

Abstract
Background
Molecular studies have suggested that the true diversity of Leucocytozoon (Apicomplexa: Haemospororida) species well exceeds the approximately 35 currently described taxa. Further, the degree of host-specificity may vary substantially among lineages. Parasite distribution can be influenced by the ability of the parasite to infect a host, vector preferences for certain avian hosts, or other factors such as microhabitat requirements that increase the probability that vertebrate hosts and vectors are in frequent contact with each other. Whereas most studies of haemosporidians have focused on passerine hosts, sampling vectors in the same habitats may allow the detection of other lineages affecting other hosts.
Methods
We sampled abundant, ornithophilic black flies (Simuliidae) across a variety of sites and habitats in the Colorado Rocky Mountains throughout the summer of 2007. Black flies were screened with PCR using Leucocytozoon-specific primers that amplify a portion of the cytochrome b gene, and the sequences were compared to the haplotypes in the MalAvi database. Infections of Leucocytozoon from birds sampled in the same area were also included.
Results
We recovered 33 unique haplotypes from the black flies in this study area, which represented a large phylogenetic diversity of Leucocytozoon parasites. However, there were no clear patterns of avian host species or geography for the distribution of Leucocytozoon haplotypes in the phylogeny.
Conclusions
Sampling host-seeking vectors is a useful way to obtain a wide variety of avian haemosporidian haplotypes from a given area and may prove useful for understanding the global patterns of host, parasite, and vector associations of these ubiquitous and diverse parasites.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-015-0952-9) contains supplementary material, which is available to authorized users.
Keywords: Haemosporidian, Leucocytozoon, Simulium, cytb haplotypes, Black fly, Parasite, Malaria
Background
Members of the genus Leucocytozoon are globally distributed avian haemosporidian parasites [1, 2]. These parasites are transmitted among avian hosts by black fly vectors (Diptera: Simuliidae). Although at least 35 morphologically defined species have been described to date [1], molecular studies suggest that the true diversity well exceeds this number e.g. [2–4]. Several of the molecular lineages have demonstrated that the degree of host-specificity varies; some lineages are found consistently only in one bird species, whereas other lineages develop successfully in a wide range of taxonomically varied hosts [2, 3]. Further, species of Leucocytozoon can complete development across a large range of simuliid vectors [1, 5]. Because both the vertebrate host and dipteran vector are essential for the complete development of Leucocytozoon species [1], the distribution of parasites will be influenced by the ability of the parasite to infect a particular vertebrate host, active vector preferences for certain avian hosts, and other factors such as climatic variables and microhabitat requirements that place vertebrate hosts and vectors in frequent contact with each other [6].
The objective of this study was to sample a set of host-seeking black flies across a variety of sites and habitats in an alpine ecosystem and to use molecular methods to assess the diversity of Leucocytozoon parasites in these vectors. Our hypotheses were that the flies would contain a wider variety of parasite lineages than would be obtained by sampling the avian community, and parasite distributions would be structured by avian host or habitat type.
Methods
Field site description and study design
Black flies were collected from two field sites within the vicinity of the Rocky Mountain Biological Laboratory (RMBL) in Gothic, Gunnison County, Colorado, U.S.A between May 19 and July 31, 2007. One field site, East River Valley, was located approximately 2 km south from RMBL (UTM: N 4312713 E 327700) and the other field site was located in the adjacent, Washington Gulch valley (UTM: N 4311531 E 325807). Elevation ranges from 2902 m to 2987 m asl. Both field sites were a mosaic of alpine meadows, forest stands, and riparian willow thickets. Meadow habitats consisted of a diversity of herbaceous vegetation across a range of elevations. Forest habitats were composed of conifer (Picea engelmannii) and aspen (Populus tremuloides) stands at higher elevations. Willow thickets were dominated by bog birch (Betula glandulosa), mountain alder (Alnus tenuifolia), and several species of willow (Salix spp.). To ensure a representative sample of the black fly communities, each field site was stratified by the following habitat types: willow, meadow, and forest. We took a random sample of habitat patches from each field site to ensure we sampled across two patches of each habitat type per field site. The following permits were required for this research: Federal Banding Permit, 2328; Colorado Fish and Wildlife Service License, 09TRb1094; and University of Michigan Animal Care Committee Permit, #9077.
