

Knowledge concerning acute lymphoblastic leukemia (ALL) has increased greatly,1 and personalized medicine has become a reality. More sophisticated diagnostic procedures, including immunophenotyping, cytogenetics, molecular genetics, and new genomics, have allowed the definition of new ALL sub-entities which, in some cases, has translated into specific therapies. A great achievement is the possibility of evaluating minimal residual disease (MRD), which can now be done in about 95% of ALL patients. MRD is the most important prognostic factor and thus a major component of a personalized treatment algorithm. Progress has also come from targeted therapies, extending the existing backbones of chemotherapy and stem cell transplantation (SCT). Targeted therapy in Philadelphia chromosome-positive ALL (Ph+ ALL) with tyrosine kinase inhibitors (TKI) and immunotherapy with monoclonal antibodies targeting surface antigens expressed on leukemic blast cells have extended the armamentarium. A new promising approach is the activation of patients’ T cells directed against their own leukemic blast cells either through a bi specific antibody, or chimeric antigen receptor modified T cells.
Diagnostics
Immunophenotyping is still the most important diagnostic feature, separating B-lineage ALL (~75%) from T-lineage ALL (~25%), and their subtypes according to the stage of maturation/differentiation (Table 1).
Table 1.
Diagnostics of major ALL subtypes.
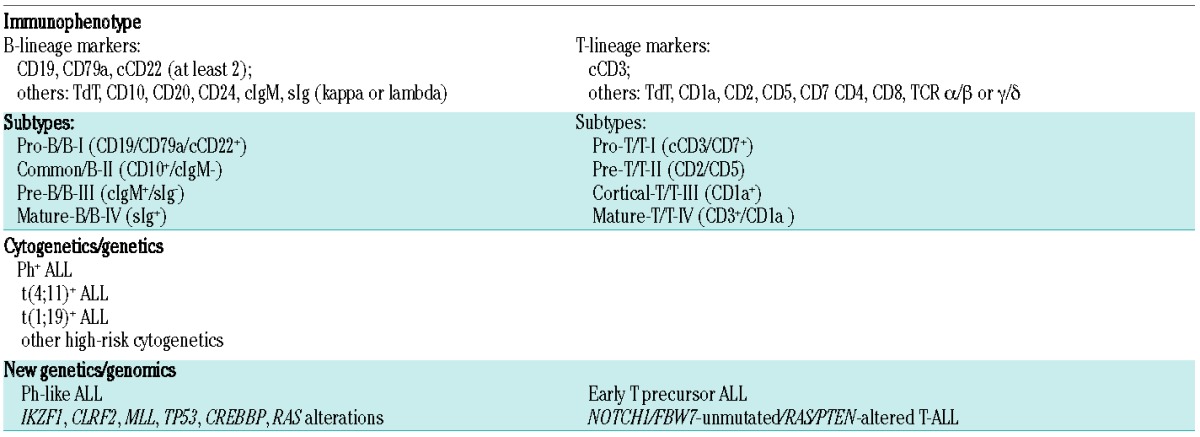
Other diagnostic techniques are standard cytogenetics, fluorescence in situ hybridization, and reverse transcriptase polymerase chain reaction. These methods allow the detection of Ph+ ALL, with the chromosomal translocation t(9;22)(q34;q11), and the detection of the corresponding BCR-ABL1 gene rearrangement. Further ALL entities that have been identified are t(4;11)(q21;q23)/MLL-AFA4, abn11q23/MLL, and t(1;19) (q23;p13)/PBX-E2A.
Gene expression profiling, single nucleotide polymorphism array analysis, array-comparative genomic hybridization, and next generation sequencing recognize newly defined ALL entities with poor prognosis: Ph-like ALL, and early T precursor ALL.
Ph-like ALL, also called BCR-ABL1-like ALL, is characterized by genetic lesions similar to Ph+ ALL, associated with IKZF1 deletion, CLRF2 overexpression and tyrosine kinase activating rearrangements involving ABL1, JAK2, PDGFRB and several other genes.2,3 The frequency is 10% in children and 25–30% in young adults, but does not increase further with age.4 Treatment could be directed at the underlying genetic pattern with BCR-ABL inhibitors (e.g. dasatinib) or JAK2 inhibitors (e.g. ruxolitinib).5
Early T precursor ALL is characterized by lack of CD1a and CD8, weak CD5 expression, at least one myeloid/stem cell marker, a specific transcriptional profile and the possible involvement of several critical genes.6 No new treatment approaches are currently available for this subtype, and thus SCT in first complete remission is the preferred option.
