

Abstract
Hox homeobox transcription factors drive leukemogenesis efficiently only in the presence of Meis or Pbx proteins. Here we show that Pbx3 and Meis1 need to dimerize to support Hox-induced leukemia and we analyze the molecular details of this cooperation. In the absence of Pbx3, Meis1 was highly unstable. As shown by a deletion analysis Meis1 degradation was contingent on a motif coinciding with the Pbx-binding domain. Either deletion of this sequence or binding to Pbx3 prolonged the half-life of Meis1 by preventing its ubiquitination. Meis1 break-down could also be blocked by inhibition of the ubiquitin proteasome system, indicating tight post-transcriptional control. In addition, Meis1 and Pbx3 cooperated genetically as overexpression of Pbx3 induced endogenous Meis1 transcription. These functional interactions translated into in vivo activity. Blocking Meis1/Pbx3 dimerization abrogated the ability to enhance proliferation and colony-forming cell numbers in primary cells transformed by Hoxa9. Furthermore, expression of Meis1 target genes Flt3 and Trib2 was dependent on Pbx3/Meis1 dimerization. This correlated with the requirement of Meis1 to bind Pbx3 in order to form high affinity DNA/Hoxa9/Meis1/Pbx3 complexes in vitro. Finally, kinetics and severity of disease in transplantation assays indicated that Pbx3/Meis1 dimers are rate-limiting factors for Hoxa9-induced leukemia.
Introduction
The 39 clustered HOX-homeobox genes in mammals are best known as important determinants of embryonic development. Yet, the intricate control circuits that establish segment identity during gestation are “recycled” again to control organogenesis. HOX proteins, in particular those encoded by the “A” and “B” cluster, are major protagonists governing development and differentiation of hematopoietic stem and precursor cells. In their role as controllers of hematopoiesis they influence the decision between self-renewal and differentiation. It is not, therefore, surprising that HOX gene/protein hyperactivity is associated with malignant transformation and acute leukemia (for reviews on this topic1–3). In an experimental setting overexpression of most HoxA-cluster genes is sufficient to block differentiation of primary hematopoietic stem and precursor cells in vitro.4 Yet, cells transformed solely by Hox genes elicit leukemia only sporadically after long periods of latency. In contrast efficient leukemogenesis is obtained in the presence of cofactors that belong to the “three loop amino-acid-loop extension” (TALE) homeobox family.5,6
The TALE group includes Pbx (homolog to extradenticle in Drosophila) and Meis (homothorax in fly) proteins. Early in vitro studies on artificial DNA templates suggested that “labial-type” Hox proteins of paralog groups 1–9 interact with Pbx7 whereas “abdominal-like” Hoxa9 to 13 bind to Meis18 to increase affinity and specificity for a particular recognition sequence. The essential role of Meis1 as a rate-limiting cofactor for Hox-induced leukemia was corroborated in various settings. Meis1 and Hoxa9 are almost always coactivated by retroviral integration in the BXH2 mouse leukemia model9,10 and MEIS1 and HOX expression levels are well correlated in clinical samples.11 In experimental leukemia models co-expression of Meis1 converted the slowly developing myeloproliferative-like disease elicited by Hoxa9 into an aggressive and fully-penetrant acute leukemia.5,6 At a genetic level cooperation of Meis1 and Hoxa9 enabled the transcription of a new set of genes, such as Flt3, which was not efficiently activated by Hoxa9 alone.12 Structure-function analyses narrowed down the essential contributions of Meis1 to a transactivating function and a domain that can interact with Pbx proteins.13,14
Next to Hox/Meis dimers, DNA-dependent and -independent Meis/Pbx complexes could be detected in in vitro electrophoretic mobility shift assay experiments on artificial optimized binding templates.15,16 In analogy to the situation in the fly, Meis binding was necessary for the nuclear import of Pbx proteins. Yet, in contrast to the situation with Meis1, a potential role of Pbx family members in accelerating Hox-initiated leukemia remained obscure because overexpression of Pbx1 did not accelerate or exacerbate Hoxa9-induced malignancies.5 In addition, neither PBX1 nor PBX2 expression was related to HOXA9 levels in patients’ samples.17 Interest in Pbx proteins as Hox cofactors was rekindled with the discovery that another Pbx family member, Pbx3, cooperated with Hoxa9 in experimental leukemogenesis and that knock-down of Pbx3 could impair transformation induced by Hoxa9.18,19 In a clinical setting PBX3 expression was an independent predictor of poor survival in leukemia patients and the presence of PBX3 was well correlated with HOX status within leukemic cells. Yet, the molecular mechanism behind these in vivo observations was not investigated further.
Here we examine the molecular basis by which Pbx3 augments the ability of Meis1 to support Hox-mediated leukemogenesis. We demonstrate that Pbx3 protects Meis1 from proteasomal degradation. Additionally, Pbx3 increases Meis1 affinity for Hoxa9 and it induces endogenous Meis1 transcription.
