

Abstract
It is well established that Toll-like receptors (TLRs) play a critical role in the generation of innate immune responses and thereby also play an important, indirect role in the initiation of subsequent adaptive T cell responses. However, T cells also express certain TLRs, and we have focused on the physiological importance of direct TLR signaling in T cells. TLRs can function as co-stimulatory receptors that complement TCR-induced signals to enhance effector T cell proliferation, survival and cytokine production. We also found that TLR signaling pathways in T cells are required for the effective clonal expansion of antigen-specific T cells during infection in vivo. Thus, the importance of TLRs in T cell-mediated immunity reflects both T cell-extrinsic and T cell-intrinsic components, which warrants a reconsideration of the dogma that restricts germ-line encoded pattern recognition to cells of the innate immune system.
Keywords: TLR, MyD88, PAMP, CD4 T cell, CD8 T cell, Co-stimulation, Inflammation, Clonal expansion
Introduction
The mammalian Toll-like receptor (TLR) family is a major class of germ-line encoded pathogen recognition receptors that play an important role in host defense. The family is known to include at least 11 members, which detect a broad range of pathogen-associated molecular patterns (PAMPs) [1]. TLRs 2, 4, 5 and 11 are expressed on the cell surface and recognize bacterial lipoproteins, lipopolysaccharide (LPS), flagellin and profilin, respectively. TLRs 3, 7, 8 and 9 are expressed in endosomal compartments and are involved in the recognition of viral and bacterial nucleic acids. While TLRs have generally been considered a means to distinguish “non-self” from “self”, it is interesting to note that endogenous molecules may also act as TLR ligands. Heat shock proteins (HSPs) and extracellular matrix components, such as fibronectin and hyaluronan are released during stressful tissue injury and have been reported to activate the innate immune system through TLR2 and TLR4 [2–5], though the potential contamination of these endogenous ligands with PAMPs is a concern in some of these studies [6, 7]. In addition, chromatin complexes released from damaged cells contain nucleic acids that may engage TLR3, TLR7 and TLR9 [8, 9]. In analogy to PAMPs, these putative endogenous ligands may be considered damage-associated molecular patterns (DAMPs) [10].
Toll-like receptors contain a leucine-rich repeat motif, which serves as a ligand binding domain, and a cytoplasmic Toll/interleukin-1 receptor (TIR) domain that initiates signal transduction [11]. The super-family of TIR-domain-containing receptors also includes the IL-1R/IL-18R family. These receptors act by recruiting and homodimerizing with TIR domain-containing adaptors proteins. Myeloid differentiation protein 88 (MyD88) is a major adaptor protein linking these receptors to downstream signaling molecules. MyD88 is required for signaling through all TLRs except TLR3, which signals through the adaptor molecule Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF), and TLR4, which can signal through both MyD88- and TRIF-dependent pathways. Recruitment of MyD88 leads to the activation of IL-1 receptor-associated kinases (IRAKs) and TNF receptor-associated factor 6 (TRAF6), ultimately resulting in the activation of transcription factors, including NFκB. In addition to this sequence of events, MyD88 has also been shown to activate the PI-3 kinase pathway, to induce IFN regulatory factor IRF-1, IRF-5 and IRF-7 and to play a role in Fas-mediated and IFNγ receptor-mediated signaling [12–17].
Toll-like receptors are highly expressed on myeloid cells of the innate immune system, such as macrophages and dendritic cells, and have been studied most extensively in these cell types. TLR engagement has been found to enhance antigen presentation, upregulate co-stimulatory molecule expression and promote the production of pro-inflammatory cytokines [18]. Thus, TLRs have traditionally been considered to play an important role in indirectly controlling T cell responses through the innate immune system. However, we and others have found that T cells themselves express TLRs, and we have focused on whether TLR signaling in T cells also contributes directly to T cell-mediated immune responses.