Black fly collection methods
During the summer of 2007, we captured host-seeking black flies with carbon dioxide-baited Centers for Disease Control (CDC) miniature light traps (John W. Hock Company, No. 512 fine mesh collection cups, Gainesville, FL, USA) (Service, 1976). CDC traps were set and checked every 24 h. Due to the possibility of trap failure, traps were paired approximately 50 m from each other within each sampled patch. We trapped each habitat patch for two consecutive nights over an interval of eight days. After each two-day trapping session, traps were pulled and rotated to the other field site to minimize any potential effects of season or weather on trap success. Sampling occurred May 19–May 26, June 8–June 17, July 1–July 12, and July 22–July 31 in 2007.
To immobilize captured dipterans, we placed them in a bag and exposed them to cotton soaked in triethylamine (Fisher Scientific, Amber Glass, No. BP616-500, Pittsburgh, PA, USA) for five minutes in a well-ventilated area. We then separated the black flies from other biting dipterans and stored them in 95 % ethanol for future identification and parasite DNA analyses. Female black flies were identified to species or species complex based on structural characters [7]. Identifications were facilitated by genitalia preparations of selected specimens. The females of Simulium silvestre and Simulium craigi are not reliably distinguished by morphological criteria, but for the purposes of this paper, we will refer to them simply as S. silvestre. Likewise, Simulium exulatum and Simulium pilosum are not easily distinguishable and those flies will be referred to as S. exulatum. Representative specimens have been deposited in the Clemson University Arthropod Collection, Clemson, South Carolina.
DNA extraction, amplification, and sequencing methods
Black flies were separated into vials corresponding to morphological identification and sampling date and location. Because of the large number of flies in some traps, we opted to extract DNA from pools of five flies for downstream molecular screening. Each black fly was minced with sterile razorblades. Total DNA was extracted using the DNeasy Animal Tissue extraction kit QIAGEN (Valencia, CA, USA), eluting in 200 μl total of Buffer AE. To screen the black fly samples for Leucocytozoon parasites, we conducted a polymerase chain reaction (PCR) on the extracted DNA, using the nested primer set developed by Waldenström et al. [8], which amplifies a 479 bp fragment of the mitochondrial cytochrome b gene with outer primers that are general to the three common genera of avian haemosporidians and nested primers that are specific to Leucocytozoon. PCRs were conducted in 25 μl reactions using 10 μl of TopTaq MasterMix (QIAGEN, Valencia, CA, USA), 1 mM of each primer and 2 μl of DNA extracted from the pooled black fly samples. PCR conditions followed recommended protocols by Waldenström et al. [8]. To determine if our sample of black flies had recently fed, and to describe any potential vector feeding patterns, we used vertebrate primers L14816 and H15173 [9] to amplify any vertebrate DNA present in blood meals [10]. Negative and positive controls were always included in PCR reactions to detect possible contamination. Amplified DNA was visualized on a 1.5 % agarose gel with CyberSafe (10 %; Invitrogen, Carlsbad, CA; 2 μl per 100 μl of gel), and positive amplifications were cleaned with the AMPure reagent (Agencourt, Beverly, Massachusetts) and sequenced in both directions using BigDye v. 3.1 (Applied Biosystems, Foster City, California) with the same primers that were used in amplification. Sequences were cleaned with CleanSeq (Agencourt, Beverly, Massachusetts) and run on an ABI 3730×l automated sequencer. We re-amplified and re-sequenced any samples that revealed ambiguous or poor-quality base calls. Black fly pools that showed evidence of multiple infections after sequencing (via multiple peaks in electropherograms) were re-amplified and PCR products were cloned using the TOPO® TA cloning kit for sequencing (Invitrogen, Carlsbad, CA), to separate products. To avoid erroneously incorporating chimeric sequences into our phylogenetic analyses, only sequences from these cloned products that showed base calls matching those in the corresponding directly sequenced products were analyzed.
