

Abstract
Controversies remain regarding the precise cell type from which prostate cancers originate. In the last 2 years, two separate studies have arrived at apparently conflicting models for the cell type involved in prostate cancer initiation. However, these results are not mutually exclusive: there are potential solutions, and alternative views on the initiating cell derivation of prostate tumors also exist.
It has been suggested that tumor-initiating cells of solid tumors originate from primitive tissue-based stem cells, as these are cells which survive long enough to accumulate a series of genetic and epigenetic alterations, possess inherent self-renewal ability, and are able to differentiate into a number of resident cell types. Research during the last few years has produced a great deal of information regarding the stem cell biology of prostatic epithelia and its association with cancer.1,2 Cells in the basal compartment of mouse and human prostate possess self-renewal capability2,3 and can generate both basal and luminal cells.2–4 This supports a model in which basal stem cells can self-renew and also generate progenitor cells (that is, intermediate or transient amplifying cells) with an intermediate phenotype, whose progeny differentiate into mature columnar luminal cells. Other studies indicate that intermediate cells, or other cells within the luminal compartment, can also possess prostate epithelial stem cell characteristics.1,2,5,6
In their study, Wang et al.6 used genetic lineage mapping in the mouse prostate and identified rare multipotent luminal cells that could generate both basal and luminal cells in vivo. These cells, designated castration-resistant Nkx3.1-positive cells, developed into adenocarcinoma when the tumor suppressor gene Pten was disrupted. Conversely, Goldstein et al.3 used gene transfer into isolated human prostate cells and found that basal cells which expressed low or absent levels of the androgen receptor could be tumor-initiating cells, but that androgen-receptor-positive luminal cells could not.3 After transduction of myristoylated-AKT (a serine/threonine protein kinase) and ERG (an ets family transcription factor), lesions resembling human prostatic intraepithelial neoplasia (PIN), a precursor to invasive prostatic adenocarcinoma, developed. These lesions contained both basal cells and transformed luminal-like cells. When AR was also transduced, microinvasive adeno-carcinomas developed that demonstrated a lack of basal cells—a hallmark of the human disease.3 In both studies, the authors suggest that a tissue stem cell is the cell of origin for prostate cancer, but in each case a different tissue stem cell was identified as the tumor-initiating cell.
How can these two disparate conclusions be reconciled? Neither group claims that the cell type that they identified is the exclusive stem cell in the prostate, nor that it is the only tumor-initiating cell. Perhaps both cell types could be active in the prostate and produce tumors with different biological properties.1,2,5
It is also interesting to consider how these findings fit with our knowledge of the human disease. Numerous observations indicate that in high-grade PIN, only luminal-like cells possess the somatic genomic changes found in invasive carcinomas.7 Silencing of the GSTP1 promoter by CpG island hyper-methylation occurs in approximately 90% of prostatic carcinomas and around 70% of PIN. In PIN, only the androgen-receptor-positive luminal cells lack GSTP1 protein. Furthermore, androgen-receptor-positive luminal cells are the only cells in PIN in which somatic telomere shortening (which can lead to chromosomal instability) occurs. TMPRSS2-ERG rearrangements, resulting in ERG mRNA and protein overexpression, occur in around 50% of prostatic carcinomas; and in ERG-positive PIN lesions it is only the androgen-receptor-positive luminal cells which stain positively for ERG protein.8 The oncogenic transcription factor MYC is also overexpressed in most PIN lesions in the androgen-receptor-positive luminal compartment.7 Thus, while basal cells which are low or negative for the androgen receptor may be stem cells they do not contain the molecular genetic and epigenetic ‘hits’ which are characteristic of PIN or prostate adenocarcinoma, even within basal compartments of PIN lesions. Alternatively, it may be that such genetic and epigenetic changes have thus far eluded detection.