Methods
Plasmids, retroviral constructs, antibodies and cell culture
Meis1 (accession NM_010789.3), Hoxa9 (NM_010456.3), Pbx1 (NM_183355.3), PBX2 (NM_002586.4) and Pbx3 (NM_016768.2) and derivatives thereof were cloned by polymerase chain reaction using murine or human (PBX2) cDNA from the IMAGE repository as templates. Amplicons were inserted into either pMSCV retroviral backbones (Clontech, TaKaRa, Mountain View, CA, USA) or the pcDNA3 expression vector (LifeTechnologies, Darmstadt, Germany) and supplemented with N-terminal epitope-tags to allow immunological detection. All clones were confirmed by sequencing. Retroviral packaging was done in the Phoenix-E packaging line.20 Anti-flag (M2) and anti-HA antibodies for western blot detection were purchased from Sigma (Taufkirchen, Germany). FACS reagents were supplied by BD-Bioscience (Heidelberg, Germany). Replating or colony-formation cell (CFC) assays were done essentially as described elsewhere.21 For these experiments primary cells were transduced with pMSCV-based retroviral constructs. After transduction, cells were selected with antibiotics ensuring a 100% transduction rate. For CFC determination the cells were replated twice in methocel and colony numbers achieved in the third round of plating were counted. Cell proliferation was measured by MTT assays according to the instructions of the manufacturer (Promega, Mannheim, Germany)
Coimmunoprecipitation
For immunoprecipitation studies, HEK293T cells were transiently transfected by the standard calcicum-phosphate technique. In control experiments with a green fluorescent protein (GFP) expression construct, transfection efficiencies were >90%. Extracts from transfected cells expressing epitope-tagged proteins were prepared by treatment with triton-lysis (TL) buffer (20 mM HEPES pH 7.5, 0.5 mM EDTA, 0.1% Triton X-100, 0.5 mM sodium vanadate, 2 mM NaF, 2 mM DTT, 0.2 mM PMSF, 20 μg/mL leupeptin, 0.4 μg/mL aprotinin and 40 μg/mL pepstatin A) supplemented with 300 mM NaCl. This procedure elutes cytoplasmic and nuclear proteins, but does not touch tightly DNA-associated proteins such as histones. This avoids the release of viscous DNA that interferes with clean immunoprecipitation. Extracts were diluted with TL to 150 mM salt and digested with benzonase after addition of 2 mM MgCl2 to remove residual contaminating nucleic acid. Precipitation was done with immobilized anti-tag antibodies (anti-flag, anti-HA agarose from SIGMA, Taufkirchen, Germany). Precipitating material was washed eight times with TL buffer + 300 mM NaCl and analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis and immunoblotting. Depending on the batch and the storage time of the antibody-coupled agarose reagents, occasionally heavy chain was co-eluted with precipitated material from the beads. In these instances the molecular weight allowed discrimination of contaminating heavy chain. To detect DNA-dependent interactions benzonase treatment was omitted and 1 pmol of an equimolar mixture of a double-stranded oligo containing a Hox/Meis consensus site (5′-ccagatctgacagttttacgacagatctcc-3′)8 and a Hox/Pbx (5′-ctgcgatgatttacgaccgc-3′) binding site was added. The Hox/Pbx site was designed by replacing the Meis binding half-site by the known Pbx site present in the EphA2 enhancer.22 This oligo had been previously shown to bind Hoxa9 in combination with Pbx2.23 DNA-containing complexes were washed three times in phosphate-buffered saline only.
Determination of protein half-life
For determination of protein half-life, 24 h after transfection cells were treated with 50 μg/mL cycloheximide (SIGMA, Taufkirchen, Germany) to stop protein translation. Samples were drawn at 0 h, and after 1 h, 2 h, and 4 h of incubation. Protein abundance was determined by anti-tag western blots with β-actin as normalization control.
Transplantation experiments, gene expression and statistics
Transplantation studies were done in syngenic Balb/C mice. Three days before transplantation animals were treated with 1.1 g/L neomycin and 1×106 units/L polymyxin added to drinking water. Twenty-four hours after myeloablation by total body irradiation (8 Gy), 0.5×106 transformed cells created by transduction with subsequent antibiotic selection, and 1×106 total bone marrow cells for short-term rescue were transplanted by retro-orbital injection. Animals that developed predefined termination criteria (hunched posture, ruffled fur, labored breathing, loss of body weight, pale and/or cold extremities) were euthanized. Spleen weight was recorded as an objective surrogate marker of leukemia burden. White blood cells were counted in peripheral blood after red cell lysis. Engraftment of cells was confirmed by polymerase chain reaction on genomic DNA isolated from peripheral blood of transplanted animals. Amplification was done with intron spanning primers detecting the transduced HOXA9 cDNA (fw: 5′-ctgtcccacgcttgacactcac-3′, rev: 5′-gagcgcgcatgaagccagttg-3′). All animal procedures were performed according to the regulations of the local and institutional authorities (FELASA recommendations, University of Erlangen, Dept. of Animal Welfare, regional licensing authority, license # TS-99/01, 54-2532.2-5-12)
Gene expression was measured by real time polymerase chain reaction of cDNA generated from transduced cells with the following primers: Meis1: fw, 5′-cctctgcactcgcatcagtac-3′; rev, 5′-gtttggcgaacaccgctatatc-3′; Pbx3: fw, 5′-ctcccaaattctggggacatg-3′; rev, 5′-atccacctgtgactgcacattg-3′; Flt3: fw, 5′-atccttccccaacctgacttc-3′; rev, 5′-gttgccacccatgttctgatac-3′; Trib2: fw, 5′-ctggagggagaccacgttttc-3′; rev, 5′-tccgtgatttggttgatgttgc-3
Where appropriate a Student t-test was performed to probe for statistical significance, which was assumed for P-values <0.05.
Results
The Pbx interaction domain of Meis1 is a destabilizing element
To begin we wanted to probe whether all three Pbx family members, including Pbx3, form complexes with Meis1 in the absence of DNA. For this purpose co-immunoprecipitation of Meis1 with Pbx1, PBX2 and Pbx3 was performed (Online Supplementary Figure S1A). For PBX2 the human cDNA was used because a complete mouse clone could not be obtained and human and mouse PBX2/Pbx2 are 98% identical (421 of 430 amino acids). In these experiments cellular extracts were exhaustively digested with benzonase to remove any contaminating nucleic acid that might have acted as a tether between the individual proteins. Precipitation of Meis1 brought down all three Pbx proteins indicating the formation of stable, DNA-independent Meis1/Pbx complexes. GST pulldowns also confirmed a tight interaction of Meis1 and Pbx3 in the absence of DNA (Online Supplementary Figure S1B). As a control, interaction of Hoxa9 with Pbx family members was tested under DNA-free conditions (Online Supplementary Figure S1A, lower panel). In contrast to Meis1, precipitation of Hoxa9 did not lead to copurification of Pbx molecules corroborating the need for nucleic acid to enable Hox-Pbx binding. During these experiments we noted a dramatic buildup of Meis1 whenever it was co-expressed together with a Pbx protein in cells (Online Supplementary Figure S1C). A reciprocal but less pronounced effect could be detected for Pbx1 and PBX2. Pbx3 protein levels did not change regardless of whether Meis1 was present or not.