Sequence analysis and phylogenetic analyses
Raw sequences were assembled into contigs and edited in Geneious v 6.1.3 (http://www.geneious.com) [11]. The clean sequences generated were incorporated into two phylogenetic analyses. First, all Leucocytozoon sequences that were available in the MalAvi database [12] were downloaded on January 30, 2015 (a total of 500 sequences) along with 5 sequences from Haemoproteus (Parahaemoproteus) species to serve as an outgroup. Sequences were aligned in Geneious using the Geneious Alignment algorithm and default parameters. Although the resulting alignment contained several single-base-pair gaps, likely to be the result of sequencing errors in the deposited sequences, we did not manually edit these gaps in an effort to not introduce additional misalignments in the analysis. The first phylogeny was produced using the RaxML GUI interface [13], with nodal support assessed using the software’s rapid bootstrap algorithm. Following this large analysis, we created a smaller set of MalAvi sequences representing the top BLAST hits for each Leucocytozoon haplotype recovered from our black fly samples (Table 1). This reduced data set included any haplotype that had been associated with a named Leucocytozoon species in the MalAvi database. Also included in this data set, were nine sequences isolated from seven different passerine hosts. These sequences were generated with the same primers and conditions as for sampled black flies, and came from birds that were sampled across the same field sites and visually or molecularly identified as having single-lineage infections [14]. Nodal support was obtained by using the thorough bootstrap search strategy in RAxML. The phylogeny in Fig. 1 was plotted in RStudio version 0.98.507 using the “ape” package [15].
Table 1.
Best MalAvi database BLAST hits for Leucocytozoon haplotypes found in Colorado black flies
| Haplotype | Detected in: | Top BLAST hit | E value |
|---|---|---|---|
| COLBF01 | G. denaria 9 Jun (2-W) | GALLUS08 | 5E-152 |
| S. silvestre 26 Jul (2-W) | |||
| S. silvestre 28 Jul (2-F) | |||
| S. silvestre 31 Jul (1-F) | |||
| S. silvestre 31 Jul (1-M) | |||
| COLBF02 | S. exulatum 14 Jun (2-F) | STOCC13 | 0.0 |
| COLBF03 | S. silvestre 30 Jun (1-W) | PARUS25 | 0.0 |
| COLBF04 | S. silvestre 30 Jun (1-W) | PARUS25 | 0.0 |
| COLBF05 | S. silvestre 2 Jul (1-W) | CB1 | 0.0 |
| S. silvestre 9 Jul (1-M) | |||
| S. silvestre 10 Jul (1-M) | |||
| S. silvestre 11 Jul (2-M) | |||
| S. silvestre 19 Jul (1-F) | |||
| S. silvestre 19 Jul (1-F) | |||
| S. silvestre 28 Jul (2-F) | |||
| S. silvestre 30 Jul (1-F) | |||
| S. silvestre 30 Jul (1-W) | |||
| S. silvestre 31 Jul (1-M) | |||
| COLBF06 | S. silvestre 2 Jul (1-W) | EUSE2 | 0.0 |
| S. silvestre 9 Jul (1-M) | |||
| COLBF07 | S. silvestre 6 Jul (2-W) | TABI09 | 0.0 |
| COLBF08 | S. silvestre 7 Jul (2-M) | TUFAL01 | 0.0 |
| COLBF09 | S. silvestre 7 Jul (2-W) | GALLUS08 | 2E-150 |
| COLBF10 | S. silvestre 7 Jul (2-W) | TUMER02 | 0.0 |
| S. silvestre 9 Jul (1-M) | |||
