

Abstract
The prognosis of older patients with acute myelogenous leukemia is generally poor. The interleukin-3 receptor α-chain (CD123) is highly expressed on the surface of acute leukemia cells compared with normal hematopoietic stem cells. CSL362 is a fully humanized, CD123-neutralizing monoclonal antibody containing a modified Fc structure, which enhances human natural killer cell antibody-dependent cell-mediated cytotoxicity. Six continuous acute myelogenous leukemia xenografts established from patient explants and characterized by cell and molecular criteria, produced progressively lethal disease 42-202 days after transplantation. CSL362 alone reduced engraftment of one of four and three of four acute myelogenous leukemia xenografts in the bone marrow and peripheral organs, respectively. A cytarabine and daunorubicin regimen was optimized using this model to identify potentially synergistic interactions with CSL362. Cytarabine/daunorubicin improved the survival of mice engrafted with four of four acute myelogenous leukemia xenografts by 31–41 days. Moreover, CSL362 extended the survival of cytarabine/daunorubicin-treated mice for two of two acute myelogenous leukemia xenografts, while augmentation of natural killer cell-deficient NSG mice with adoptively transferred human natural killer cells improved survival against a single xenograft. Interestingly, this enhanced CSL362 efficacy was lost in the absence of chemotherapy. This study shows that acute myelogenous leukemia xenografts provide a platform for the evaluation of new therapeutics, simulating complex in vivo interactions, and that the in vivo efficacy of CSL362 supports continued clinical development of this drug.
Introduction
Contemporary protocols for the treatment of acute myelogenous leukemia (AML) are able to cure 10–60% of patients, with those in older age groups having a poorer prognsois.1,2 Standard therapy includes cytarabine (AraC) for 7 days and daunorubicin (DNR) for 3 days (the “7 + 3” regimen). The addition of novel drugs to the 7 + 3 regimen, such as fludarabine,3 cyclosporine4,5 and zosuquidar,6 has produced varied clinical benefit, and new therapies are required. Antibody targeting therapy is a promising approach with high specificity and low toxicity.
The interleukin-3 receptor α-chain (CD123) is highly expressed on AML stem cells but not hematopoietic stem cells,7–11 providing a target for antibody-based therapy. 7G3 is a mouse neutralizing monoclonal antibody targeting human CD123,12 which demonstrated activity against AML-leukemic stem cells, reduced AML burden and improved survival of engrafted mice.8 The human chimeric version of this antibody, CSL360, was tested in AML patients in a phase I clinical trial in which it demonstrated specificity and safety but did not have clear anti-leukemic activity.13 While CD123 remains a therapeutic target for AML, its neutralization was not sufficient for clinical benefit. CSL362 is based on the CSL360 antibody but is fully humanized, affinity matured, and contains specific Fc-domain point modifications to enhance binding affinity with Fcγ receptors (FcγR)14 and augment anti-leukemic activity via antibody-dependent cellular cytotoxicity (ADCC) against leukemic blasts and leukemic stem cells.15
Human natural killer (NK) cells express CD16 (FcγRIIIa), which binds the Fc portion of antibodies.16 Once ligated, CD16 associates with FceRIγ or CD3ζ resulting in transduction of an activation signal.17 NK cell responses to target cells are governed by the balance of cell surface activating and inhibitory receptors. There is evidence to indicate that NK cells play an important role in leukemia eradication in patients. In cases of haploidentical stem cell transplants that had mismatched inhibitory ligands to the NK cell receptors, improved survival of patients was observed.18–20 Adoptive transfer of NK cells in mice also delayed the progression of cancer cell lines in vivo.21–23
In this study, six continuous AML xenografts with defined cytogenetics and mutations, which produced clinically relevant progressive lethal disease in immune-deficient mice, were established from AML apheresis samples. A regimen of AraC and DNR modeling the clinical 7 + 3 schedule was optimized, which improved the survival of AML engrafted mice, providing a platform for preclinical examination of CSL362. Since the compromised NK cell function of immune-deficient mice may underestimate the efficacy of CSL362, human buffy coat-derived NK cells were expanded ex vivo for adoptive transfer into AML xenografted mice. CSL362 demonstrated additional efficacy against AML xenografts in combination with chemotherapy and adoptively transferred human NK (huNK) cells, supporting its further development in the clinic.
Methods
Samples from patients with acute myelogenous leukemia, xenograft cells, and cell lines
Apheresis samples were collected from AML patients after informed consent and studies were approved by the Royal Adelaide Hospital Human Ethics Committee, Melbourne Health Human Research Ethics Committee and South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee in accordance with the Declaration of Helsinki. The demographics, cytogenetics and clinical data of the AML patients are described in Table 1. The Epstein-Barr virus-transformed B lymphoblastoid (SMI-LCL)24 and K562-mIL15-41BBL22 cell lines were cultured as previously described.
Table 1.
Patients’ characteristics, cytogenetics, and mutations of AML xenografts.
Xenotransplantation and in vivo drug treatments
All mice were maintained under protocols approved by the Animal Care and Ethics Committee of the University of New South Wales. Transplantation of human cells into female NOD/SCID (NOD.CB17-Prkdcscid/J) or NSG (NOD.Cg-PrkdcscidIl2rgtm1WjI/SzJ) mice was performed as previously described, and patients’ samples that produced aggressive and highly infiltrative disease were prioritized for development into continuous xenografts.8 Briefly, mice were exposed to sub-lethal irradiation (250 cGy, delivered by X-RAD 320, Precision X-Ray, CT, USA) 1 day before intravenous (IV) injection of 2–10×106 human AML cells via a heat-dilated tail vein. Engraftment levels were quantified by flow cytometry and expressed as the percentage of human CD45+ (hCD45+) cells to total hCD45+ and mouse CD45+ (mCD45+) cells in the tissue sample.25 AML cells were distinguished from huNK cells by dual staining for CD45 and CD33. Hematology was analyzed on a Coulter® AcT·diff™ Analyzer (Beckman Coulter, CA, USA). Blood was obtained either by cardiac puncture or tail vein bleeds. In vivo drug treatment protocols are described in the Online Supplement.
