genotype being a significant covariate on both irinotecan and SN-38 clearance terms. This effect may be due to decreased hepatic and renal elimination, since P-gp is expressed on both the canalicular membrane of hepatocytes and the apical brush-border membrane of the proximal renal tubules [23]. It was reported that although Mdr1a/b−/− mice have 40% decreased biliary excretion of irinotecan, they show similar biliary excretion of SN-38 to wild-type mice [11]. Therefore, the decreased SN-38 elimination we observed in Mdr1a/b−/− mice may be due to decreased renal elimination. However, this study was not designed to assess the specific pathway of elimination that is regulated by P-gp.
Several pharmacogenetic studies have shown relationships between ABCB1 polymorphisms and irinotecan pharmacokinetics or toxicity. The ABCB1 1236C>T polymorphism, which is a synonymous SNP affecting expression of P-gp, was associated with exposure to both irinotecan and SN-38 {Mathijssen, 2003 #7569}. Patients with two copies of the variant T allele had an irinotecan exposure that was 1.29-fold higher and SN-38 exposure that was 1.47-fold higher than patients with one or two copies of the C allele. In a group of Korean patients, those with the ABCB1 2677GG/3435CC haplotype had higher SN-38 AUC and a trend toward higher irinotecan AUC and those with the ABCB1 2677GG genotype had more severe neutropenia {Han, 2007 #7568}. A recent study did not replicate the role of the ABCB1 1236C>T SNP, but found that the ABCB1 IVS9 −44A>G SNP was associated with reduced SN-38 AUC {Innocenti, 2009 #7680}. Although there is conflicting evidence on the importance of specific polymorphisms, our data support the findings that genetic variations in P-gp will influence SN-38 exposure and toxicity in cancer patients [21, 22].
Mrp4 can localize to both the apical and basolateral membranes of polarized cells, and has been identified in the basolateral membranes of liver cells where efflux activity has been shown {Rius, 2003 #7685}. Despite in vitro data showing interactions between SN-38 and MRP4, we observed no effect of Mrp4 expression on systemic disposition of irinotecan or SN-38 lactone. The plasma concentration-time profiles of Mrp4−/− and wild-type mice were similar at both dosage levels, suggesting little effect of Mrp4 on irinotecan and SN-38 lactone systemic disposition. Although the in vitro studies were performed with cells overexpressing human MRP4 and the in vivo studies performed in mice, this is unlikely to account for the differing results since the murine and human genes are highly homologous [24]. The effect of Mrp4-deficiency on the disposition of other drugs has been conflicting. One study showed that systemic clearance of the nucleotide analog PMEA was decreased in Mrp4-/- mice [26], whereas another study showed no difference in serum levels of PMEA between Mrp4-/- and wild-type mice [27]. Similarly, no differences were seen in plasma methotrexate concentrations after oral administration in Mrp4-/- and wild-type mice [28]. These differences may be due to sex-specific renal expression patterns of Mrp4, since female mice are reported to have higher renal expression of Mrp4 [29]. The present study used female mice, , and yet we still saw noeffect of Mrp4 on irinotecan/SN-38 disposition. It is possible other transporters may compensate for the deletion of Mrp4.
The gene for MRP4 is highly polymorphic in humans, and some polymorphisms were shown to affect expression or activity in vitro [25]. To our knowledge, effects of MRP4 polymorphisms on the clinical pharmacokinetics of irinotecan have not been examined. However, the MRP4 4131T>G polymorphism was shown to affect the pharmacokinetics of the MRP4 substrate lamivudine-triphosphate in HIV patients{Anderson, 2006 #7684}. Heterozygous patients had 15% higher and homozygous variant patients had 20% higher plasma levels than patients with patients with wild-type alleles. Based on our preclinical data, genetic variants may contribute little to the high degree of interpatient variability in the clinical pharmacokinetics of irinotecan and SN-38. Although Mrp4 may not affect the systemic disposition of irinotecan/SN-38, MRP4 may still prevent SN-38 anti-tumor activity through cellular drug resistance or by reducing penetration to protected sites, such as the CNS [16, 27].
In conclusion, although irinotecan and SN-38 are substrates for both MRP4 and P-gp in vitro, P-gp impacts the systemic disposition of irinotecan and SN-38, whereas no effect of Mrp4 was seen on the disposition of irinotecan and SN-38 in mice.