TLR expression on T cells
A number of studies have examined the expression of TLRs 1–10 on various subsets of purified T cells. In two studies, sorted CD4+CD45RBhigh T cells from C57/BL6 (B6) mice were found to express TLR1, 2, 3, 6, 7 and 8, but low to undetectable levels of TLR 4, 5 and 9 mRNA [19, 20]. Naïve CD8+ T cells from B6 mice were reported to express mRNA for TLR1, 2, 6 and 9 but not TLR4 [21, 22]. We have found that naïve CD4+ T cells from BALB/c mice express mRNA for TLR3, 4, 5 and 9 [23]. Regarding protein expression, TLR2, 3, 4 and 9 protein have been reported to be found on CD4+ T cells via flow cytometry [24], and TLR2 protein has been detected by flow cytometry on CD8+ T cells [21, 22]. Lastly, human T cells isolated from peripheral blood have also been reported to express mRNA for most TLRs, though there is a considerable variation in the reported expression levels in these studies [25–27]. Protein expression of TLR2, 3, 4, 5 and 9 has also been detected by flow cytometry [26, 28–30].
Interestingly, TLR expression on T cells also appears to be regulated by TCR-dependent activation. We have found that naïve BALB/c CD4+ T cells upregulate TLR3 and TLR9 mRNA in response to TCR stimulation by αCD3 antibodies [23]. Furthermore, while naïve BALB/c T cells were shown to express intracellular TLR2, expression on the cell surface was only readily detectable by flow cytometry following a CD3-induced activation [31]. Similarly, B6 CTLs express higher levels of TLR2 mRNA than naïve CD8+ T cells [21], and TCR stimulation increases surface expression of TLR2 protein as detected by flow cytometry [22]. Antigen-experienced T cells appear to continue to maintain higher levels of TLR expression, since mouse CD4+CD45RBlow memory cells have also been shown to express higher mRNA levels of most TLRs than naïve CD4+CD45RBhigh T cells [19] and human CD45RO+ memory CD4+ T cells express higher levels of TLR2 than naïve T cells [29].
To summarize, while TLR expression on various T cell subsets has been independently confirmed by a number of studies, the specific patterns of expression that have been reported vary considerably. Thus, there may be significant species, and even strain, -specific differences in TLR expression. It is also possible that these differences in expression result from technical variations in different studies. It is important to note that mRNA levels do not always correlate with protein expression, and TLR expression is known to be regulated at the protein level by ubiquitin-mediated degradation. Furthermore, minimal contamination of purified T cells by APCs could significantly influence results. In addition, studies that have examined TLR protein levels in T cells are limited by the reliability of available antibody reagents. Indeed, our own experience with reagents to detect murine TLRs via flow cytometry has been quite variable (A. Gelman, D. LaRosa, A. Rahman and L. Turka, unreported data). These caveats may partially explain the considerable variability in the TLR expression patterns that have been reported on T cells. Given the limitations in examining expression levels of TLRs, we have considered the ability of known TLR ligands to activate signaling pathways [23] as more reliable indicators of expression.
TLR ligands can directly co-stimulate T cell subsets
Effector T cells
Effective T cell activation requires a primary signal delivered through the TCR and secondary signals delivered through co-stimulatory receptors. CD28 is generally considered to be the principle co-stimulatory receptor involved in initial T cell activation. CD28-mediated signals synergize with TCR-induced signals to promote the synthesis of pro-survival molecules and enhance IL-2 production. We and others have found that a number of TLR ligands can act directly on highly purified B6 T cells in vitro, to provide co-stimulatory signals in the absence of CD28 engagement. Thus, we found that ligands for TLR2, 3 and 9 can complement TCR-induced activation to enhance effector functions, such as IL-2 production, in purified B6 CD4+ T cells (Fig. 1). Similarly, ligands for TLR2, 5 and 7/8 have been shown to enhance proliferation and IL-2 and IFNγ production in human CD4+ T cells, particularly in CD45RO+CCR7− memory cells [29, 32]. It is important to note that these TLR-induced signals in T cells are strictly co-stimulatory, in that TLR ligands do not appear to produce functional responses in naïve T cells in the absence of concurrent TCR stimulation ([22]; A. Rahman, D. LaRosa and L. Turka, unpublished observations). This may reflect the importance of preventing TLR signals from inducing non-specific T cell activation.
Fig. 1.