To examine this phenomenon further, interaction-defective mutants of Meis1 and Pbx3 were constructed. For these and all following experiments we concentrated on Pbx3 as it is the family member for which there is most evidence for an involvement in human leukemia. The Pbx binding domain in Meis1 has been mapped to a leucine zipper in the N-terminus.13 Five hydrophobic amino acids of this region were exchanged against alanine and the Pbx interacting capacity of this mutant (MeisΔ) was tested in co-immunoprecipitation studies (Figure 1A). As intended, MeisΔ completely lost its ability to interact with Pbx3. Unexpectedly, this mutation also increased the inherent stability of MeisΔ. In cellular extracts MeisΔ on its own was almost as abundant as wild-type (wt) Meis1 in the presence of Pbx3 (Figure 1B). This pointed to a role of the Pbx interaction motif in controlling Meis1 protein turnover.
Figure 1.

Binding of Pbx3 or deletion of the Pbx-binding domain stabilizes Meis1 by prolonging protein half-life and blocking proteasomal degradation. (A) Generation of a Pbx-binding defective Meis1 mutant. Five alanine substitutions were introduced into the Pbx-binding domain of Meis1 to create a MeisΔ construct as schematically indicated. The lower panels demonstrate the results of immunoprecipitation experiments confirming that MeisΔ lost detectable affinity for Pbx3. f = flag tag, h = HA tag. (B) Deletion of the Pbx3-binding domain in MeisΔ leads to protein stabilization. Extracts from cells transfected with equal amounts of MeisΔ or wt Meis1 constructs were tested for expression of flag-tagged MeisΔ and wt Meis1 in immunoblots (upper panel). β-actin served as a loading control (lower panel). (C) Construction of a Meis1-interaction defective Pbx3. A Pbx3Δ mutant was cloned introducing a small deletion within the N-terminal Meis-interaction domain. In immunoprecipitation experiments this mutant lost its ability to bind to Meis1 (upper right panel). Concomitant with the loss of Meis1 binding capacity Pbx3Δ also did not stabilize Meis1 protein (lower left panel). (D) Comparison of the Meis1 stabilization effect achieved by co-expression of Pbx3 or by deletion of the Pbx-binding domain. Wt Meis1 or the Pbx-binding defective MeisΔ mutant were expressed alone or in combination with Pbx3 or together with Pbx3Δ that has lost Meis-binding capability. Accumulated Meis protein was detected by immunoblotting. (E) Instability of Meis1 is due to short half-life. Protein decay rates were determined for Meis1 and a combination of Meis1 + Pbx3 in a time course after shut-off of synthesis by addition of cycloheximide (CHX). Samples were drawn at the indicated time points and analyzed by western blot for the remaining quantity of Meis1. Please note that protein amounts and exposure times were adjusted to start with an equal signal intensity for Meis1 and Meis1 + Pbx3 samples. (F) Half-life determination of Meis1. Quantification of detectable Meis1 protein after shutting down protein synthesis was done in a parallel experiment as described in (B) by densitometric analysis. Representative values for one out of two experiments are given. (G) Small molecule inhibitor of the proteasome system phenocopies the presence of Pbx3. Meis1 alone or in combination with Pbx3 was expressed in cells incubated for 24 h after transfection with increasing amounts of the proteasome inhibitor MG132 as indicated. Flag-tagged protein was detected by immunoblot with β-actin as a loading control.
In an additional experiment the influence of Pbx3 on Meis1 stability was tested with a Pbx3 deletion mutant (Pbx3Δ) that has lost its Meis1 binding domain. Residues important for Meis1 binding have been mapped to the N-terminus of Pbx proteins.15 We checked a series of N-terminal deletion mutants and found that removal of 22 residues (amino acids 78 to 100) just reaching beyond the hitherto known Meis1 binding motif (amino acids 1-89) abrogated the Pbx3-Meis1 interaction in immunoprecipitation studies (Figure 1C). Correspondingly, this Pbx3Δ mutant had no stabilizing effect on Meis1 when co-expressed in cells. In summary, either physical occupation by Pbx binding or removal of the Pbx-binding domain leads to stabilization of Meis1.
Pbx3 prolongs Meis1 half-life and blocks entry into proteasome-mediated degradation
To clarify the role of the Meis1/Pbx3 interaction in controlling protein turnover rates we first compared the individual effects of deleting the Pbx3-interaction domain and/or co-expression of Pbx3/Pbx3Δ on Meis1 stability side-by-side in a single experiment (Figure 1D). Again, co-expression of Pbx3 but not the interaction-defective mutant or deletion of the Pbx3 binding domain in Meis1 substantially increased detectable protein levels. To investigate whether this phenomenon was due to alterations in protein synthesis or degradation rates we determined the half-life of Meis1 protein (Figure 1E). Protein levels of Meis1 and of a combination of Meis1 with Pbx3 were determined at regular intervals after blocking protein synthesis with cycloheximide. Whereas Meis1 by itself was rapidly degraded it became more stable in the presence of Pbx3. In a second experiment that also included MeisΔ, protein decay was quantified by densitometry of the respective western blots (Figure 1F). It was found that only 10% of Meis1 remained intact 1 h after shutting down translation. In contrast 90% of the original Meis1 level was still present once Pbx3 was added. MeisΔ had an intermediate behaviour with about 75% of the protein remaining after 1 h of cycloheximide treatment.