Mutation analyses of acute myelogenous leukemia xenografts
The mutant:wild-type allelic burden of FLT-internal tandem duplication (FLT3-ITD) and nucelophosmin (NPM1) in genomic DNA was analyzed using modifications of the methods of Murphy et al.26 and Huang et al.27 Polymerase chain reaction products were resolved by capillary electrophoresis (AB 3130 analyzer, Applied Biosystems). For FLT3-ITD, mutant to wild-type peak area ratios ≥0.05 were considered positive and are expressed as percentages in Table 1. The mixed lineage leukemia-partial tandem duplication (MLL-PTD) was determined using a reverse transcriptase real-time quantitative polymerase chain reaction method, in which the MLL-PTD was quantified as the ratio of the MLL-PTD concentration (fg/μL) to ABL1 gene concentration (ag/μL) × 10−3.
A multiplexed matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) genotyping approach (Sequenom MassARRAY Compact System) was used to detect mutations affecting c-KIT D816, DMNT3A R882, FLT3-TKD, IDH1 R132, IDH2 R140/R172, JAK2 V617, KRAS G12/13, MPL W515, NPM1 W288, and NRAS G12/13 and Q61 using custom designed sequences (available on request). Polymerase chain reaction products were prepared using Sequenom iPLEX Pro following published methods, spotted onto a SpectroChip II matrix and resolved using mass spectrometry.28
Isolation and expansion of human natural killer cells and the antibody-dependent cellular cytotoxicity assay
NK cells were isolated from human buffy coats (Australia Red Cross Blood Service, Sydney, Australia), expanded and activated as previously described,24, 29 with modifications as described in the Online Supplement.
Additional details of the methods are provided in the Online Supplement.
Results
Development and characterization of a panel of continuous acute myelogenous leukemia xenografts
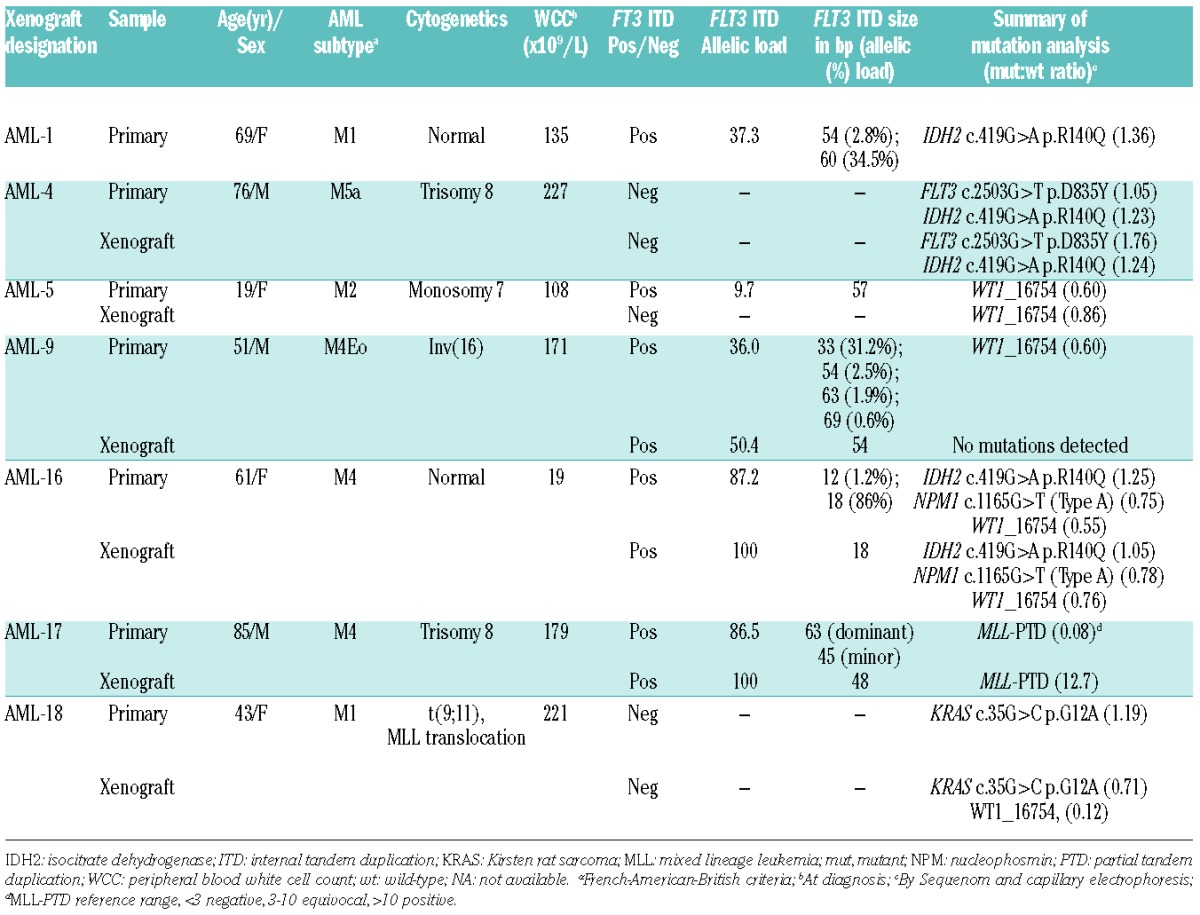
A panel of six continuous xenografts was established in immune-deficient mice from AML patients’ explants with heterogeneous clinical characteristics, cytogenetics and mutations, although four of the six harbored alterations in the FLT3 gene (Table 1). AML apheresis samples known to produce highly infiltrative disease were inoculated into NSG or NOD/SCID mice and expanded for up to six serial passages by transferring human leukemia cells harvested from the spleens, bone marrow, and livers of engrafted mice into secondary recipient mice (Figure 1A, Online Supplementary Figure S1A). A sufficient number of human leukemia cells could not be routinely harvested from mice inoculated with AML-1 in order to establish a continuous xenograft.
Figure 1.
AML xenograft engraftment in blood and immunophenotypes in immunodeficient mice. (A) The engraftment of different passages of AML xenografts in the blood over time post-transplantation. NSG mice were used for all AML xenografts except AML-5 for which NOD/SCID mice were used. Each line represents the median engraftment level for an individual experiment with 2–16 mice per experiment. (B) Representative scatter plots of immunophenotypes of the original patients’ samples and respective xenografts harvested from mouse spleens, based on the expression of CD34, CD38 and CD123, gated on human CD45+ cells. Quadrant lines were based on fluorescence levels of corresponding isotype controls.