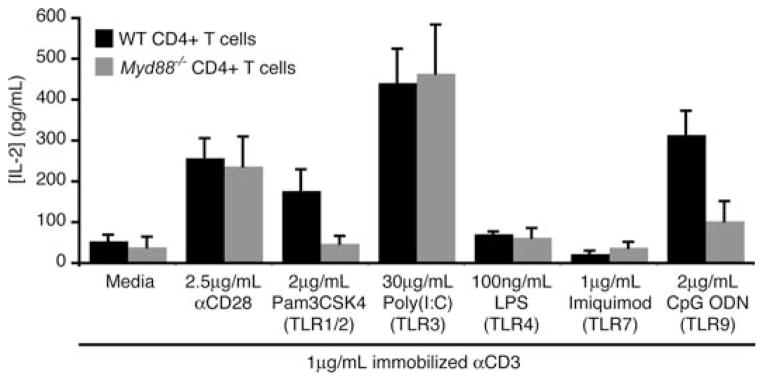
TLR ligands can directly co-stimulate IL-2 production in CD4+ T cells. CD4+CD25− T cells were sort-purified from WT or Myd88−/− B6 mice, and equal numbers were cultured separately under the indicated stimulation conditions. Cell-free supernatants were collected after 24 h and the IL-2 concentration was determined by ELISA. Data show mean ± SD of three mice
We have found that the functional responses of TLR9-induced co-stimulation in CD4+ T cells can be attributed to two main signaling pathways mediated by the adaptor molecule, MyD88. Following stimulation with CpG oligonucleotides (ODN), MyD88 induces NF-κB activation in CD4+ T cells through homotypic death domain interactions with IRAKs. This NF-κB-dependent pathway upregulates expression of the anti-apoptotic molecule Bcl-xL, and promotes activated CD4+ cell survival [23]. TLR2 stimulation has been found to similarly enhance survival and Bcl-xL expression in activated CD8+ T cells [22]. While NF-κB activation appears to be solely required for CpG-ODN to induce anti-apoptotic signals in T cells, we have found that this pathway synergizes with a PI-3-kinase-dependent pathway in promoting proliferative responses and IL-2 production [33]. In contrast to the death domain interactions by which MyD88 activates NF-κB, MyD88-dependent PI-3 kinase activation involves a tyrosine residue located in the TIR domain. This allows MyD88 to associate with the p85 regulatory subunit of PI-3 kinase and induce Akt phosphorylation in response to TLR stimulation. Thus, specific outcomes of TLR-mediated stimulation in T cells may be associated with discrete MyD88 motifs and signaling pathways.
Interestingly, we have found that stimulation of CD4+ T cells with TLR ligands also results in enhanced expression of secondary co-stimulatory receptors that are generally upregulated following initial T cell activation (Fig. 2). Certain co-stimulatory receptors, such as CD40L, play an important role in allowing CD4+ T cells to promote B cell immune responses, which may relate to our finding that MyD88 expression in CD4+ T cells is critical in supporting B cell-class switching to IgG production in an in vivo model of CpG DNA-induced antibody responses against a T cell-dependent antigen [33]. Secondary co-stimulatory receptors can also play an important role in supporting sustained T cell responses. For example, signals delivered through OX-40 serve to enhance CD4+ T cell effector responses, survival and IL-2 production [34]. Given that T cell responses induced by these co-stimulatory receptors are quite similar to those that occur following TLR stimulation, it is possible that, in addition to the direct TLR-induced signaling pathways discussed earlier, some of the functional outcomes of TLR stimulation on T cells may reflect secondary consequences of enhanced signaling through these induced co-stimulatory pathways.
Fig. 2.