Acknowledgements
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM071321], the National Institutes of Health Nation Cancer Institute, [Grants CA21765, CA23099], and by American, Lebanese, Syrian Associated Charities (ALSAC).
Abbreviations
- ABC
ATP-binding-cassette
- CPT-11
irinotecan
- HPLC
high performance liquid chromatography
- LLOQ
lower limit of quantification
- MRP
multidrug resistance protein
- P-gp
P-glycoprotein
- SN-38
7-ethyl-10-hydroxycamptothecin
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clinical Cancer Research. 2001;7(8):2182–2194. [PubMed] [Google Scholar]
- 2.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Research. 1991;51:4187–4192. [PubMed] [Google Scholar]
- 3.Arimori K, Kuroki N, Hidaka M, Iwakiri T, Yamsaki K, Okumura M, Ono H, Takamura N, Kikuchi M, Nakano M. Effect of P-glycoprotein modulator, cyclosporin A, on the gastrointestinal excretion of irinotecan and its metabolite SN-38 in rats. Pharm Res. 2003;20(6):910–7. doi: 10.1023/a:1023847521767. [DOI] [PubMed] [Google Scholar]
- 4.Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, Kamihira S, Doyle LA, Ross DD, Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem.Biophys.Res.Commun. 2001;280(5):1216–1223. doi: 10.1006/bbrc.2001.4267. 2001.Feb.9.;280.(5):1216.-23. [DOI] [PubMed] [Google Scholar]
- 5.Nakatomi K, Yoshikawa M, Oka M, Ikegami Y, Hayasaka S, Sano K, Shiozawa K, Kawabata S, Soda H, Ishikawa T, Tanabe S, Kohno S. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem.Biophys.Res.Commun. 2001;288(4):827–832. doi: 10.1006/bbrc.2001.5850. [DOI] [PubMed] [Google Scholar]
- 6.Sugiyama Y, Kato Y, Chu X. Multiplicity of biliary excretion mechanisms for the camptothecin derivative irinotecan (CPT-11), its metabolite SN-38, and its glucuronide: role of canalicular multispecific organic anion transporter and P-glycoprotein. Cancer Chemother.Pharmacol. 1998;42(Suppl):S44–9. doi: 10.1007/s002800051078. S44-S49. [DOI] [PubMed] [Google Scholar]
- 7.Maliepaard M, van Gastelen MA, Tohgo A, Hausheer FH, van Waardenburg RC, de Jong LA, Pluim D, Beijnen JH, Schellens JH. Circumvention of breast cancer resistance protein (BCRP)-mediated resistance to camptothecins in vitro using non-substrate drugs or the BCRP inhibitor GF120918. Clin Cancer Res. 2001;7(4):935–941. [PubMed] [Google Scholar]
- 8.Tian Q, Zhang J, Tan TM, Chan E, Duan W, Chan SY, Boelsterli UA, Ho PC, Yang H, Bian JS, Huang M, Zhu YZ, Xiong W, Li X, Zhou S. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm.Res. 2005;22(11):1837–1853. doi: 10.1007/s11095-005-7595-z. [DOI] [PubMed] [Google Scholar]
- 9.Luo FR, Paranjpe PV, Guo A, Rubin E, Sinko P. Intestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters Pgp, cMOAT, and MRP1. Drug Metab Dispos. 2002;30(7):763–70. doi: 10.1124/dmd.30.7.763. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto W, Verweij J, de Bruijn P, de Jonge MJ, Takano H, Nishiyama M, Kurihara M, Sparreboom A. Active transepithelial transport of irinotecan (CPT-11) and its metabolites by human intestinal Caco-2 cells. Anticancer Drugs. 2001;12(5):419–32. doi: 10.1097/00001813-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Iyer L, Ramirez J, Shepard DR, Bingham CM, Hossfeld DK, Ratain MJ, Mayer U. Biliary transport of irinotecan and metabolites in normal and P-glycoprotein-deficient mice. Cancer Chemother Pharmacol. 2002;49(4):336–41. doi: 10.1007/s00280-001-0420-4. [DOI] [PubMed] [Google Scholar]
- 12.Gupta E, Safa AR, Wang X, Ratain MJ. Pharmacokinetic modulation of irinotecan and metabolites by cyclosporin A. Cancer Research. 1996;56:1309–1314. [PubMed] [Google Scholar]