Minimal residual disease
MRD is the detection of residual leukemic cells, not detectable by light microscopy.
Methods for determining MRD are based on the detection of leukemia-specific aberrant immunophenotypes by flow cytometry, the evaluation of leukemia-specific rearranged immunoglobulin or T-cell receptor sequences by real-time quantitative polymerase chain reaction, or the detection of fusion genes associated with chromosomal abnormalities (e.g., BCR-ABL, MLL-AF4). The detection limit with these methods is 10−3–10−5 (0.1%–0.001%). The phenotypic aberrations are unique to each patient with ALL and can be detected in up to 95% of individuals.
Methods for MRD evaluation and standardization of MRD quantification have been extensively described.7,8
Minimal residual disease response and terminology
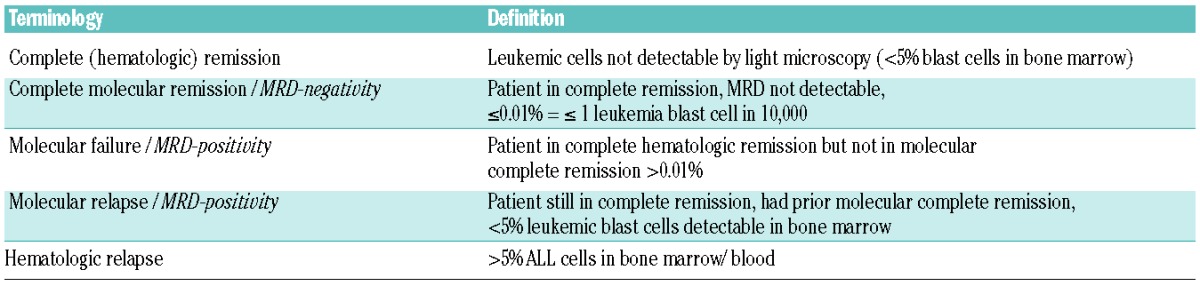
Molecular response can be evaluated only for patients in complete cytological remission, with one marker or more for MRD analysis and samples available at diagnosis and followed at specific time points during the course of disease. Results are classified as presented in Table 2.
Table 2.
Response parameters according to MRD.
Molecular response after induction therapy and impact on outcome
Achievement of molecular complete response/molecular remission is the most relevant independent prognostic factor for disease-free survival and overall survival. Patients with molecular complete remission after induction therapy had significantly superior outcome in several studies, with a disease-free survival of 54–74%, compared to 17–40% for MRD-positive patients.9–12 Patients with molecular failure after induction should proceed to allogeneic hematopoietic SCT.10
Prognostic factors, risk stratification, and minimal residual disease
The aim of identification of prognostic parameters at diagnosis, which include age, white blood cell count, specific immunophenotypes, and cytogenetic and genetic aberrations, is to stratify patients into risk groups: standard-risk patients without any risk factor, with a good chance of cure by chemotherapy, and high-risk patients with one or more of those risk factors. High-risk patients are most often candidates for a SCT in first complete remission.
Will minimal risk disease evaluation replace pre-therapeutic risk factors?
The question arises as to whether the evaluation of MRD overcomes all of those pre-therapeutic risk factors, or whether they should be combined.1,9,13 A practical approach is to enter the conventional prognostic factors and MRD into a decision algorithm. Thereby defined standard-risk patients who will achieve molecular remission (about 90–95%), will remain as standard-risk patients, whereas those who are MRD-positive will be defined accordingly as high-risk patients. Clinically defined high-risk patients are potential candidates for a SCT in first complete remission. However, it is not clear how to proceed if they achieve a complete molecular remission, since some studies suggest a lack of benefit from SCT. If MRD information is not available, the risk stratification should rely on clinical risk factors evaluated at diagnosis.
Unfortunately, 20–30% of adult ALL patients who are MRD-negative after induction will relapse. Potential reasons include loss of sensitivity, evolution of leukemic subclones, and extramedullary origin of disease.
Treatment principles
The goal of induction therapy is achievement of a complete remission, or even better, a molecular complete remission, mostly evaluated within 6–16 weeks of starting chemotherapy. With current regimens the complete remission rate has increased to 80%–90%, being higher for standard-risk patients (90% or more), and lower for high-risk patients (~80%). The outcome of ALL is strictly related to the age of a patient, and treatment protocols considering the age of an individual patient have emerged.