Mutations affecting c-KIT, DNMT3A, FLT3-ITD, FLT3-TKD, IDH1, IDH2, JAK2, c-MPL, NPM1, NRAS, KRAS, and MLL-PTD were assessed in the xenografts and diagnostic primary samples (Table 1). AML-4 appeared to be derived from an ancestral clone, with the same FLT3-TKD and IDH2 mutations present in both xenograft and primary samples, although the FLT3 mutant:wild-type ratio was notably higher in the xenograft. Similarly, AML-5 also appeared to be derived from an ancestral clone expressing the WT1 synonymous single nucleotide polymorphism rs16754, although a low level FLT3-ITD subclone detected in the primary sample was lost in the xenograft. The AML-9 primary sample contained at least four separate FLT3-ITD subclones, with a minor FLT3-ITD subclone selected in the xenograft along with loss of WT1 rs16754. Similarly, the AML-16 primary and xenograft expressed identical mutations in IDH2, NPM1, and WT1, although the primary sample contained two FLT3-ITD subclones, the larger of which was selected in the xenograft. The AML-17 primary sample consisted of dominant (63 bp) and minor (45 bp) FLT3-ITD subclones, although the xenograft expressed a unique 48 bp FLT3-ITD subclone that could not be detected in the primary sample even after two rounds of polymerase chain reaction amplification. The AML-17 xenograft also harbored an MLL-PTD, which was detected at an extremely low level (0.08) in the primary sample. Lastly, the AML-18 primary sample consisted of a dominant KRAS-mutated clone, which was also dominant in the xenograft, but with the appearance of a minor WT1 rs16754 subclone. These results indicate that the overall mutational spectrum of these AML samples was conserved upon xenografting, along with variability in the selection of subclones depending on the xenograft.
AML xenografts produced progressive disease that was monitored in the peripheral blood, and engraftment rates accelerated in subsequent passages in five of six AML xenografts (Figure 1A, Online Supplementary Figures S1A and S2 and Online Supplementary Table S1). Engraftment of the AML-17 biopsy did not reach the predefined event point after 163 days, although cells harvested from the bone marrow subsequently produced a continuous xenograft.
Cell surface expression of CD33 and CD45 was maintained on the AML xenograft cells (Figure 1B, Online Supplementary Figure S1B). Moreover, there were no overt changes in cell surface CD34, CD38 and CD123 expression between the patients’ samples and the respective xenografts except for AML-4 and AML-17 xenografts. AML-17 acquired a CD123hi population and AML-4 became CD34−. In patients’ biopsies AML-4, -9, -16, and -17, the CD33− population was CD3+ (data not shown), which disappeared upon secondary transplantation.
NSG mice are more receptive hosts for human xenografts than NOD/SCID mice30 and this was observed with AML-9 and AML-18 xenografts (Online Supplementary Figure S3 and Online Supplementary Table S1). Furthermore, for AML-9 and AML-17, only three of five and three of six NOD/SCID mice, respectively, developed a human AML disease due to the early manifestation of spontaneous murine thymic lymphoma inherent in this mouse strain.31 In contrast, all NSG mice inoculated with AML-9, -17 and -18 developed AML.
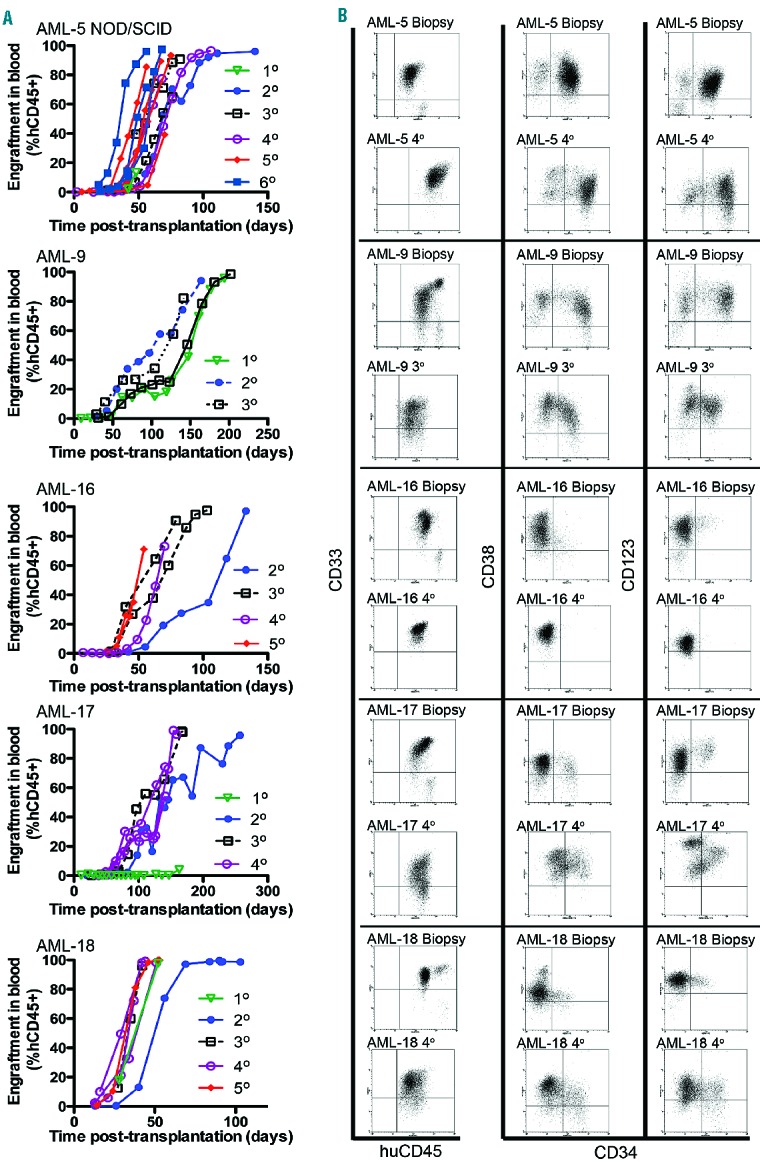
We also tested the requirement for sub-lethal irradiation of NOD/SCID mice for engraftment using AML-5. While four of four irradiated mice reached an event at a median of 63 days (Figure 2A and Online Supplementary Table S1), AML cells were not detected in the peripheral blood of two of three non-irradiated mice at the termination of the experiment, and only one of the mice had trace levels of leukemia in the bone marrow, spleen and liver (data not shown). Surprisingly, when we repeated the study with NSG mice, irradiation significantly decreased the rate of engraftment of three of three different AML xenografts tested (Figure 2B and Online Supplementary Table S1), albeit in small numbers of mice per AML xenograft.