To determine whether Meis1 is subject to proteasomemediated degradation either Meis1 alone or a combination of Meis1 and Pbx3 was expressed in the presence of increasing amounts of the proteasome inhibitor MG132 (Figure 1G). Blocking the 26S proteasome increased Meis1 concentrations substantially. The addition of 1 μM MG132 elicited nearly the same effect as co-expression of Pbx3. Once complexed with Pbx3 addition of MG132 did not further increase Meis1 levels.
Pbx3 protects Meis1 from ubiquitination
Because proteasome activity is dependent on prior ubiquitination, the function of the Meis Pbx-binding domain as an ubiquitin acceptor was examined. Poly-ubiquitin chains are attached to lysine residues by E3-ligase enzymes. The intermediate poly-ubiquitinated products designated for degradation can be stabilized by treatment of cells with MG132. If tagged ubiquitin is expressed in the cells this tag will be transferred to the protein of interest allowing visualization by western blot. An expression construct coding for HA-tagged ubiquitin was, therefore, co-transfected with vectors producing wt Meis1, MeisΔ and an additional mutant that exchanged lysine 161 within the Pbx-binding domain against alanine (MeisK161A). After transfection cells were treated for 24 h with 1 μM MG132. Meis1 and all potentially ubiquitinated derivatives were specifically precipitated by anti-flag immunoprecipitation. Ubiquitinated and hence HA-tagged Meis1 derivatives were detected by anti-HA immunoblot (Figure 2A). These experiments demonstrated that ubiquitination was almost absent in the MeisΔ construct and that it was significantly reduced for the MeisK161A samples. Thus an intact Pbx-binding domain was essential for efficient ubiquitin modification. Lysine 161, however, cannot be the sole ubiquitin acceptor residue because the K161A mutant still accumulates substantial amounts of ubiquitin side chains. The influence of Pbx3 on ubiquitination of Meis1 was examined in a similar experiment. Allowing complex formation with Pbx3 completely prevented the generation of HA-ubiquitin-modified Meis1 (Figure 2B). The most straightforward explanation for these results is that Pbx3 and the responsible E3 ubiquitin ligase share common binding requirements within Meis1. Thus Pbx3 would compete with the E3-enzyme for association with Meis1, preventing ubiquitin addition.
Figure 2.

Ubiquitination of Meis1 is blocked by interaction with Pbx3. (A) Mutations of the Pbx3-binding domain reduce ubiquitination of Meis1. Ubiquitination of Meis1, MeisΔ, and MeisK161A that carries an alanine substitution of the putative ubiquitin lysine acceptor within the Pbx3-binding domain, was investigated by co-expression with HA-modified ubiquitin (HA-Ub) in cells treated with MG132 to stabilize the transient ubiquitin-modified degradation intermediates. Meis-derivatives were specifically precipitated by anti-flag IP and analyzed for ubiquitin addition by an anti-HA immunoblot. The presence of Meis modified by addition of ubiquitin chains of varying length was indicated by the appearance of high molecular weight, HA-reactive material in western blotting (Meis-Ub). (B) Binding of Pbx3 blocks ubiquitination of Meis1. Meis1 alone or in combination with Pbx3 was co-expressed with HA-modified ubiquitin in cells treated with 1 μM proteasome inhibitor. Upon anti-flag precipitation ubiquitination was detected as described for (A) by anti-HA immunoblot.
Pbx binding is necessary for an efficient interaction of Meis1 with Hoxa9
To test whether protection against proteasomal degradation is the only feature of Pbx3 that is important for Meis1 function, Meis1 and MeisΔ were compared in CFC and proliferation assays. Meis1 augments the leukemogenic activity of Hoxa9-transformed cells, a feature that reads out in higher CFC numbers and accelerated proliferation of transformed hematopoietic stem and precursor cells. If stability is the sole decisive factor, MeisΔ that is intrinsically degradation-resistant should work similarly to or even better than wt Meis1. In addition, the consequences of co-expression of Pbx3, Pbx3Δ, and a combination of Meis1 and Pbx3 were recorded in the same assays (Figure 3A). Unexpectedly, MeisΔ lacked any appreciable activity in these experiments. Pbx3 alone had no significant influence on Hoxa9-induced CFC numbers although proliferation was slightly increased. This effect was largely reversed by loss of the Meis interaction motif in Pbx3Δ. Adding Pbx3 as well as Meis1 had an additive effect on proliferation but not on CFC numbers, which seemed to have reached a limit already in the presence of Meis1 alone. In the absence of Hoxa9 a combination of Meis1 and Pbx3 did not cause CFC activity (data not shown). In summary these results support the hypothesis that Pbx3-Meis1 dimers are the actual Hoxa9 cooperating entity but they also indicate that Meis1-Pbx3 dimerization must convey a function beyond simple stabilization of Meis1.
Figure 3.

Pbx3 binding is required for cooperation of Meis1 and Hoxa9 in vivo and in vitro. (A) Colony formation assay (CFC, upper panel) and proliferation rates measured by MTT incorporation (lower panel) of primary hematopoietic stem and precursor cells transduced with Hoxa9 in combination with Meis1, MeisΔ, Pbx3, Pbx3Δ, as well as with Meis1 and Pbx3 as indicated. Data represent averages and standard deviations of biological triplicates. Significance between control (vector) and test data was analyzed by the Student t-test. *P<0.05 compared to vector control. (B) The Pbx-binding site in Meis1 is necessary for efficient formation of a Hoxa9/Meis1/Pbx3/DNA quaternary complex. Meis1 or Meis∆ was precipitated from extracts of cells co-expressing Hoxa9 and Pbx3 and supplemented with oligonucleotides coding for optimal Hoxa9/Meis1 and Hoxa9/Pbx binding sites. Hoxa9 was efficiently co-purified after precipitation of Meis1 but not of MeisΔ with anti-flag reagents. Because MeisΔ was less abundant in cell extracts and consequently also in the precipitations a second experiment was added separating 20-fold more MeisΔ sample than Meis1 probe per lane (right lower panel). The graphic depiction indicates the interactions within the trimeric Hox/Meis/Pbx protein complex that are targeted by the individual mutants. (C) Hoxa9-Pbx3 contacts contribute to Meis1-Hoxa9 cooperation in vivo. CFC and proliferation assays were performed as described for (A) except exchanging Hoxa9 for a mutant (Hoxa9ΔPbx) that can no longer bind to Pbx proteins because of a deletion of the corresponding hexapeptide binding motif. (D) Hoxa9-Pbx3 contacts provide avidity for the Meis1-Hoxa9 interaction. Affinity of Meis1 for Hoxa9 and Hoxa9ΔPbx was tested in coimmunoprecipitation experiments in the presence of DNA, analogous to (B). The numbers below the panels indicate arbitrary intensity units quantifying specific bands (Meis1, upper panel; Hoxa9, lower panel) by scanning the western blot.