Pediatric-inspired therapies
Pediatric-inspired therapies for adolescents and young adults provide increased drug intensity at several stages of treatment, including larger cumulative doses of drugs such as corticosteroids, vincristine, L-asparaginase, and consequent central nervous system-directed therapy, which should be strictly adhered to, thereby reducing the role of SCT in such cases. In a 2012 meta-analysis of 11 trials including 2489 adolescents and young adults, pediatric-inspired regimens were superior to conventional adult chemotherapy.14 However none of the trials was a randomized comparison. In recent studies of pediatric-inspired therapies for adolescents and young adults, survival rates at 5 years were 67% to 78% compared to 34% to 41% with the former protocols.
Treatment in adults and elderly patients with acute lymphoblastic leukemia
The treatment results for adult ALL patients have also improved. The overall survival of 38% (54% at 5 years - 27% at 9 years) has improved for standard-risk adult ALL patients to 50–70% with chemotherapy alone, and the outcome for high-risk patients from 20–30% to >50% when they receive an allogeneic SCT in first complete remission.1 There has also been treatment progress in elderly ALL patients. Since palliative treatments and intensive chemotherapy regimens have failed, with low complete remission rates, high early death rates, and short survival, elderly-specific ALL protocols have been initiated, with less intensive therapy (avoiding anthracyclines and alkylating agents). In nine prospective studies for older ALL patients (55–81 years), with less intensive protocols the complete remission rate was 71% (43–90%), early death decreased to 15% (0–36%) and overall survival was 33 months (range, 16–71).15
Targeted therapies with tyrosine kinase inhibitors in Philadelphia-positive acute lymphoblastic leukemia
Patients with Ph+ ALL constitute approximately 25% of adult B-lineage ALL patients, with the incidence increasing to about 50% among elderly patients. In the pre-imatinib era complete remission rates were 60–70%, the survival in patients treated with chemotherapy alone was ~10%, and that of patients undergoing allogeneic SCT was ~30%. The results improved substantially when the first-generation TKI inhibitor imatinib became available, with complete remission rates of 80–90%, but particularly the rate of molecular remissions (BCR-ABL-negativity) increased from 5% to ≥50%, and the 5-year survival to 50% or more.1,16–18
Treating adult Ph+ ALL with an allogeneic SCT in first complete remission is still the best treatment option. However, in current studies patients not undergoing SCT receiving only chemotherapy plus a TKI also had improved outcome. Thus, a Ph+ group with lower relapse risk, not needing SCT, has to be identified, e.g. by MRD response, absence of additional chromosomal abnormalities, or IKZF1 gene deletion. Faster and deeper molecular responses can probably be achieved with second-generation TKI (dasatinib, nilotinib).19 A third-generation TKI is ponatinib, which targets the T315I mutation either present at diagnosis, or developing/remaining after treatment with other TKI.20 Administration of a TKI after SCT is now standard practice although the optimal duration of this treatment has not yet been established.
Immunologically based treatments with monoclonal antibodies or activated T cells
B-lineage blast cells express a variety of specific antigens, such as CD19, CD20, and CD22. Recently monoclonal antibodies have been developed to target these antigens.21,22 The anti-CD20 monoclonal antibody rituximab has substantially improved the outcome of patients with Burkitt leukemia/lymphoma. With repeated short cycles of intensive chemotherapy, combined with rituximab the overall survival of such patients increased from 60% to >80%.23 Monoclonal antibodies directed against CD22, linked to cytotoxic agents, such as calicheamicin (inotuzumab ozogamicin), or to plant or bacterial toxins (epratuzumab) are being explored for use in refractory/relapsed childhood and adult ALL. In a trial of patients with relapsed/refractory ALL treated with inotuzumab ozogamicin, the complete response rate (including responses without blood cell count recovery) was 66%, and of those 78% achieved a molecular complete remission.24 Targeting CD19 is of great interest, as this antigen is expressed in all B-lineage cells, most likely including early lymphoid precursor cells. A new promising approach is the bi-specific antibody blinatumomab, which combines single chain antibodies to CD19 and CD3, such that T cells lyse the CD19-bearing B cells. This antibody was effective in patients with positive MRD, and 80% converted to MRD-negativity.25 In a trial of adult patients with refractory/relapsed ALL, the rate of complete remissions/complete remissions with partial recovery of peripheral blood counts with blinatumomab was 43% and the MRD response rate was 82%,26 leading to a recent approval of this agent by the Food and Drug Administration. Another promising new approach is chimeric antigen receptor modified T cells, targeting CD19+ B-lineage ALL cells.27 When these genetically engineered T cells were given to children with refractory/relapsed ALL, the complete remission rate was 90%, with an event-free survival of 63% at 6 months, and an overall survival of 78%.28
Conclusion and future directions
Progress in the diagnosis of ALL with identification of genomic-defined sub-entities, the evaluation of MRD, and new targeted therapies have led to a substantial realization of personalized medicine in adult ALL. Current options, such as less intensive chemotherapy, reduction of SCT, incorporation of targeted therapies and optimal combinations of treatments require prospective, cooperative research, hereby further refining the individualized approach to each patient.