Figure 2.
Effects of sub-lethal whole body irradiation on AML engraftment in NOD/SCID and NSG mice, and effects of engraftment on hematology. (A) Engraftment levels of AML-5 in the blood over time of irradiated and non-irradiated NOD/SCID mice and respective event-free survival curves. Each line represents the engraftment level of an individual mouse (B). Event-free survival curves of AML-4, AML-16, and AML-17 transplanted NSG mice with or without irradiation before transplantation. (C) Changes in hematologic parameters with AML-5 and AML-16 xenograft progression. Each individual point represents data from an independent mouse blood sample obtained at different time points; four to eight mice per experiment, results for AML-5 were from two independent experiments. The dashed lines represent respective hematologic parameters obtained from non-irradiated, non-engrafted control mice from four independent experiments with three to five mice per experiment.
Mice engrafted with continuous AML xenografts developed “clinical” symptoms and signs associated with leukemia, such as lethargy, loss of body mass, breathing difficulties, and paleness, which if allowed to progress would have resulted in death. The red blood cell count, hematocrit, hemoglobin concentration, and platelet count all correlated inversely (P<0.0001) with the %hCD45+ cells in the blood of AML-5 and AML-16 engrafted mice (Figure 2C). The white cell count initially decreased due to the disruption of normal hematopoiesis, and subsequently increased as the leukemia blast cells become numerically dominant. The disruption of hematopoiesis by AML engraftment was further highlighted by a decrease in platelet count before leukemia cells appeared in the blood (Figure 2C). At autopsy, splenomegaly, hepatomegaly, and occasional thymic enlargement were apparent, dependent on the specific AML xenograft. For example, AML-9 consistently formed tumors around the bladder, ovaries and small intestinal regions with thickening of stomach walls, while AML-16 produced exceedingly large spleens (Online Supplementary Figure S4). The surface expression of markers associated with hematopoietic cell “stemness” (CD34, CD38 and CD123) was assessed on AML xenograft cells harvested from various organs. Remarkably, within a specific engrafted mouse, there were substantial differences in the immunophenotype of AML cells recovered from different organs. For example, the proportion of CD34+CD38− AML-5 cells was significantly higher in the liver, blood and spleen than in the bone marrow, while AML-17 CD123 expression was significantly higher in the bone marrow than in the liver, blood and spleen (Online Supplementary Figure S5). Taken together these results indicate that the %hCD45+ cells in the blood provides a surrogate indicator of overall disease burden in the mice.
Optimization of an induction-type, cytarabine/daunorubicin chemotherapy regimen in mice xenografted with human acute myelogenous leukemia
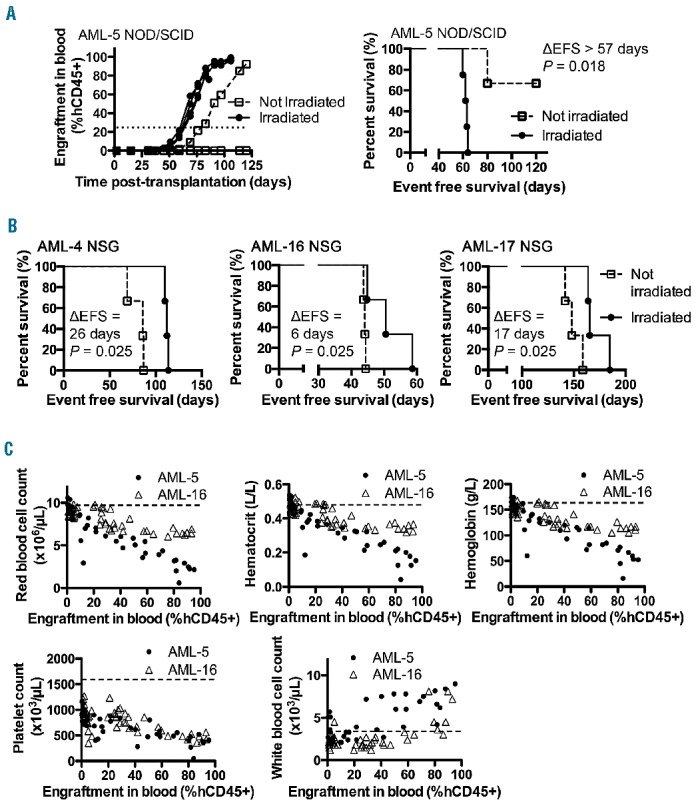
In order to identify possible synergistic or antagonistic interactions of novel treatments in combination with established therapy for AML, we optimized an induction-type “4+1” regimen for the xenograft models. The rationale behind the development of this regimen was to induce temporary remissions in leukemia progression (30–40 days) that could then be used as a platform to combine with novel therapies. Engraftment of AML-1 in the murine bone marrow was significantly reduced following treatment with this AraC (12.5–50 mg/kg/dose) and DNR (0.32–1.25 mg/kg/dose) regimen (Figure 3A). This regimen was further tested on the panel of continuous AML xenografts at a dose of 25 mg/kg AraC and 0.63 mg/kg DNR for 2–3 weeks, which caused substantial decreases in absolute hCD45+ and mCD45+ cell counts in the blood. After the termination of treatments, mCD45+ cell counts rapidly recovered relative to human blast counts, producing a remission-like state. However, the AML blasts eventually overtook the murine hematopoietic recovery producing the final signs and morbidity of the disease (Online Supplementary Figure S6). This dynamic interplay between normal hematopoietic cells and leukemic blasts can be represented as %hCD45+ engraftment, and this regimen of AraC/DNR resulted in significant delays in leukemia progression, and prolonged event-free survival for four of four xenografts (Figure 3B).
Figure 3.
AML xenograft responses to a “4+1” regimen of AraC and DNR in immunodeficient mice. (A) Engraftment levels of AML-1 xenograft cells in the bone marrow (BM) of NOD/SCID mice in response to AraC/DNR dose combinations. 100% represents 50 mg/kg of AraC and 1.25 mg/kg of DNR; 50% (25 mg/kg AraC, 0.63 mg/kg DNR) and 25% (12.5 mg/kg AraC, 0.32 mg/kg DNR) doses were also tested. The mice received three different doses of the AraC/DNR regimen for 2 weeks over days 14–25 after transplantation, and the mice were sacrificed on day 28. Each point represents data from a single mouse; four mice per group. Horizontal bars represent median values. (B) Engraftment levels in blood and respective event-free survival curves for four different AML xenografts treated for 2–3 weeks with AraC (25 mg/kg) and DNR (0.63 mg/kg) in NOD/SCID (AML-17), and NSG (AML-5, 16, and 18) mice. LGD = leukemia growth delay, Horizontal bars underneath the graphs represent time of treatment, each line represents results from an individual mouse (6–15 mice per group). *P<0.05; ***P<0.001; and ****P<0.0001.