To elucidate the reason why the auto-stable MeisΔ would not be able to cooperate with Hoxa9 in vivo, the affinity of MeisΔ for Hoxa9 binding was biochemically assessed by co-immunoprecipitation in the presence of DNA. Extracts of cells expressing either MeisΔ/Pbx3/Hoxa9 or wt-Meis1/Pbx3/Hoxa9 were supplemented with oligonucleotides coding for optimized Hox/Meis and Hox/Pbx binding sites to allow the formation of DNA-dependent Hox/Meis and Hox/Pbx interactions. Wt Meis1 accumulated to even higher levels than the auto-stable MeisΔ in cells expressing all three proteins and wt Meis1 co-precipitated efficiently with Hoxa9 (Figure 3B). In contrast MeisΔ did not interact with Hoxa9 under identical conditions, suggesting that additional Pbx-Hox contacts in the trimeric complex contribute to overall stability of the Hoxa9-Meis1-Pbx3 assembly (see graphical representation of the interaction below Figure 3B). To corroborate this conclusion the accelerating effect of Meis1 was tested again in CFC and proliferation assays with Hoxa9 exchanged for a previously described23 mutant (Hoxa9ΔPbx) that has lost its Pbx interaction capability due to a deletion of the hexapeptide Pbx interaction motif. The loss of Hoxa9-Pbx contacts weakened the transforming activity of Hoxa9ΔPbx on its own. Moreover, even a triple combination of Hoxa9ΔPbx, Meis1, and Pbx3 did not reach the same activity in CFC and proliferation assays as a similar combination including wt Hoxa9 (Figure 3C). This result was paralleled in biochemical experiments (Figure 3D). The interaction of Pbx3 with Hoxa9 contributed indirectly to the stability of the Meis1-Hoxa9 interaction within the quaternary DNA/Meis1/Hoxa9/Pbx3 complex because less Hoxa9ΔPbx than Hoxa9 co-precipitated with Meis1. In summary, these results suggest that a Meis1/Pbx3 dimer has to be tethered to Hoxa9 through Meis1 and Pbx3 for optimal activity.
Pbx3 cooperates with Meis1 to induce target gene transcription
Because biological activity is a consequence of alterations in gene expression the effect of Meis1 and/or Pbx3 on transcription of individual downstream targets was tested. When endogenous Meis1 and Pbx3 transcripts were measured in Hoxa9-0020and Hoxa9DPbx-transformed cells Meis1 RNA was present at low but detectable levels (ΔCtactin = 20.8 cycles). Pbx3 mRNA was 24-fold more abundant, as determined with primers calibrated against plasmid standards. Interestingly, Pbx3 over-expression caused a 100-fold induction of Meis1 transcription (Figure 4A). Again, this effect was not seen with an interaction-defective Pbx3Δ mutant and it was also absent in cells transformed by Hoxa9ΔPbx. In contrast, the Pbx3 promoter was insensitive towards Meis1.
Figure 4.
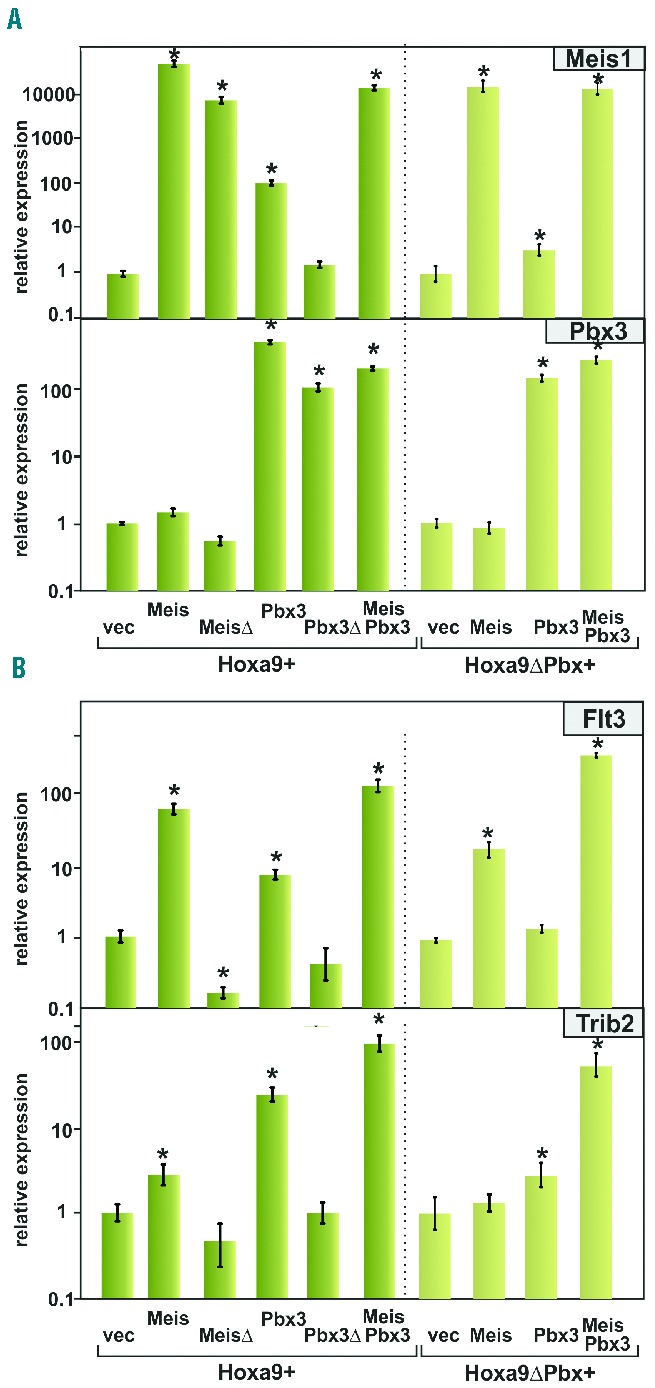
A Meis1-Pbx3 interaction is required for optimal induction of target genes. (A) Pbx3 overexpression can induce Meis1 transcription. Primary cells transformed either by Hoxa9 or Hoxa9ΔPbx in combination with Meis1, MeisΔ, Pbx3, Pbx3Δ, as well as Meis1+Pbx3 were used for RNA isolation. Relative concentrations of Meis1 and Pbx3 transcripts were quantified by real-time quantitative polymerase chain reaction normalized to actin and expressed relative to a vector control. Values indicate averages and standard deviations of polymerase chain reaction triplicates. *P<0.05 compared to vector control. Please, note the log10 scale. (B) Induction of Meis1 target genes is contingent on undisturbed an Meis1-Pbx3 interaction. Experiment performed as in (A) quantifying relative concentrations of the known Meis1 target genes Flt3 and Trib2.