| S. silvestre 11 Jul (2-M) | |||
| S. silvestre 12 Jul (2-W) | |||
| S. silvestre 24 Jul (2-M) | |||
| S. silvestre 29 Jul (2-W) | |||
| COLBF11 | S. silvestre 9 Jul (1-M) | CNEORN01 | 0.0 |
| COLBF12 | S. silvestre 9 Jul (2-M) | CB1 | 0.0 |
| S. silvestre 19 Jul (1-F) | |||
| COLBF13 | S. silvestre 10 Jul (1-W) | ZOLEU02 | 0.0 |
| COLBF14 | S. silvestre 10 Jul (1-W) | GALLUS08 | 2E-150 |
| COLBF15 | S. silvestre 10 Jul (1-M) | TABI09 | 0.0 |
| COLBF16 | S. silvestre 11 Jul (2-M) | PARUS25 | 0.0 |
| S. silvestre 28 Jul (2-W) | |||
| S. silvestre 30 Jul (1-F) | |||
| COLBF17 | S. silvestre 18 Jul (1-F) | TRPIP2 | 0.0 |
| COLBF18 | S. silvestre 18 Jul (1-F) | EMPALN01 | 0.0 |
| S. silvestre 31 Jul (1-M) | |||
| COLBF19 | S. silvestre 19 Jul (1-F) | TRPIP2 | 0.0 |
| COLBF20 | S. silvestre 24 Jul (1-F) | BUVIR03 | 0.0 |
| COLBF21 | S. silvestre 24 Jul (1-F) | EMPALN01 | 0.0 |
| COLBF22 | S. silvestre 24 Jul (1-F) | HGR1 | 0.0 |
| COLBF23 | S. silvestre 24 Jul (2-M) | TROEAD09 | 0.0 |
| S. silvestre 25 Jul (1-M) | |||
| S. silvestre 30 Jul (1-W) | |||
| COLBF24 | S. silvestre 25 Jul (1-M) | GALLUS08 | 1E-152 |
| S. silvestre 28 Jul (2-F) | |||
| COLBF25 | S. silvestre 27 Jul (2-W) | CB1 | 0.0 |
| COLBF26 | S. silvestre 28 Jul (2-F) | CARFLA04 | 0.0 |
| COLBF27 | S. silvestre 29 Jul (2-W) | BUVIR03 | 0.0 |
| S. silvestre 31 Jul (1-F) | |||
| COLBF28 | S. silvestre 30 Jul (1-M) | STUR1 | 0.0 |
| COLBF29 | S. silvestre 30 Jul (1-F) | EUSE1 | 0.0 |
| COLBF30 | S. silvestre 30 Jul (1-F) | EUSE1 | 0.0 |
| COLBF31 | S. silvestre 30 Jul (1-W) | TRPIP2 | 0.0 |
| COLBF32 | S. silvestre 31 Jul (1-W) | STUR1 | 0.0 |
| COLBF33 | S. silvestre 31 Jul (1-F) | HYPHI28 | 0.0 |
Sample designation include species of blackfly, date collected in 2007, site (1 = East River Valley, 2 = Washington Gulch), and habitat type (W = willow, F = forest, M = meadow)
Fig. 1.
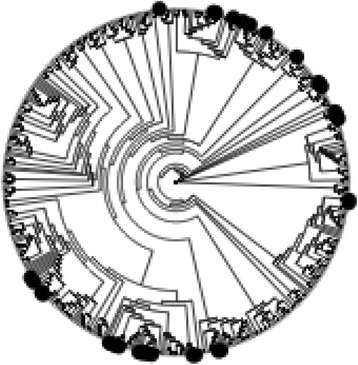
Maximum likelihood phylogeny of cox1 sequences of Leucocytozoon parasites amplified from Colorado black fly pools in the context of all Leucocytozoon haplotypes in the MalAvi database. The phylogeny is rooted with five sequences of Haemoproteus (Parahaemoproteus). Samples from the Colorado flies are indicated with large black dots
Results and discussion
Parasite prevalence and blood meal analysis
During the summer of 2007, we collected a total of 2921 black flies including 1929 ornithophilic species specimens belonging to six species/species complexes (Additional file 1). We subsampled from three of five genera and screened a total of 174 pools of black flies with PCR. We detected Leucocytozoon cytb amplicons from 67 out of 145 (46.2 %) pools of S. silvestre, two out of 14 (14.3 %) pools of Greniera denaria, one of the two Simulium exulatum pools and no positives from any of the 13 mammalophilic Prosimulium spp. pools. Twenty-one of the amplicons from the S. silvestre pools and one of the G. denaria pools yielded sequences with multiple peaks in the electropherograms. PCR products from these pools were subsequently cloned.