Efficacy of cytarabine/daunorubicin or CSL362 as monotherapy against acute myelogenous leukemia xenografts in vivo
The effects of AraC/DNR on AML engraftment in bone marrow, spleen, liver and blood were measured in AML-5 and AML-18 engrafted NSG mice once the disease was established. Two to 3 weeks of the AraC/DNR regimen strongly reduced AML engraftment in all organs (Figure 4A–B). AML-18 cells caused extensive destruction of the bone marrow space and engraftment data are unavailable. This is consistent with aggressive AML xenografts32 and was reproducible for AML-5 and AML-18 (data not shown). Interestingly, AraC/DNR specifically reduced the absolute AML-5 blast count, with no effects on the mouse hematopoietic cell count in the bone marrow (Online Supplementary Figure S7).
Figure 4.
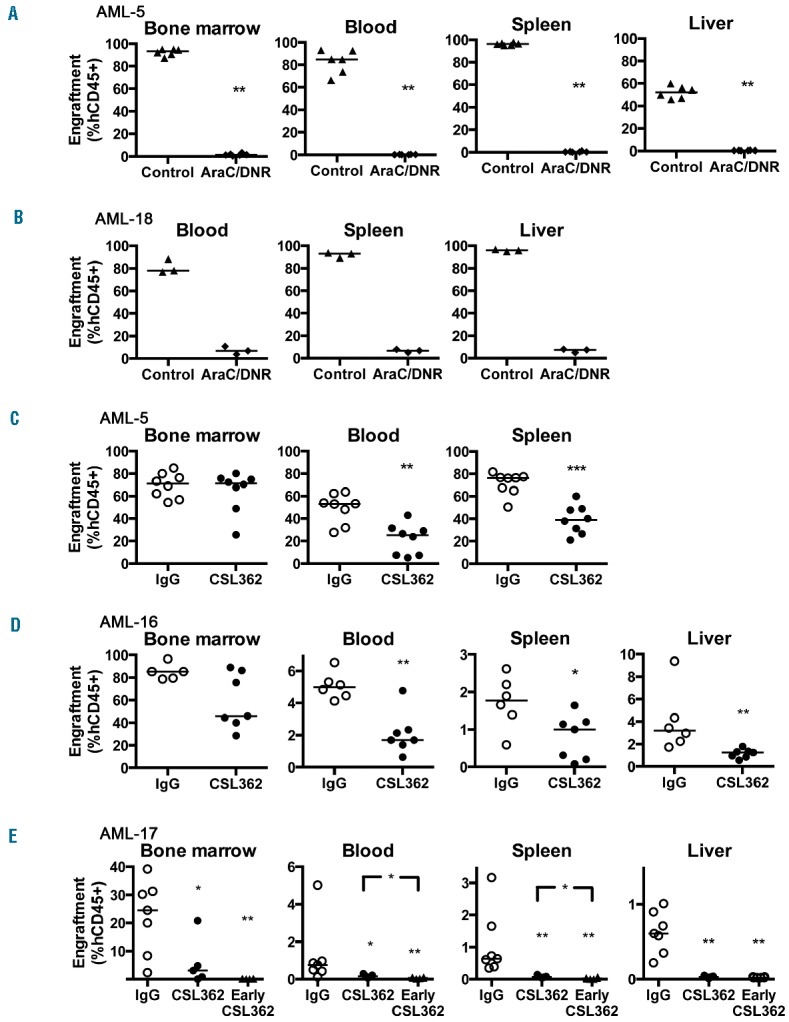
Engraftment levels of AML xenografts in vivo in animals treated with the AraC/DNR regimen or CSL362 as monotherapy. (A, B) AML engraftment in various organs of NSG mice treated with AraC (25 mg/kg) and DNR (0.63 mg/kg). (A) For AML-5 engrafted mice, 3 weeks of treatment was initiated on day 14 after-transplantation. (B) For AML-18 engrafted mice treatment was initiated on day 21 after-transplantation and was given for 2 weeks. (C-E) NOD/SCID mice were treated for 3 weeks with CSL362 or isotype control (IgG). For (C) AML-5 and (D) AML-16, treatment was initiated 3 and 6 week after transplantation, respectively. (E) For AML-17 engrafted mice, treatment was initiated either 6 weeks or 5 days (Early CSL362 group) after transplantation. Each point represents an individual mouse (3–8 mice per group), and horizontal bars represent the median. *P<0.05; **P<0.01; and ***P<0.001.
In order for CSL362 to mediate additional clinical benefits against AML, it must demonstrate anti-leukemic effects in the bone marrow and other organs. NOD/SCID mice were used because of their residual immunity when compared with NSG mice.30 Consistent with this, CSL362 efficacy against AML-5 was attenuated in NSG mice compared with NOD/SCID mice (Online Supplementary Figure S8). CSL362 significantly reduced engraftment of AML-5, -16, and -17 in the blood, spleen and liver, but was only significantly effective against AML-17 in the bone marrow (Figure 4C–E). CSL362 was not effective against AML-18 despite initiation of treatments early at 6 days after-transplantation (Online Supplementary Figure S9). When CSL362 treatment was initiated early in AML-17 engrafted mice, in a simulation of minimal residual disease, the results tended towards improved efficacy in the bone marrow and other organs (Figure 4E).
In vitro antibody-dependent cellular cytotoxicity of natural killer cells against acute myelogenous leukemia xenografts with CSL362
The ability of CSL362 to cause AML xenograft cell lysis in vitro in the presence of huNK cells was also tested in ADCC assays. Following 21 days of expansion from donor buffy coats, the average increase in NK cell numbers ranged from 622- to 725-fold at >95% purity with very high expression of CD56, CD16 and positivity for NKp46 (Online Supplementary Table S2, Online Supplementary Figure S10). Four separate huNK cell expansions were tested against five AML xenografts, and huNK cells in the absence of CSL362 increased AML cell death by up to 40% (Figure 5A). CSL362-mediated cell death in the presence of huNK cells was significantly increased against AML-5, -9 and -17 compared with isotype controls, with AML-16 also approaching significance (Figure 5A,B). These results confirm the potency of expanded huNK cells in mediating CSL362-induced cell death.