Next, transcription of two well-characterized down-stream targets of Meis1, Flt312 and Trib224 was examined (Figure 4B). Both genes generally followed the overall pattern observed before. Optimal induction required Meis1 and Pbx3. Replacement of Meis1 by the Pbx-interaction-defective MeisD completely abrogated the stimulatory effect on target transcription. Interfering with Pbx3-Hoxa9 interactions by supplementing Hoxa9ΔPbx instead of wt Hoxa9 also reduced Trib2 transcription.
Meis1 and Pbx3 coordinately regulate leukemogenesis
In order to score the requirement for Meis1 and Pbx3 interaction during leukemia induction, primary bone marrow cells were transduced with Meis1, MeisΔ, Pbx3, Pbx3Δ and with Meis1+Pbx3 in combination with either Hoxa9 or Hoxa9ΔPbx according to the scheme used before. After transplantation syngenic recipient animals were scored daily according to an institutionally approved scale for signs of distress (behavior, ruffled fur, hunched posture, labored breathing, weight loss). Upon reaching a preset clinical score the recipients were euthanized and a post-mortem examination for signs of leukemia (pale organs, enlarged spleen and lymph nodes) was performed. Cohorts of five animals were transplanted but occasionally an animal was lost early in the experiment due to post-conditioning mortality. Survival times closely matched the behavior predicted from the in vitro results (Figure 5A). Despite the fact that a combination of Hoxa9 and Meis1 already induced fulminant leukemia with a very short latency (mean ± SD; 37 ± 0.8 days) additional co-transduction of Pbx3 still exacerbated the disease (mean ± SD; 33.6 ± 2.1 days; P=0.02). Cooperation between Meis1 and Pbx3 became more obvious in Hoxa9ΔPbx-transformed cells in which leukemogenicity was generally more attenuated. Whereas Hoxa9ΔPbx/Meis1/Pbx3 mice died at 43.4±2.7 days after transduction, it took 141.7±2.3 days for animals receiving Hoxa9ΔPbx/Meis1 cells to develop clear symptoms. All diseased animals had a spleen weight in the pathological range, being heavier than 100 mg. Within the Hoxa9 and Hoxa9ΔPbx cohorts, average spleen size was larger for animals receiving a Meis1 + Pbx3 graft compared to those transplanted with Meis1 only (Figure 5B). Expression of Hoxa9 or Hoxa9ΔPbx alone or in combination with MeisΔ, Pbx3, and Pbx3Δ did not induce disease within 180 days. At that point, the presence of transplanted cells in peripheral blood was checked by polymerase chain reaction-based detection of Hoxa9 and Hoxa9ΔPbx, respectively (Figure 5C). Transplanted cells could be detected in hematopoietic cells of all recipients with the exception of a single animal in the Hoxa9/vector cohort that had lost the graft. In essence, introduction of Pbx3 exacerbated leukemia development while blocking Meis1/Pbx3 dimer formation by disabling the respective interaction domains completely prevented disease. A reduction of the affinity of Meis1/Pbx3 dimers for Hoxa9 by interfering with the Pbx3/Hoxa9 contact also retarded disease development. In summary, these data are best reconcilable with a physiological function of Meis1 and Pbx3 as a dimer.
Figure 5.