The majority of black flies sampled for Leucocytozoon parasites had not fed recently; no sampled black flies had visible blood in their abdomens and only four of the S. silvestre pools that were screened with vertebrate-specific primers showed any amplification. These results suggest that parasite amplification could reflect either of the following two scenarios: 1) established and potentially infectious parasite stages rather than parasites present in the blood meal that would not be transmitted, or 2) persisting parasite DNA from abortive sporogony [16]. Of the pools positive for vertebrate DNA, three (SS46d, SS42a, and SS39E) yielded sequences identified as Dusky Grouse (Dendragapus obscurus). The fourth pool (SS44A) yielded a sequence identified as an American Crow (Corvis brachyrhynchos).
Diversity of Leucocytozoon sequences in Colorado black flies
We obtained 33 unique cytb sequences from 60 of the 70 positive black fly pools that were successfully sequenced (Table 1, GenBank Accession Numbers KR052933-KR052965). Twenty-three of the 33 haplotypes were observed only from a single trapping event; however, the other 10 haplotypes occurred in more than one, with several haplotypes detected in different sites, in different habitat types (meadow, forest, or willow) and at different dates throughout the sampling period. Most notably, haplotype COLBF01 was detected in both S. silvestre and G. denaria and was present in the system from the first sampling period until the last sampling event. Figure 1 presents the full phylogeny of these sequences in the context of the MalAvi Leucocytozoon haplotypes from around the globe and from 13 different host orders, rooted with five Haemoproteus (Parahaemoproteus) sequences. The tips of this tree corresponding to sequences observed in the Colorado black flies are shown with large dots. Although we did not observe Colorado black fly-borne Leucocytozoon haplotypes in each of the major clades of the full Leucocytozoon tree, Fig. 1 demonstrates that there is a great diversity of Leucocytozoon parasites present in this particular vector community, with no evidence that limited in situ diversification has occurred.
Figure 2 shows a more detailed phylogeny of the 33 Leucocytozoon haplotypes that we obtained from the black flies. This phylogeny was analyzed in the context of the nine sequences of Leucocytozoon from passerine birds captured at the same site [14]. Also included were the top BLAST matches for each of the black-fly-borne parasites in the MalAvi database and a set of MalAvi haplotypes that came from morphologically identified species of Leucocytozoon. Overall, the nodal support of this phylogeny is low, owing to the limited diversity and small fragment size analyzed. However, a few patterns from the analysis are worthy of mention. First, in no case, did any of the black-fly-borne Leucocytozoon haplotypes that we obtained exactly match haplotypes detected in birds from the same site; this suggests that the potential vector(s) feed(s) primarily on non-passerines, which comprise the majority of the birds sampled for blood parasites on these field sites. However, one haplotype (COLBF_10) did cluster with the parasites sequenced from a Mountain Bluebird at RMBL, and several of the fly-borne haplotypes (COLBF_11, COLBF_13, and COLBF_23) were part of a clade that included a haplotype isolated from a local Lincoln’s Sparrow, as well as haplotypes detected in both Alaskan and Peruvian birds.
Fig. 2.
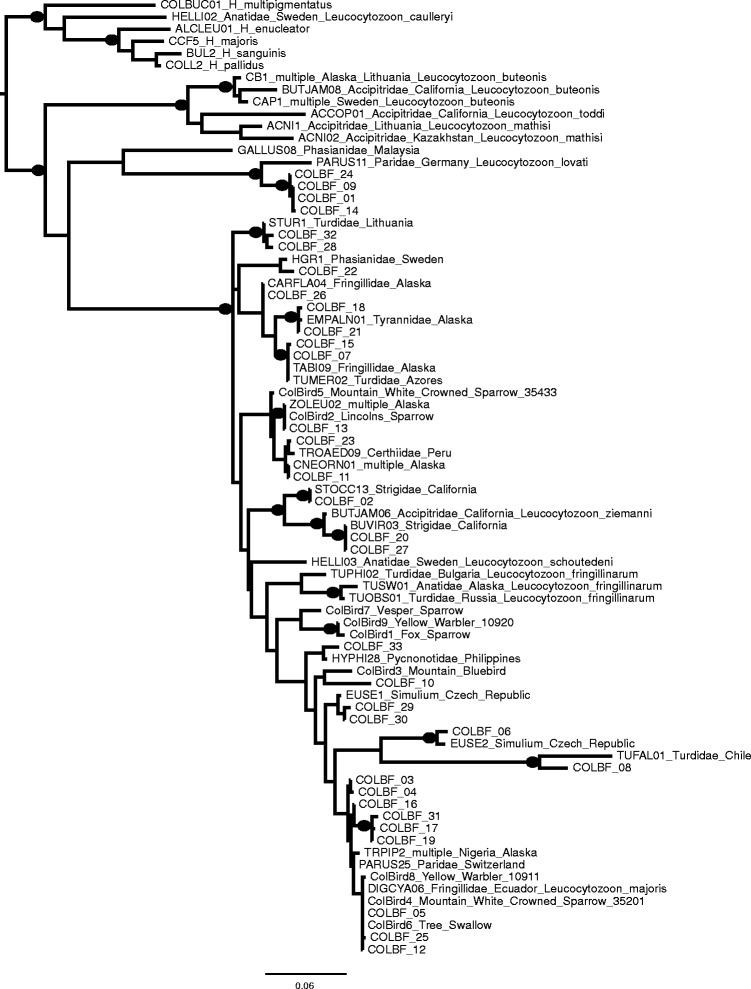