Perhaps key carcinogenic hits do occur in basal cells, but the consequences manifest only when the cells lose basal cell features—for example, expression of p63—and undergo differentiation to luminal cells. This could explain the lack of basal cells seen in prostatic carcinoma. Analogy to some leukemias suggests that a proliferative or survival advantage from such hits would be expected to produce an expansion of this precursor basal cell population. While this expansion was found in one mouse model of prostate cancer,1 an expansion of basal cells in prostate cancer or PIN has not been identified in humans. Another possibility is that when oncogenic stress occurs in basal cells, they rapidly differentiate into luminal-like cells which appear neoplastic, leaving no trace of their previous basal state.1,2 Regardless of the cell in which the initial oncogenic or tumor suppressive hits occur, some of the resulting neoplastic luminal-like cells do not fully differentiate to become ‘post-mitotic’. Recently, plausible mechanisms responsible for this differentiation block have arisen, and overexpression of MYC or ERG both seem capable of inducing such a block.7–9
There are alternative views to the notion that tissue stem cells are the sole direct target cells of tumor initiation. Data have accumulated indicating that certain tumor types do not originate from multipotent tissue stem cells, but rather from differentiated progenitor cells that co-opt self-renewal programs from the tissue stem cells. It has been suggested that such cells are responsible for initiating development of prostate tumors. For example, the human prostate contains regions of focal atrophy—often associated with variable degrees of inflammation—in which many of the atrophic luminal cells have an intermediate phenotype.7 These focal atrophy lesions appear in the prostate in response to diverse carcinogenic insults, including exposures to dietary heterocyclic amines, estrogens, and various pathogens. Furthermore, these regions of expanded proliferating intermediate cells commonly show transitions to PIN and, at times, to adenocarcinoma, supporting the notion that neoplastic transformation may commence in intermediate cells.7
Perhaps the focus thus far has been misdirected somewhat. Recent findings indicate that four transcription factors (MYC, KLF4, Oct4, and SOX2) can reprogram fully differentiated cells into induced pluripotent stem cells. Thus, it is now plausible that even mature differentiated postmitotic prostatic luminal cells that lack inherent self-renewal capabilities could be ‘reprogrammed’ by oncogenic pressure into tumor-initiating PIN cells.7 In addition, the self-renewal programs co-opted by the transformed cell need not originate in cognate tissue stem cells. Rather, certain oncoproteins may induce aberrant self-renewal programs foreign to the tissue stem cells, such as those found in embryonic stem cells.10 In the prostate, the oncogenic activation could occur in any of the cell compartments, although we favor one of the luminal cell types (Figure 1). The resultant tumor-initiating cell would probably express a phenotype reminiscent of a luminal cell, but with no precisely matched ‘normal cell’. The intense research activity in this field should help resolve these apparent disagreements, which will undoubtedly produce a deeper understanding of the biology of prostate cancer.
Figure 1.
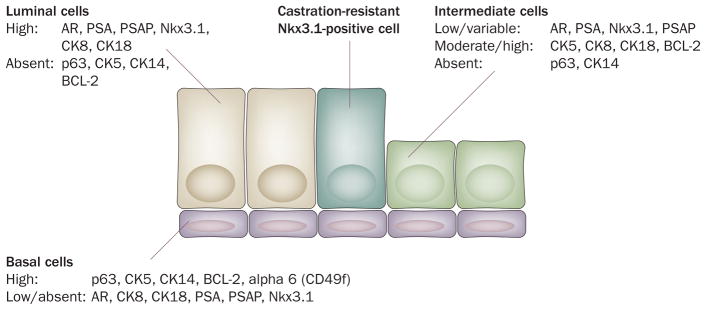
Phenotype of prostatic epithelial cells. In the normal prostate, basal cells express abundant CK5, CK14 (found in around 50% of cells), p63, and integrins (such as CD49f/alpha 6 integrin); and very low or no CK8, CK18, AR, PSA, Nkx3.1, and PSAP. The luminal cells express abundant CK8, CK18, AR, Nkx3.1, PSA, and PSAP; and lack nuclear p63, CK5, CK14, and most integrins. The apoptosis regulator BCL-2 is highly expressed in basal cells and is absent or present at very low levels in luminal cells. Intermediate cells have intermediate or high levels of CK5, CK8, CK18 and BCL-2; and low or variable levels of AR, Nkx3.1, PSA and PSAP, yet lack p63 and CK14. AR-negative neuroendocrine cells are scattered throughout the prostate (not shown). PIN cells and prostate cancer cells most closely resemble luminal cells. Castration-resistant Nkx3.1-positive cells are rare, comprising only 0.7% of prostate luminal epithelial cells; and are present in the luminal compartment. Although they share features with luminal cells they are distinguished from most other luminal cells by their retention of Nkx3.1 protein after castration. Abbreviations: AR, androgen receptor; CK, cytokeratin; PIN, prostatic intraepithelial neoplasia; PSAP, prostate-specific acid phosphatase.
Acknowledgments
The authors would like to thank Donald S. Coffey, Alan K. Meeker, Michael Haffner and members of the De Marzo lab from Johns Hopkins for their helpful comments and discussions regarding this work. A. M. De Marzo is supported in part by the Patrick C. Walsh Prostate Cancer Research Fund as the Peter J. Sharp Foundation Scholar.
Footnotes
Competing interests
W. G. Nelson declares that he is the owner of a patent (#5552277). The other authors declare no competing interests.
References
- 1.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor RA, Toivanen R, Risbridger G. Stem cells in prostate cancer—treating the root of the problem. Endocr Relat Cancer. 2010;17:R273–R285. doi: 10.1677/ERC-10-0145. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamb LE, Knudsen BS, Miranti CK. E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci. 2010;123:266–276. doi: 10.1242/jcs.054502. [DOI] [PubMed] [Google Scholar]
- 5.Moscatelli D, Wilson EL. PINing down the origin of prostate cancer. Sci Transl Med. 2010;2:43ps38. doi: 10.1126/scitranslmed.3001445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koh CM, et al. MYC and prostate cancer. Genes Cancer. 2010;1:617–628. doi: 10.1177/1947601910379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furusato B, et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis. 2010;13:228–237. doi: 10.1038/pcan.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong DJ, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]