Figure 5.
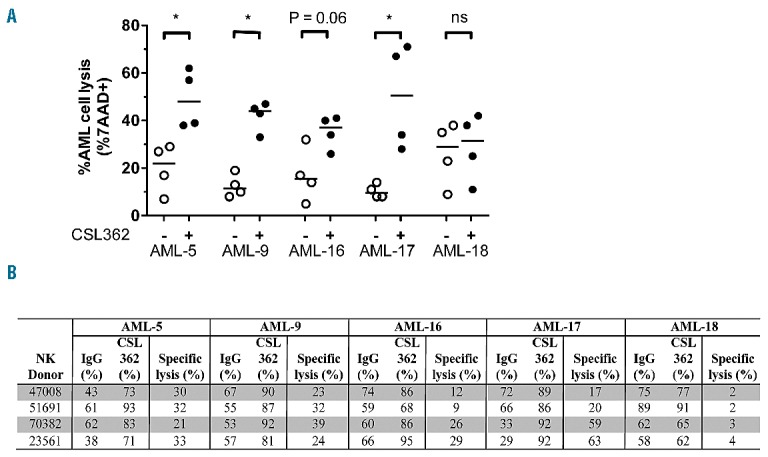
Cytotoxic killing of AML cells by expanded huNK cells and CSL362 in ADCC assays. (A) Cell lysis of AML cells mediated by huNK cells harvested from ex vivo expansions, with 10 μg/mL of CSL362 or IgG1 control. Each dot represents an experiment conducted with an independent huNK donor (triplicates were performed per experiment). Background AML cell death was subtracted from all values. Horizontal lines represent the median. *P<0.05; ***P<0.001; and n.s.: non-significant. (B) Summary of % cell lysis in ADCC assays with or without CSL362 against AML xenografts with expanded NK cells from different donors, without correction for background AML cell death. Specific lysis refers to the ADCC mediated specifically by the addition of CSL362.
In vivo efficacy of CSL362 combined with adoptive transfer of human natural killer cells and chemotherapy
We next carried out in vivo experiments to determine whether adoptive transfer of huNK cells could increase the efficacy of CSL362 against AML xenografts in NSG and NOD/SCID mice. Overall, CSL362 and the addition of huNK cells resulted in no significant delays in leukemia progression against AML-5 (two independent experiments), and AML-17 (Online Supplementary Figure S11 and Online Supplementary Table S3). CFSE-labeled huNK cells were detectable in the peripheral blood, bone marrow, spleen, and liver 1 day after inoculation (Online Supplementary Figure S12), indicating that the huNK cells could enter relevant leukemia niches.
CSL362 is currently being tested in AML patients in remission following chemotherapy. This scenario was simulated in the AML xenograft models in two different approaches in which CSL362 and huNK cell treatment was initiated: (i) 1 day after transplantation (Figure 6A–D); and (ii) after the leukemia had been established and subsequently treated with chemotherapy (Figure 6E–H). In both cases, the leukemia burden simulated a minimal residual disease setting. AML-16-engrafted mice were treated for 3 weeks with CSL362 ± huNK cells and this did not delay leukemia progression (Figure 6A–D and Online Supplementary Table S3). However, treatment with CSL362 following chemotherapy significantly delayed leukemia progression (P=0.02; Figure 6E–H, Online Supplementary Table S3). The addition of huNK cells with CSL362 further delayed AML-16 progression compared with either CSL362 or huNK cells alone following chemotherapy (P=0.0008 and P=0.0004, respectively). A similar experiment was carried out with AML-17, in which the huNK cells were delivered over six doses (Online Supplementary Figure S13A) instead of two, to avoid adverse reactions associated with the combination of large numbers of huNK cells after chemotherapy. CSL362 administration after chemotherapy significantly extended the event-free survival of mice compared with the control (P=0.03; Online Supplementary Figure S13 and Online Supplementary Table S3). However, the adoptive transfer of huNK cells was unable to extend the delay in leukemia progression.
Figure 6.
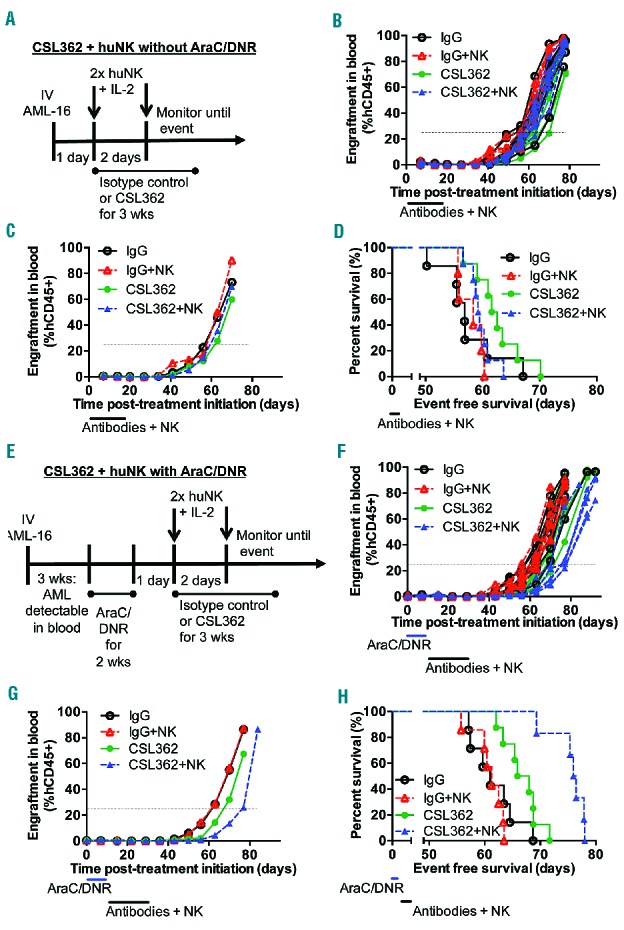
Chemotherapy enhances the efficacy of CSL362 and huNK cell against AML-16 xenografts in NSG mice. (A–D) In vivo efficacy of CSL362 and/or adoptive transfer of huNK cells against AML-16 without chemotherapy. (A) Schematic illustrating the treatment of AML-16 engrafted mice without chemotherapy. (B) AML engraftment levels in the blood of individual mice over time. (C) Median engraftment levels in the blood over time. (D) Event-free survival curves. (E–H) In vivo efficacy of CSL362 and/or adoptive transfer of huNK cells against AML-16 with chemotherapy. (E) Schematic illustrating the treatment of AML-16 engrafted mice after chemotherapy. (F) AML engraftment levels in the blood of individual mice over time. (G) Median engraftment levels in the blood over time. (H) Event-free survival curves. Blue bars mark the time of treatment with AraC/DNR (25 mg/kg AraC, and 0.63 mg/kg DNR); black bars mark the time of treatment with antibodies (300 μg/dose, three times per week) and huNK cells (20 × 106 huNK cells, 500 IU interleukin-2 per injection). Five to eight mice per group.