Maximum likelihood phylogeny of Leucocytozoon cyt b sequences obtained from Colorado black fly species (indicated as COLBF), their best BLAST match in the MalAvi database, named Leucocytozoon species in the database and sequences from Leucocytozoon from nine birds (indicated as ColBird) sampled at the same Colorado location. Nodal support values > 95 % are indicated with a black circle on the node
Several of our black fly-borne parasite sequences fell within clades that contained haplotypes of Leucocytozoon reported in a recent study that sampled four types of blood parasites in passerine birds over a latitudinal gradient in Alaska [17]. For example, COLBF_26 was extremely similar to haplotype CARFLA04 (Table 1), which was obtained from a Redpoll, a bird that is partially migratory. COLBF_07 and COLBF_15 were similar to a sequence from a parasite in a Two-barred Crossbill (Table 1), another bird that is a resident in both Alaska and Colorado. Although these results could suggest that parasite genotypes move between these locales in either migratory birds or spreading populations, there are also numerous cases where close relatives come from geographically distant locales, such as the clustering of COLBF_33 with a parasite sequence detected in the Philippines or two haplotypes (COLBF_28 and COLBF_32) with their closest relative in our tree being from a parasite in a bird sampled in Lithuania. Overall, these results suggest that some common haplotypes have wide geographic ranges and have turned up in locations where a large amount of sampling for avian haemosporidians has occurred (e.g. Lithuania, Sweden), a pattern that has also been observed for the other genera infecting birds, such as the GRW4 lineage of Plasmodium relictum [18].
Parasite-vector associations
We obtained a large diversity of Leucocytozoon haplotypes from S. silvestre, and the distribution of parasite haplotypes was not strongly affected by avian host, habitat, or time of season. The large diversity of parasite haplotypes recovered from S. silvestre suggests that it is probably an important vector for a multitude of Leucocytozoon species on our field sites. The degree of overlap in emergence of S. silvestre with bird breeding on these sites, its ornithophilic nature, and its abundance across all habitat types and throughout much of the summer season reinforce the vector potential of this black fly species and might explain why there were no strong effects of avian host, habitat, and time of season on the distribution of Leucocytozoon haplotypes. Both S. silvestre and S. craigi feed on a variety of avian hosts [reviewed in 7]. Based on the sheer number of individuals recovered from light traps, S. silvestre was the most abundant ornithophilic black fly on our field sites, and its abundance was not restricted by habitat type. Further, S. silvestre reaches peak abundance during the time of season (June 25 – July 14) when most bird species are hatching and feeding susceptible young of the year on our field sites [14]. Other studies have implicated S. silvestre as vectors for a number of Leucocytozoon species [summarized in 7].
The presence of parasite haplotypes amplified from G. denaria, a black fly species that reaches peak abundance early in the summer season [14], further indicates that these parasite haplotypes may come from non-passerine bird hosts we were unable to sample in the field (i.e. resident species or bird species in different orders). Hosts of Greniera species are poorly known; the only host record for the entire genus is the Black Grouse (Tetrao tetrix) from a study in northern Finland [19]. A wider sampling of the avian and ornithophilic black fly communities is needed to achieve a concrete understanding of how avian hosts, black fly vectors, and environmental factors may shape the distribution of Leucocytozoon species on our field sites and globally.