Direct stimulation of CD4+ T cells with TLR9 ligands upregulates OX-40 expression. CD4+CD25− T cells were sort-purified from WT, TLR9−/− and Myd88−/− B6 mice. Equal numbers of cells were cultured with 1 μg/mL of immobilized αCD3 alone, or with the addition of 2.5 μg/mL immobilized αCD28, 6 μg/mL CpG or 100 ng/mL LPS, as indicated. After 48 h, surface expression of OX-40 was examined by flow cytometry. OX-40 expression following the indicated stimulation conditions is shown (black line) relative to αCD3 alone (shaded histogram). Data are representative of four mice
Our studies have focused on survival, proliferation and effector cytokine production as readouts of functional TLR responses in T cells. However, in considering other aspects of T cell physiology it has been found that LPS treatment results in increased adhesion of mouse and human T cells to fibronectin and inhibited chemotaxis [35]. Human T cells were also reported to respond similarly to the endogenous ligand HSP60 through TLR2, though these results could reflect potential contamination of commercially available HSP60 with bacterial TLR2 ligands [36]. Thus, in addition to enhancing effector functions, TLR ligands may also play a role in controlling T cell trafficking. The finding that T cells may potentially respond to endogenous TLR ligands is particularly intriguing, since this presents the possibility that DAMPs may act to recruit T cells and support their responses at sites of tissue inflammation and necrosis. These findings also suggest that T cells may respond to a broader range of TLR ligands than has been thus far been considered, since functional outcomes may differ in responses to specific TLR ligands.
Regulatory T cells
The studies discussed thus far indicate that TLR ligands can promote more effective T cell-mediated immunity by acting directly on CD4+ and CD8+ T cells to enhance their respective effector functions. However, TLR ligands can also influence CD4+CD25+ regulatory T cells. Tregs are known to inhibit T cell effector responses in vitro and are critical in maintaining immune tolerance and preventing the development of autoimmunity in vivo. As in the case of effector T cells, TLR ligands indirectly modulate regulatory T cell responses by promoting inflammatory cytokine production in APCs, which can inhibit the suppressive capacity of Tregs [37]. However, it is now clear that TLR ligands can also act directly on Tregs, though there are differences in the reported outcomes of these actions.
Pam3CysSK4 can act directly on mouse Tregs through TLR2 to promote their expansion both in vitro and in vivo, which may relate to the reduced numbers of Tregs found in Myd88−/− and Tlr2−/− mice [38]. Yet while promoting increased proliferation of Tregs, Pam3CysSK4 also abrogates their suppressive activity, which correlates with a transient suppression of Foxp3 expression [31]. It has also been found that direct TLR8 triggering of human Tregs similarly prevents their suppressive activity [39]. It should be noted though that, in contrast to these studies, others have found that signals through TLR2 and TLR5 can enhance the suppressive function of Tregs as well as Foxp3 expression [28, 40].
While TLR ligands may influence suppression in co-culture experiments, this potentially reflects the combined result of distinct effects on both conventional T cells and regulatory T cells. By using co-culture suppression assays, in which either the responder or Treg population lacked MyD88 expression, we showed that this is indeed the case as CpG DNA can act directly on Tregs to block their suppressive activity, but also acts on effector T cells to render then resistant to suppression [41]. Interestingly, once TLR ligands are removed, Tregs fully regain their suppressive phenotypes [31, 38]. Overall, these findings suggest a model, whereby TLR ligands can act on both effector and regulatory T cells to directly promote T cell-mediated immune responses by enhancing T cell effector functions and clonal expansion through increased proliferation, survival and a transient loss of suppression. These TLR ligands also serve to concurrently expand the Treg population, which regains its suppressive capacity following clearance of the TLR ligands and can then act to regulate the expanded effector T cell population at the end of the immune response.
The importance of MyD88 in T cells during in vivo immune responses
Toll-like receptors play an important role in host defense, and mice lacking the adaptor protein MyD88 have greatly increased susceptibility to a number of eukaryotic, prokaryotic and viral pathogens. In these infection models, the immune impairments associated with MyD88-deficiency have generally been attributed to the importance of MyD88 in the innate immune system. However, the studies discussed earlier demonstrated that TLR ligands can directly modulate T cell effector functions. Therefore, we have recently considered the possibility that defective adaptive immune responses in MyD88-deficient mice also reflect a T cell-intrinsic role for MyD88. To this end, we have employed bone marrow chimeras and adoptive transfer systems to examine immune responses of MyD88-deficient T cells in vivo, in the context of an otherwise unimpaired immune system.