The AML-5 xenograft did not develop long-term resistance to CSL362
When CSL362 was tested as a single agent against AML-5-engrafted NOD/SCID mice it produced a leukemia growth delay of 16 days (P<0.01, n=6 per group) (Figure 7A,B). The expression of surface CD123 was significantly down-regulated on AML-5 cells in the blood during and after CSL362 treatments, with some recovery of CD123 expression (Figure 7C) after CSL362 was no longer binding (data not shown). These changes in CD123 expression after CSL362 treatments had been observed for other AML xenografts (data not shown).
Figure 7.
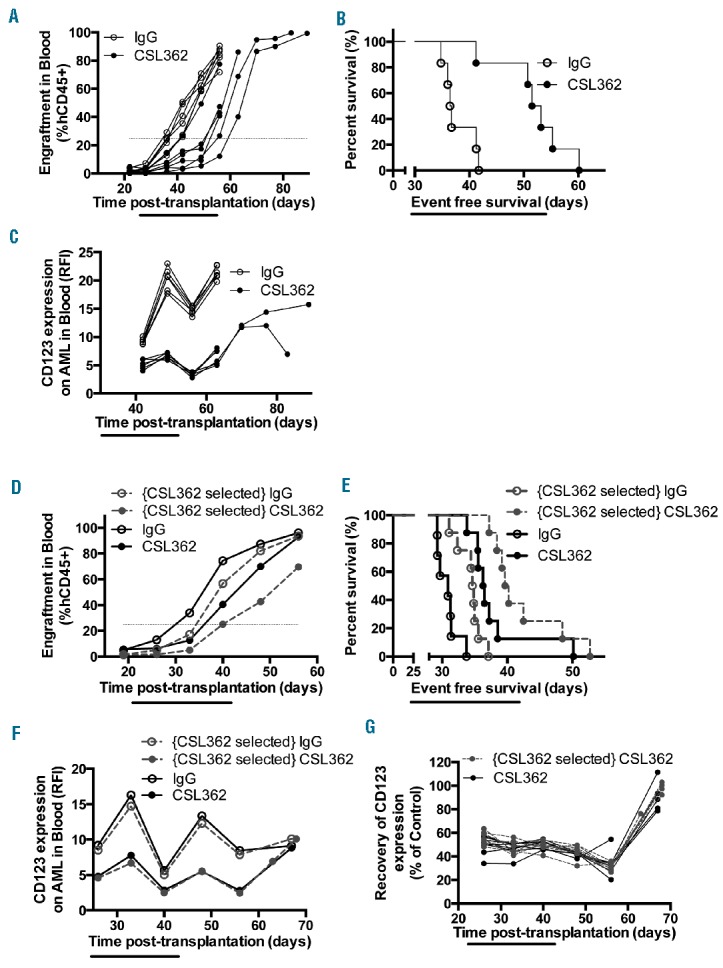
CSL362 treatment does not select for subsequent resistance. (A) Engraftment levels in the blood of individual AML-5 engrafted mice over time and (B) event-free survival curves after treatment with isotype control or CSL362. (C) Changes in surface CD123 expression on AML-5 cells in the blood of mice treated with isotype control or CSL362. (D–G) AML-5 cells were harvested from mice treated with isotype control or CSL362 (identified as “CSL362-selected” and transplanted into secondary recipient mice, then further treated with isotype control or CSL362. (D) Median AML-5 engraftment levels (7–8 mice per group) and (E) Event-free survival curves in secondary recipient mice. (F) Changes in surface CD123 expression levels on AML-5 cells in the blood of secondary recipient mice during and after treatment. (G) Recovery of surface CD123 expression levels as a % of respective isotype control on AML-5 cells in the blood of CSL362-treated mice engrafted with CSL362-selected or naïve AML-5 cells.
To investigate whether CSL362 exerted selection pressure on AML-5 based on CD123 expression, thus potentially decreasing CSL362 efficacy in regimens with multiple treatment cycles, cells from isotype control (i.e. “CSL362 naïve”) and CSL362-treated mice (identified as “CSL362-selected” in Figure 7D–G) were collected from spleens after CSL362 was no longer binding (data not shown). These two lines of AML-5 were transplanted into secondary recipient NOD/SCID mice and re-treated with isotype control or CSL362. In mice engrafted with “CSL362 naïve” and “CSL362-selected” AML-5 cells the leukemia growth delay was 5.5 days (P<0.0001, n=7–8 per group) and 5.2 days (P<0.0001, n=8 per group), respectively, when compared with their corresponding isotype controls (Figure 7D–E). CD123 expression was again consistently and statistically lower for CSL362-treated mice, regardless of the history of the cells transplanted (Figure 7F, P<0.001 when comparing CSL362-treated mice with their respective controls at all time-points before day 67, n=4–8). CD123 expression on AML-5 cells in CSL362-treated mice recovered to the same level as that in isotype controls 25 days after the last CSL362 injection (Figure 7G, P>0.33 between isotype control and CSL362-treated groups, n=4–8). These results suggest that AML-5 xenografts did not develop resistance to CSL362.
Discussion
In this study, we developed a panel of continuous AML xenografts in immune-deficient mice that produced clinically relevant disease pathology, exhibited reproducible engraftment, and responses to an induction-type AraC/DNR chemotherapy regimen that allowed combination testing with novel treatments such as CSL362. Continuous AML xenografts engrafted more rapidly and reproducibly in later passages, suggesting selection of AML subclone(s), Specific evidence for clonal selection upon xenografting was obtained by molecular analysis of candidate genes. In particular to FLT3-ITD, loss of a minor subclone (AML-5), selection of a dominant subclone from multiple subclones present at diagnosis (AML-9 and AML-16), emergence of a new subclone (AML-17), and increased mutant levels (AML-9 and AML-16) are all consistent with disease evolution from diagnosis to relapse.33–35 These observations suggest that the xenograft model selects for more aggressive, self-supporting clones of the disease.