Conclusions
To our knowledge, this is the first study to amplify parasite DNA from non-engorged, host-seeking black flies. Previous studies on field-captured black flies amplified a diversity of Leucocytozoon DNA from blood-fed individuals [6]. However, parasite DNA amplified from the abdomens of blood-fed individuals does not provide proof of vector competence. Not all Leucocytozoon species ingested with a bloodmeal can develop infectious stages in the salivary glands of a given black fly species and successfully transmit to the next avian host. These results present only tentative links among S. silvestre, a variety of alpine birds, and potential transmission of Leucocytozoon parasites. The black flies in our study were collected in CO2-baited CDC light traps and were not blood engorged; therefore, most can be presumed to be host-seeking individuals. Thus, parasite sequences amplified from these individuals may potentially be generated from parasite transmission stages, indicating that S. silvestre is a competent vector for these Leucocytozoon strains. Thus, sampling avian haemosporidian parasites via the collection and screening of host-seeking vectors may provide a means of obtaining a broader sampling of the genetic diversity of these parasites within a locality.
Acknowledgments
We would like to thank Stanley Kovak, Morgan Graham, and Nathaniel Schafrick for assistance in the field, Ian Billick and the RMBL staff for lab space and support in the field, and Bryan Falk and Stephen Gaughran for assistance in the lab. We also would like to thank Betsy Foxman, Bobbi Low, Emily Silverman, and Johannes Foufopoulos, as well as the Low, Silverman, and Foufopoulos lab members, for advice on experimental design and analysis. Funding for this study comes from grants provided by the University of Michigan Rackham discretionary funds, Interdisciplinary Perspectives on Infectious Diseases Training Grant, and School of Natural Resources & Program in the Environment Alumni Incentive, and the Sackler Institute for Comparative Genomics.
Abbreviations
- RMBL
Rocky mountain biological labs
- CDC
Centers for disease control
- PCR
Polymerase chain reaction
- Cytb
Cytochrome oxidase subunit b
Additional file
Trapping data for the black fly species captured in Colorado and the number of individuals for each species sampled in 2007 by field site. Species in bold type are ornithophilic.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed sampling design and collected samples: CCM. Performed identifications of black flies: PHA. Performed molecular screening: CCM, JF, and SLP. Performed phylogenetic analyses: SLP. Wrote the paper: CCM and SLP. All authors read and approved the final version of the manuscript.
Authors’ information
CCM is an Assistant Professor in the Department of Infectious Diseases, College of Veterinary Medicine, and the Odum School of Ecology at the University of Georgia. Her current work focuses on understanding parasite/pathogen-vector-host interactions, what environmental drivers are important for transmission, and how future changes in climate and land use will affect vector-borne disease emergence and transmission. PHA is a Professor of Entomology in the Department of Agricultural and Environmental Sciences at Clemson University. His research is focused on the genetics, systematics, and ecology of black flies and their symbionts. JF was an undergraduate volunteer working in the lab of SP. He was mainly interested in describing parasite diversity using genomic and phylogenetic methods during his research at the American Museum of Natural History. SP is an Associate Curator in the Division of Invertebrate Zoology Sackler Institute for Comparative Genomics at the American Museum of Natural History. In regards to this study, SP’s research interests include the systematics, biogeography, and molecular evolution of the protozoan parasites that cause malaria.
Contributor Information
Courtney C. Murdock, Email: cmurdock@uga.edu
Peter H. Adler, Email: padler@clemson.edu
Jared Frank, Email: jaredrf@umich.edu.