Myd88−/− mice display significantly greater mortality following infection by the protozoan pathogen, Toxoplasma gondii [42]. This increased susceptibility is associated with reduced Th1 effector responses, which have been attributed to defective innate immune activation and reduced levels of IL-12 production in these mice. To examine whether MyD88-dependent signaling in T cells also directly contributes to reduced Th1 responses, we generated mixed bone marrow chimeras in which the T cell compartment was MyD88-deficient, but MyD88-dependent innate immune responses were shown to be intact. These mice demonstrated comparable levels of IL-12 production and APC activation as WT control chimeras following T. gondii infection, but had reduced numbers of IFNγ-producing effector T cells and were highly susceptible to toxoplasmic encephalitis, succumbing to infection with similar kinetics to full Myd88−/− mice [43]. These findings establish that MyD88 expression in T cells is required for prolonged resistance to T. gondii, even in the setting of an intact innate immune response. However, the precise mechanism by which MyD88 supports T cell responses in this model remains unclear.
MyD88-dependent signals in T cells also play a role in CD4+ T cell-mediated colitis. When CD4+CD45RBhigh T cells are transferred into RAG1/2-deficient animals, the recipients rapidly develop wasting disease associated with clinical signs of colitis. This experimental colitis depends on the presence of commensal intestinal microflora and TLRs from these microorganisms are thought to activate innate immune cells in the gut and thereby indirectly support colitogenic T cell responses [44]. However, it has recently been reported that purified MyD88−/− CD4+CD45RBhigh T cells transferred into RAG-deficient recipients do not induce as severe colitis as WT T cells [20, 24]. Using competitive transfer systems, Myd88−/− colitogenic CD4+ T cells were shown expand less effectively than WT cells and this correlated with reduced production of effector cytokines. In particular, MyD88−/− CD4+ T cells isolated from the lamina propria produced significantly less IL-17. Furthermore, it was shown that Myd88−/− CD4+ T cells did not effectively differentiate into IL-17-producing cells in vitro, suggesting that MyD88 may play a role in Th17 polarization [24]. Relevant to this, Myd88−/− mice are highly resistant to the induction of experimental autoimmune encephalitis (EAE), which is an IL-17-mediated autoimmune disease [45]. While reduced numbers of Myd88−/− Th17 cells in this model have been attributed to decreased production of IL-6 and IL-23 by Myd88−/− dendritic cells, it would be interesting to examine whether they also reflect a T cell-intrinsic role for MyD88 in supporting Th17 responses.
These studies using T. gondii and colitis models have shown that MyD88-dependent signals in T cells directly contribute to T cell responses in vivo. However, since they have only examined overall polyclonal T cell responses, these studies have been limited in their ability to directly compare antigen-specific populations of WT and MyD88−/− effector T cells. Thus, while reduced levels of CFSE dilution and BrdU uptake have been identified in MyD88−/− colitogenic CD4+ T cell populations, it remained unclear whether this represented reduced proliferation of antigen-specific T cell-clones or increased cell death resulting in a smaller proportion of dividing colitogenic cells. Similarly, it was unclear whether impaired cytokine production by MyD88−/− T cell populations truly represented reduced cytokine secretion by antigen-specific responders, or instead reflects a relative smaller proportion of cytokine-producing effector cells.
To more closely investigate these issues, we have recently examined the role of MyD88-dependent signals in CD8+ T cells during acute lymphocytic choriomeningitis virus (LCMV) infection. Acute LCMV infection elicits a dramatic expansion of CD8+ T cells that has been very well characterized. The majority of LCMV epitopes have been identified and the availability of viral peptides and MHC class I tetramers allow the detection of antigen-specific effector cells, making this an ideal model for examining in vivo T cell responses to a natural, viral pathogen [46].
Myd88−/− mice generate greatly reduced numbers of antigen-specific effector CD8+ T cells following LCMV infection [47, 48]. APCs from these mice have been found to produce significantly lower levels of inflammatory cytokines [48], and we found that this partially contributes to the reduced CD8+ T cell expansion that occurs in response to LCMV infection [49]. However, using reciprocal adoptive transfer experiments and bone marrow chimeras, we found that MyD88 also plays a critical, T cell-intrinsic role in supporting the expansion of antigen-specific CD8+ T cells.