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic Leukemia. J Clin Oncol. 2011;29(5):532–543. [DOI] [PubMed] [Google Scholar]
- 2.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herold T, Baldus CD, Gökbuget N. Ph-like acute lymphoblastic leukemia in older adults. N Engl J Med. 2014;371(23):2235. [DOI] [PubMed] [Google Scholar]
- 5.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371(11):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009;10(2):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brüggemann M, Schrauder A, Raff T, et al. Standardized MRD quantification in European ALL trials: Proceedings of the Second International Symposium on MRD assessment in Kiel, Germany, 18–20 September 2008. Leukemia. 2010;24(3):521–535. [DOI] [PubMed] [Google Scholar]
- 8.Campana D. Minimal residual disease in acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:7–12. [DOI] [PubMed] [Google Scholar]
- 9.Beldjord K, Chevret S, Asnafi V, et al. Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL). Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739–3749. [DOI] [PubMed] [Google Scholar]
- 10.Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–1876. [DOI] [PubMed] [Google Scholar]
- 11.Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–4162. [DOI] [PubMed] [Google Scholar]
- 12.Ribera JM, Oriol A, Morgades M, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol. 2014;32(15):1595–1604. [DOI] [PubMed] [Google Scholar]
- 13.Hoelzer D, Gökbuget N. Change in prognostic factors. Leukemia Supplements 2012;1:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ram R, Wolach O, Vidal L, Gafter-Gvili A, Shpilberg O, Raanani P. Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric-inspired regimens: systematic review and meta-analysis. Am J Hematol. 2012;87(5):472–478. [DOI] [PubMed] [Google Scholar]
- 15.Gökbuget N. How I treat older patients with ALL. Blood. 2013;122(8):1366–1375. [DOI] [PubMed] [Google Scholar]
- 16.Dombret H, Gabert J, Boiron JM, et al. Groupe d’Etude et de Traitement de la Leucémie Aiguë Lymphoblastique de l’Adulte (GET-LALA Group). Outcome of treatment in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia -results of the prospective multicenter LALA-94 trial. Blood. 2002;100(7):2357–2366. [DOI] [PubMed] [Google Scholar]
- 17.Fielding AK, Rowe JM, Richards SM, et al. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood. 2009;113(19):4489–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeifer H, Goekbuget N, Völp C, et al. Long-term outcome of 335 adult patients receiving different schedules of imatinib and chemotherapy as front-line treatment for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2010;116:173 [Abstract]. [Google Scholar]
- 19.Foà R, Vitale A, Vignetti M, et al. GIMEMA Acute Leukemia Working Party. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118(25):6521–6528. [DOI] [PubMed] [Google Scholar]
- 20.Cortes JE, Kim DW, Pinilla-Ibarz J, et al. PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoelzer D. Novel antibody-based therapies for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011: 243–249. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian H, Thomas D, Wayne AS, O’Brien S. Monoclonal antibody-based therapies: a new dawn in the treatment of acute lymphoblastic leukemia. J Clin Oncol. 2012;30(31):3876–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoelzer D, Walewski J, Döhner H, et al. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood. 2014;124(26):3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13(4):403–411. [DOI] [PubMed] [Google Scholar]
- 25.Topp MS, Kufer P, Gokbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–2498. [DOI] [PubMed] [Google Scholar]
- 26.Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. [DOI] [PubMed] [Google Scholar]
- 27.Russell Cruz, Bollard CM. T-cell and natural killer cell therapies for hematologic malignancies after hematopoietic stem cell transplantation: enhancing the graft-versus-leukemia effect. Haematologica. 2015;100(6)709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:(177):177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]