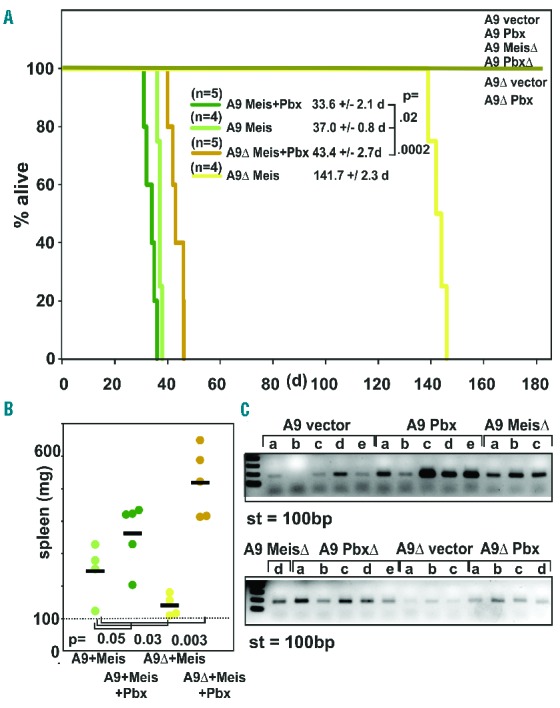
Meis1 and Pbx3 cooperate in leukemogenesis. (A) Meis1 heterodimerization with Pbx3 is rate-limiting for leukemogenesis. Kaplan-Meier survival curve of animals transplanted with syngenic primary cells transformed by either Hoxa9 (A9) or Hoxa9ΔPbx (A9Δ) in combination with either Meis1 (Meis), Pbx3 (Pbx), Meis1Δ (MeisΔ), Pbx3Δ (PbxΔ), or Meis1 + Pbx3 (Meis+Pbx) as indicated in the legend. Animals were transplanted on day 0 and observed for 6 months. Cohort size was initially five animals, with early deaths after radiation conditioning (< day 10) censored. (B) Spleen weight of euthanized animals succumbing to leukemia according to transplantation cohorts. (C) PCR analysis of white blood cell-derived genomic DNA from animals surviving 180 days after transplantation. A primer pair specifically detecting the presence of transduced Hoxa9/Hoxa9ΔPbx cDNA was used to probe for the presence of integrated provirus in genomic DNA of white blood cells isolated from recipients. St = 100bp standard.
Discussion
In this study we demonstrate that Meis1 and Pbx3 need to interact with each other to evoke the full cooperative capability that potentiates the baseline transforming activity of Hoxa9. This observation can be explained by multiple features of the Meis1/Pbx3 interaction. Pbx3 increases Meis1 half-life by preventing ubiquitin-mediated degradation, it acts as transcription factor for the Meis1 gene and it is essential for formation of high affinity Pbx3/Meis1/Hoxa9/DNA complexes.
Our experimental results are strongly substantiated by epidemiological data. Beyond what was already recognized in a smaller study,19 the analysis of gene expression across 484 unrelated acute myeloid leukemia samples (www.oncomine.org) brought up MEIS1 and PBX3 as the two non-HOX genes most strongly correlated with HOXA9 expression (Online Supplementary Figure S2). This close correlation is highly indicative of a functional requirement for a trimeric unit during leukemogenesis. In contrast, neither PBX1 nor PBX2 appeared within the list of significant correlations, arguing for the predominant role of PBX3 as a HOX/MEIS cofactor in hematopoietic cells.
The fact that many previous studies noted an individual effect of Meis1 or Pbx3 on Hoxa9-induced transformation can be explained by the presence of endogenous Meis1 and Pbx3 in Hox-transformed cells. This allows at least a partial dimerization of exogenously added protein thus explaining the (suboptimal) efficacy of increasing either Meis1 or Pbx3 without its binding partner. It has been shown before that functional Hoxa9 and Meis1 binding sites are present in an upstream enhancer of the Meis1 gene.25 Additionally ENCODE ChIP-seq data (www.encodeproject.org) show a strong binding of PBX3 around the transcription initiation site of the human MEIS1 gene. This was noted in two independent biological replicates employing two different cell lines either derived from a B-cell malignancy or neuroblastoma. In addition we detected Hoxa9, Meis1 and Pbx3 by chromatin immunoprecipitation on the same chromatin region within the Hoxa9-responsive Vav2 promoter26 (Online Supplementary Figure S3). These data underscore the strong link between Hoxa9, Meis1 and Pbx3 also on the genetic level. No information about regulatory regions governing Pbx3 expression is presently available. Nevertheless, our experiments demonstrate that endogenous Pbx3 mRNA is clearly present in Hoxa9-transformed cells. In this regard we note that we did not observe an accelerating effect of increasing Pbx3 alone on Hoxa9-induced leukemia in transplantation studies. This result is different from that of the experiments performed by Li et al.,19 who found that Hoxa9/Pbx3 double-transduced cells caused lethal leukemia within 60 to 70 days. Most likely Li et al. achieved higher expression levels of the ectopic oncogenes by their particular vector/transduction procedure. This would explain why Li et al. observed 100% penetrant leukemia after 120 to 160 days with cells transduced by Hoxa9 alone, a finding that is in contrast to those of two previous publications5,12 describing that Hoxa9 by itself does not induce leukemia within 170 or 200 days, respectively.
Further support for our conclusions comes from a recent study investigating the tumor-suppressing function of Prep1, another member of the TALE family.27 In fibroblasts Meis1 acts as an oncogene and Prep1 antagonizes Meis1 activity. Biochemically, Prep1 competes with Meis1 for Pbx1 binding and as a consequence Meis1 is post-translationally destabilized. This is fully congruent with our postulated mechanism of Pbx-induced Meis1 stabilization. Sequestering Pbx1 from Meis1 by interaction with Prep1 would allow access of an E3 ubiquitin ligase, eventually leading to proteasomal degradation of Meis1. It was not reported whether fibroblasts expressed any Hox protein or whether Meis1 and Pbx1 formed a binary system in these cells. Nevertheless, the basic mechanism of Meis1 stabilization seems to be generally conserved across various cellular and organ systems utilizing different Pbx family members. The involvement of different Pbx proteins in diverse cell types may allow a combinatorial diversity that is likely required to specify the different target gene spectrum appropriate for the respective environment.
The most likely explanation for the current results is that Pbx3 competes for the binding of an ubiquitin ligase. Unfortunately, there are more than 600 potential ubiquitin ligases encoded in the murine genome. In addition, the Meis1-E3 ligase interaction seems to be either unstable or transient as Huang et al.28 purified endogenous Hoxa9 and Meis1 complexes and they did not report any co-purification of an ubiquitin ligase. This precludes a direct identification of the enzyme responsible for Meis1 ubiquitination.