Consistent with other models, BrdU incorporation indicated reduced turnover of the Myd88−/− T cell population following LCMV infection, but we found that this merely reflected the reduced proportion of LCMV-specific CTLs in the polyclonal Myd88−/− T cell population. By specifically comparing antigen-specific populations, we found that, despite their reduced numbers following LCMV infection, Myd88−/− CD8+ T cell-clones proliferate and differentiate into functional effector cells and long-lived memory cells similar to WT cells. However, MyD88-dependent signals are critical in supporting T cell survival and accumulation during the initial phase of antigen-driven proliferation, thereby controlling the ultimate size of the effector population at the peak of the response [49].
As discussed earlier, TLR ligands can act directly on isolated T cells in vitro to promote increased proliferation, survival and effector cytokine production in the absence of traditional co-stimulatory signals. However, it was unclear whether some of these effects may be redundant when T cells are activated by TLR-matured APCs. Our findings using the LCMV model suggest that indirect TLR signaling in APCs is sufficient to promote proliferative and effector cytokine responses in CD8+ T cells but that direct TLR signals confer important, non-redundant survival signals. This suggests that the defects associated with Myd88−/− T cells in the T. gondii and colitis models may primarily be explained by reduced effector cell survival rather than impairments in proliferation or effector cell differentiation, though it is difficult to extrapolate findings between experimental systems, since the precise nature of the MyD88-mediated signals may vary.
While in vitro studies have demonstrated that T cells can be co-stimulated through specific TLRs, it is interesting to note that we have not been able to attribute the importance of MyD88 in T cells to individual TLR pathways in our infection models. TLR1, TLR2, TLR4, TLR6 and TLR9 are individually not required for resistance to T. gondii [42, 50–52]. Furthermore, while TLR11 participates in T. gondii resistance by recognition of a pathogen-derived profilin, unlike Myd88−/− mice, Tlr11−/− mice survive acute toxoplasmosis [53]. Similarly, it has been shown that mice deficient in TLR2, TLR3, TLR4 and TLR8 do not display reduced CD8+ T cell responses to LCMV, and while LCMV-specific CTL numbers are slightly reduced in mice lacking both TLR7 and TLR9, they do not recapitulate the phenotype of Myd88−/− mice [48, 54]. MyD88 is also involved in signaling downstream of the IL-1R and IL-18R, and these cytokines have been shown to play a role in supporting T cell responses [55]. While we found that IL-18 did contribute somewhat to T cell responses following both T. gondii and LCMV infection, in neither model did IL-1R or IL-18R-mediated signaling appear to entirely account for the severely impaired T cell responses associated with MyD88-deficiency [43, 49].
Thus, while MyD88 plays an important role in T cells during in vivo immune responses, the specific upstream initiators of MyD88-mediated signaling remain unclear. These findings may reflect considerable cooperation and redundancy between different MyD88-dependent pathways. They also raise the interesting possibility that the importance of MyD88 in T cells may be associated with a function outside of its role in traditional TLR signaling.
Conclusions
Toll-like receptor ligands have long been recognized to play an important, indirect role in supporting T cell responses through myriad effects on innate immune cells, including upregulating antigen presentation, co-stimulatory molecule expression and inflammatory cytokine production. In addition to these important effects in cells of the innate immune system, it is now clear that TLR ligands can also directly act on T cells. Several in vitro studies have shown that TLRs can potentially serve as co-stimulatory receptors on T cells, and that their engagement supports TCR-mediated signals, promoting effector responses including enhanced cytokine production, proliferation and survival, in the absence of traditional co-stimulation through CD28. Furthermore, MyD88-dependent signals in T cells play a critical role in supporting in vivo T cell responses in a number of physiologically relevant disease models, in the presence of normal TLR signaling in the innate immune system. These findings suggest a model (Fig. 3) in which TLRs play an important, indirect role in promoting initial T cell activation by APCs in the lymph nodes but also act directly on T cells at the site of injury, where they provide signals that support the survival and clonal expansion of effector T cells. Thus, TLRs and the adaptor protein MyD88 play important, and largely underappreciated, T cell-intrinsic roles that are likely to be relevant in the context of multiple infectious and autoimmune diseases. This represents a significant shift in the dogma that restricts germ-line encoded pattern recognition to cells of the innate immune system.
Fig. 3.