Susan L. Perkins, Email: perkins@amnh.org
References
- 1.Valkiunas G. Avian malaria parasites and other haemosporidia. New York: CRC Press; 2005. [Google Scholar]
- 2.Hellgren O, Waldenstrom J, Perez-Tris J, Szollosi E, Hasselquist D, Krizanauskiene A, et al. Detecting shifts of transmission areas in avian blood parasites - a phylogenetic approach. Mol Ecol. 2007;16(6):1281–90. doi: 10.1111/j.1365-294X.2007.03227.x. [DOI] [PubMed] [Google Scholar]
- 3.Hellgren O. The occurrence of haemosporidian parasites in the Fennoscandian bluethroat (Luscinia svecica) population. J Ornithol. 2005;146(1):55–60. doi: 10.1007/s10336-004-0055-4. [DOI] [Google Scholar]
- 4.Sato Y, Hagihara M, Yamaguchi T, Yukawa M, Murata K. Phylogenetic comparison of Leucocytozoon spp. from wild birds of Japan. J Vet Med Sci. 2007;69(1):55–9. doi: 10.1292/jvms.69.55. [DOI] [PubMed] [Google Scholar]
- 5.Desser SS, Yang YJ. Sporogony of Leucocytozoon spp. in mammalophilic simuliids. Can J Zool. 1973;51(7):793. doi: 10.1139/z73-116. [DOI] [PubMed] [Google Scholar]
- 6.Hellgren O, Bensch S, Malmqvist B. Bird hosts, blood parasites and their vectors - associations uncovered by molecular analyses of blackfly blood meals. Mol Ecol. 2008;17(6):1605–13. doi: 10.1111/j.1365-294X.2007.03680.x. [DOI] [PubMed] [Google Scholar]
- 7.Adler PH, Currie DC, Wood DM. The black flies (Simuliidae) of North America. Ithaca, NY: Cornell University Press; 2004. [Google Scholar]
- 8.Waldenström J, Bensch S, Kiboi S, Hasselquist D, Ottosson U. Cross-species infection of blood parasites between resident and migratory songbirds in Africa. Mol Ecol. 2002;11(8):1545–54. doi: 10.1046/j.1365-294X.2002.01523.x. [DOI] [PubMed] [Google Scholar]
- 9.Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals - amplification and sequencing with conserved primers. Proc Natl Acad Sci U S A. 1989;86(16):6196–200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murdock CC, Olival KJ, Perkins SL. Molecular identification of host feeding patterns of snow-melt mosquitoes (Diptera: Culicidae): potential implications for the transmission ecology of Jamestown Canyon virus. J Med Entomol. 2010;47(2):226–9. doi: 10.1093/jmedent/47.2.226. [DOI] [PubMed] [Google Scholar]
- 11.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung MS S, Buxton S, et al. Geneious Basic: an intergrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bensch S, Hellgren O, Perez-Tris J. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour. 2009;2009:1353–8. doi: 10.1111/j.1755-0998.2009.02692.x. [DOI] [PubMed] [Google Scholar]
- 13.Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Drivers Evol. 2012;12:335–7. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- 14.Murdock CC. Studies on the ecology of avian malaria in an alpine ecosystem. Ann Arbor: University of Michigan; 2009. [Google Scholar]
- 15.Paradis E, Calude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 16.Valkiunas G, Kazlauskiene R, Bernotiene R, Palinauskas V, Iezhova TA. Abortive long-lasting sporogony of two Haemoproteus species (Haemosporida, Haemoproteidae) in the mosquito Ochlerotatus cantans, with perspectives on haemosporidian vector research. Parasitol Res. 2013;112(6):2159–69. doi: 10.1007/s00436-013-3375-6. [DOI] [PubMed] [Google Scholar]
- 17.Oakgrove KS, Harrigan RJ, Loiseau C, Guers S, Seppi B, Sehgal RN. Distribution, diversity and drivers of blood-borne parasite co-infections in Alaskan bird populations. Int J Parasitol. 2014;44(10):717–27. doi: 10.1016/j.ijpara.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Beadell JS, Ishtiaq F, Covas R, Melo M, Warren BH, Atkinson CT, et al. Global phylogeographic limits of Hawaii’s avian malaria. Proc Roy Soc London Ser B. 2006;273(1604):2935–44. doi: 10.1098/rspb.2006.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ojanen U, Ratti O, Adler PH, Kuusela K, Malmqvist B, Helle P. Blood feeding by black flies (Diptera : Simuliidae) on black grouse (Tetrao tetrix) in Finland. Entomol Fennica. 2002;13(3):153–8. [Google Scholar]