Recently, efforts have been made to target the Pbx3-Hoxa9 interaction with small peptides for therapeutic purposes.19,29 Knowledge about the involvement of a larger Hox/Meis/Pbx complex offers more structures and interfaces that could be targeted, thus increasing the likelihood of finding an effective molecule that would interfere with the strong transforming activity of this dangerous menage a trois.
Acknowledgments
We thank Renate Zimmermann for technical assistance and Constanze Breitinger, as well as Vanessa Popp, for help in early stages of this study. This work was supported by research funding from the Deutsche Forschungsgemeinschaft (SL27/8-1) to RKS and partially by the Emerging Fields Initiative of the FAU University Erlangen-Nürnberg (CYDER) to RKS.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000–1008. [DOI] [PubMed] [Google Scholar]
- 2.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766–6776. [DOI] [PubMed] [Google Scholar]
- 3.Eklund E. The role of Hox proteins in leukemogenesis: insights into key regulatory events in hematopoiesis. Crit Rev Oncog. 2011;16(1–2):65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bach C, Buhl S, Mueller D, et al. Leukemogenic transformation by HOXA cluster genes. Blood. 2010;115(14):2910–2918. [DOI] [PubMed] [Google Scholar]
- 5.Kroon E, Krosl J, Thorsteinsdottir U, et al. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17(13): 3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorsteinsdottir U, Kroon E, Jerome L, Blasi F, Sauvageau G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol Cell Biol. 2001;21(1):224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang CP, Brocchieri L, Shen WF, Largman C, Cleary ML. Pbx modulation of Hox homeodomain amino-terminal arms establishes different DNA-binding specificities across the Hox locus. Mol Cell Biol. 1996;16(4):1734–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen WF, Montgomery JC, Rozenfeld S, et al. AbdB-like Hox proteins stabilize DNA binding by the Meis1 homeodomain proteins. Mol Cell Biol. 1997;17(11):6448–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura T, Jenkins NA, Copeland NG. Identification of a new family of Pbx-related homeobox genes. Oncogene. 1996;13(10): 2235–2242. [PubMed] [Google Scholar]
- 10.Nakamura T, Largaespada DA, Shaughnessy JD, Jr, Jenkins NA, Copeland NG. Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat Genet. 1996;12(2):149–153. [DOI] [PubMed] [Google Scholar]
- 11.Drabkin HA, Parsy C, Ferguson K, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16(2):186–195. [DOI] [PubMed] [Google Scholar]
- 12.Wang GG, Pasillas MP, Kamps MP. Meis1 programs transcription of FLT3 and cancer stem cell character, using a mechanism that requires interaction with Pbx and a novel function of the Meis1 C-terminus. Blood. 2005;106(1):254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mamo A, Krosl J, Kroon E, et al. Molecular dissection of Meis1 reveals 2 domains required for leukemia induction and a key role for Hoxa gene activation. Blood. 2006;108(2):622–629. [DOI] [PubMed] [Google Scholar]
- 14.Wang GG, Pasillas MP, Kamps MP. Persistent transactivation by meis1 replaces hox function in myeloid leukemogenesis models: evidence for co-occupancy of meis1-pbx and hox-pbx complexes on promoters of leukemia-associated genes. Mol Cell Biol. 2006;26(10):3902–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shanmugam K, Green NC, Rambaldi I, Saragovi HU, Featherstone MS. PBX and MEIS as non-DNA-binding partners in trimeric complexes with HOX proteins. Mol Cell Biol. 1999;19(11):7577–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen WF, Rozenfeld S, Kwong A, et al. HOXA9 forms triple complexes with PBX2 and MEIS1 in myeloid cells. Mol Cell Biol. 1999;19(4):3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Huang H, Li Y, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012;119(10):2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson GJ, Liberante FG, Kettyle LM, et al. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica. 2013;98(8):1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Zhang Z, Li Y, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121(8):1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swift S, Lorens J, Achacoso P, Nolan GP. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001;Chapter 10:Unit 10 17C. [DOI] [PubMed] [Google Scholar]
- 21.Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16(14):4226–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Ruley HE. An enhancer element in the EphA2 (Eck) gene sufficient for rhombomere-specific expression is activated by HOXA1 and HOXB1 homeobox proteins. J Biol Chem. 1998;273(38):24670–24675. [DOI] [PubMed] [Google Scholar]
- 23.Breitinger C, Maethner E, Garcia-Cuellar MP, Slany RK. The homeodomain region controls the phenotype of HOX-induced murine leukemia. Blood. 2012;120(19):4018–4027. [DOI] [PubMed] [Google Scholar]
- 24.Argiropoulos B, Palmqvist L, Yung E, et al. Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp Hematol. 2008;36(7): 845–859. [DOI] [PubMed] [Google Scholar]
- 25.Wang QF, Li YJ, Dong JF, et al. Regulation of MEIS1 by distal enhancer elements in acute leukemia. Leukemia. 2014;28(1):138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breitinger C, Maethner E, Garcia-Cuellar MP, et al. HOX genes regulate Rac1 activity in hematopoietic cells through control of Vav2 expression. Leukemia. 2013;27(1):236–238. [DOI] [PubMed] [Google Scholar]
- 27.Dardaei L, Longobardi E, Blasi F. Prep1 and Meis1 competition for Pbx1 binding regulates protein stability and tumorigenesis. Proc Natl Acad Sci USA. 2014;111(10):E896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Sitwala K, Bronstein J, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119(2):388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aulisa L, Forraz N, McGuckin C, Hartgerink JD. Inhibition of cancer cell proliferation by designed peptide amphiphiles. Acta Biomater. 2009;5(3):842–853. [DOI] [PubMed] [Google Scholar]