TLR signaling contributes both indirectly and directly to T cell responses
References
- 1.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 2.Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2), TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 3.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 4.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. 3rd: the extra domain A of fibronectin activates toll-like receptor 4. J Biol Chem. 2001;276:10229. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 5.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsan MF, Baochong G. Pathogen-associated molecular pattern contamination as putative endogenous ligands of toll-like receptors. J Endotoxin Res. 2007;13:6. doi: 10.1177/0968051907078604. [DOI] [PubMed] [Google Scholar]
- 7.Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem. 2003;278:174. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- 8.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM, toll-like receptors. Nature. 2002;416:603. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 9.Rahman AH, Eisenberg RA. The role of toll-like receptors in systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28:131. doi: 10.1007/s00281-006-0034-3. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi ME. DAMPs, PAMPs, alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 13.Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. 2006;103:15136. doi: 10.1073/pnas.0607181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature. 2005;434:243. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 15.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A. FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Mol Cell Biol. 2007;27:7394. doi: 10.1128/MCB.00600-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 18.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- 19.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomita T, Kanai T, Fujii T, Nemoto Y, Okamoto R, Tsuchiya K, et al. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J Immunol. 2008;180:5291. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]
- 21.Sobek V, Birkner N, Falk I, Wurch A, Kirschning CJ, Wagner H, et al. Direct Toll-like receptor 2 mediated co-stimulation of T cells in the mouse system as a basis for chronic inflammatory joint disease. Arthritis Res Ther. 2004;6:R433. doi: 10.1186/ar1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. 2006;36:1684. doi: 10.1002/eji.200636181. [DOI] [PubMed] [Google Scholar]
- 23.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, et al. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 26.Mansson A, Adner M, Cardell LO. Toll-like receptors in cellular subsets of human tonsil T cells: altered expression during recurrent tonsillitis. Respir Res. 2006;7:36. doi: 10.1186/1465-9921-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarember KA, Godowski PJ. Tissue expression of human toll-like receptors and differential regulation of toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 28.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 29.Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wesch D, Beetz S, Oberg HH, Marget M, Krengel K, Kabelitz D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J Immunol. 2006;176:1348. doi: 10.4049/jimmunol.176.3.1348. [DOI] [PubMed] [Google Scholar]
- 31.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 33.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, et al. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510. [PubMed] [Google Scholar]
- 35.Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner-Fischer M, Lider O, et al. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol. 2007;179:41. doi: 10.4049/jimmunol.179.1.41. [DOI] [PubMed] [Google Scholar]
- 36.Zanin-Zhorov A, Nussbaum G, Franitza S, Cohen IR, Lider O. T cells respond to heat shock protein 60 via TLR2: activation of adhesion and inhibition of chemokine receptors. Faseb J. 2003;17:1567. doi: 10.1096/fj.02-1139fje. [DOI] [PubMed] [Google Scholar]
- 37.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 38.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 40.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.LaRosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunol Lett. 2007;108:183. doi: 10.1016/j.imlet.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 43.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, et al. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci U S A. 2008;105:3855. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sartor RB. Role of commensal enteric bacteria in the pathogenesis of immune-mediated intestinal inflammation: lessons from animal models and implications for translational research. J Pediatr Gastroenterol Nutr. 2005;40(Suppl 1):S30. doi: 10.1097/00005176-200504001-00018. [DOI] [PubMed] [Google Scholar]
- 45.Marta M, Andersson A, Isaksson M, Kampe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- 46.Masopust D, Murali-Krishna K, Ahmed R. Quantitating the magnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cell response: it is even bigger than we thought. J Virol. 2007;81:2002. doi: 10.1128/JVI.01459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, et al. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822. doi: 10.1002/eji.200425730. [DOI] [PubMed] [Google Scholar]
- 48.Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, et al. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces cytotoxic T cell response via MyD88. J Virol. 2008;82:196. doi: 10.1128/JVI.01640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahman AH, Cui W, LaRosa DF, Taylor DK, Zhang J, Goldstein DR, et al. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute LCMV infection. J Immunol. 2008;181:3804. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol. 2007;179:1129. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 51.Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immuno-privileged organs and role of toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol. 2005;7:837. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 52.Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, et al. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. 2006;176:7589. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 53.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 54.Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 55.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]