- 13.Bansal T, Mishra G, Jaggi M, Khar RK, Talegaonkar S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur J Pharm Sci. 2009;36(4-5):580–90. doi: 10.1016/j.ejps.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 14.Norris MD, Smith J, Tanabe K, Tobin P, Flemming C, Scheffer GL, Wielinga P, Cohn SL, London WB, Marshall GM, Allen JD, Haber M. Expression of multidrug transporter MRP4/ABCC4 is a marker of poor prognosis in neuroblastoma and confers resistance to irinotecan in vitro. Mol.Cancer Ther. 2005;4(4):547–553. doi: 10.1158/1535-7163.MCT-04-0161. [DOI] [PubMed] [Google Scholar]
- 15.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van DL, Smit JJ, van d. V., Voordouw AC, Spits H, Van TO, Zijlmans JM, Fibbe WE, Borst P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc.Natl.Acad.Sci.U.S.A. 1997;94(8):4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, Mercer KE, Zhuang Y, Panetta JC, Johnston B, Scheper RJ, Stewart CF, Schuetz JD. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24(17):7612–21. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morton CL, Wierdl M, Oliver L, Ma MK, Danks MK, Stewart CF, Eiseman JL, Potter PM. Activation of CPT-11 in mice: identification and analysis of a highly effective plasma esterase. Cancer Res. 2000;60(15):4206–4210. [PubMed] [Google Scholar]
- 18.Xie R, Mathijssen RHJ, Sparreboom A, Verweij J, Karlsson MO. Clinical Pharmacokinetics of Irinotecan and Its Metabolites: A Population Analysis. Journal of Clinical Oncology. 2002;20(15):3293–3301. doi: 10.1200/JCO.2002.11.073. [DOI] [PubMed] [Google Scholar]
- 19.Rouits E, Guichard S, Canal P, Chatelut E. Non-linear pharmacokinetics of irinotecan in mice. Anticancer Drugs. 2002;13(6):631–5. doi: 10.1097/00001813-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Wall ME, Wani MC, Potmesil M, Kohn KW. DNA topoisomerases in Cancer. Oxford University Press; New York, New York: 1991. Chemistry and antitumor activity of camptothecins; pp. 93–102. [Google Scholar]
- 21.Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J, Sparreboom A, McLeod HL. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res. 2003;9(9):3246–53. [PubMed] [Google Scholar]
- 22.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, Lee JE, Lee DH, Kim HT, Lee JS. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110(1):138–47. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 23.Lin JH, Yamazaki M. Role of p-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42(1):59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 24.Lamba JK, Adachi M, Sun D, Tammur J, Schuetz EG, Allikmets R, Schuetz JD. Nonsense mediated decay downregulates conserved alternatively spliced ABCC4 transcripts bearing nonsense codons. Hum Mol Genet. 2003;12(2):99–109. doi: 10.1093/hmg/ddg011. [DOI] [PubMed] [Google Scholar]
- 25.Abla N, Chinn LW, Nakamura T, Liu L, Huang CC, Johns SJ, Kawamoto M, Stryke D, Taylor TR, Ferrin TE, Giacomini KM, Kroetz DL. The human multidrug resistance protein 4 (MRP4, ABCC4): functional analysis of a highly polymorphic gene. J Pharmacol Exp Ther. 2008;325(3):859–68. doi: 10.1124/jpet.108.136523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol. 2007;71(2):619–27. doi: 10.1124/mol.106.028233. [DOI] [PubMed] [Google Scholar]
- 27.Belinsky MG, Guo P, Lee K, Zhou F, Kotova E, Grinberg A, Westphal H, Shchaveleva I, Klein-Szanto A, Gallo JM, Kruh GD. Multidrug resistance protein 4 protects bone marrow, thymus, spleen, and intestine from nucleotide analogue-induced damage. Cancer Res. 2007;67(1):262–8. doi: 10.1158/0008-5472.CAN-06-2680. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura Y, Hirouchi M, Kusuhara H, Schuetz JD, Sugiyama Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3) J Pharmacol Exp Ther. 2008;327(2):465–73. doi: 10.1124/jpet.108.140475. [DOI] [PubMed] [Google Scholar]
- 29.Maher JM, Slitt AL, Cherrington NJ, Cheng X, Klaassen CD. Tissue distribution and hepatic and renal ontogeny of the multidrug resistance-associated protein (Mrp) family in mice. Drug Metab Dispos. 2005;33(7):947–55. doi: 10.1124/dmd.105.003780. [DOI] [PubMed] [Google Scholar]