AML xenograft cells also displayed different immunophenotypes depending on the organ of infiltration. While the relevance of this observation is not clear it highlights the potential of this model to examine subtle microenvironment-leukemia interactions. The mechanism of efficacy of novel compounds is often not fully understood during pre-clinical development, and subtle regimen changes such as providing an inhibitor before, during or after chemotherapy can increase efficacy.36 The AML xenograft models with AraC/DNR treatments provide a system for optimizing a regimen before a clinical trial. In addition, AML xenografts with specific mutations or karyo types can be used to support pre-clinical development of specific agents.
In AML patients, AraC and DNR are administered at doses from 100 and 45 mg/m2, respectively.37 The body surface area-normalized doses of AraC and DNR used in our xenograft studies were 75 and 1.9 mg/m2, respectively.38 While the dosage of AraC can be increased in mice to the human equivalent, anthracyclines are substantially more toxic in mice.39,40 The AraC/DNR regimen developed in this study produced mild and temporary side effects only, thus allowing novel agents to be combined for investigation. The regimen of AraC/DNR used was sufficient to produce remission-like conditions in the bone marrow and relevant organs in engrafted mice.
Surprisingly, this study discovered that irradiation of NSG mice impaired the engraftment of three of three established xenografts tested, in contrast to the situation with NOD/SCID mice. We reason that whole body irradiation of NSG mice may have a negative impact on the murine microenvironment that supports AML homing and engraftment. Sub-lethal irradiation of mice can be associated with adverse events, and increased toxicity of chemotherapy.39 This discovery provides a more robust and reliable model for pre-clinical testing of novel therapies.
Previous results showed that the Fc region of the anti-CD123 antibody 7G3 was important in mediating efficacy8 and huNK cells were primarily responsible for this effect.15 This is supported by the decrease in CSL362 efficacy against the AML-5 xenograft in NSG mice when compared with NOD/SCID mice. The in vitro ADCC assays verified the activity of expanded huNK cells and their role in mediating CSL362-directed anti-leukemic effects. Interestingly, huNK cells alone were able to kill AML-18 cells in vitro but the addition of CSL362 had no further effect; future examination of AML-18 and other AML samples might reveal biomarkers predictive of CSL362 responses. We attempted to complement the lack of functional immune cells in NOD/SCID and NSG mice30,31,41 by adoptive transfer of huNK cells, but this did not consistently improve in vivo efficacy of CSL362. This results was similar to that of a clinical study in which patients with metastatic melanoma or renal cell carcinoma received large doses of huNK cells with no clear benefit.42 More sophisticated methodology, such as the transplantation of fetal thymic, liver tissue with hematopoietic stem cells into NSG mice, will produce a more functional human immune system,43 although it will be technically challenging to incorporate AML xenografts into such a humanized model. Despite the relative lack of immunity in NOD/SCID mice, CSL362 significantly reduced the leukemic burden in the peripheral blood, spleens and livers of three out of four AML xenografts tested, but only significantly decreased bone marrow disease in one out of four xenografts. It is unclear whether the process of establishing continuous AML xenografts affected the individual responses of these AML cells to CSL362. Moreover, we cannot exclude the possibility that the results obtained may have been influenced by the aggressive nature of the primary AML samples studied.
To assess CSL362 side effects, cynomolgus monkeys (n=12) were treated with a single dose of CSL362 (0.01–10 mg/kg). CD123 expressing basophils and dendritic cells were depleted within 6 h and subsequently recovered 4 weeks after treatment with no other significant side effects.15 Furthermore, our model indicated that AML cells do not develop long-term resistance to CSL362. These findings strongly support further examination of CSL362 in the current clinical trial in remission AML patients.
In this study we showed that the %hCD45+ cells in the murine peripheral blood provided a surrogate indicator of overall disease burden in the animals, and that the disease would result in mortality if allowed to progress. It is, therefore, important to acknowledge that measurements of mouse event-free survival in all efficacy experiments used a pre-defined endpoint of 25% hCD45+ cells in the peripheral blood or an animal exhibiting signs of leukemia-related morbidity, and event-free survival data should be interpreted accordingly.
For some AML xenografts, CSL362 alone did not improve mouse event-free survival. However, when treatments were administered after chemotherapy, CSL362 prolonged event-free survival to a modest extent for AML-16- and AML-17-engrafted mice. Bone marrow mesenchymal stem cells were reported to suppress the activity of NK cells against K562 cells in a mouse model44 and the chemotherapy in the present study may have affected these bone marrow cells, decreasing immunosuppression. Alternatively, since AraC treatment in pediatric patients increased levels of the pro-inflammatory cytokines tumor necrosis factor-α, interleukin-6 and interferon-γ,45 and similarly, doxorubicin increased the levels of interleukin-1β in mice46 and interleukin-1β, tumor necrosis factor-α and interleukin-6 in murine macrophages in vitro, the chemotherapy in this study may have generated a proinflammatory environment that potentiated the effects of the huNK cells and CSL362. Interestingly, the addition of huNK cells to CSL362 after chemotherapy prolonged the event-free survival of AML-16, but not of AML-17-engrafted mice. This result may reflect the ability of different AML to interact with the microenvironment to induce immunosuppression, or cytoprotection since the huNK cells were able to lyse a substantial proportion of AML-17 cells in vitro. HuNK cell adoptive transfer has been investigated in human patients with limited survival benefits.42,47 The use of antibody with NK cells increased ADCC or activated NK cells,48,49 and our current results supported this, but the mechanism responsible for the anti-leukemic effects of the CSL362 and huNK combination after chemotherapy, and the role of the microenvironment require further elucidation.
In conclusion, this AML xenograft model provides a platform for the testing of new therapeutics and combinations, and is especially crucial for heterogeneous diseases such as AML, in which the probability of developing resistance to a single agent is high.50 The combination of chemotherapy, CSL362 and huNK cell adoptive transfer is novel and produced some evidence of in vivo efficacy; CSL362 will be further examined in the current phase I clinical trial.
Acknowledgments
The authors wish to thank Dr. Daniel Thomas (Division of Human Immunology, Centre for Cancer Biology, Adelaide, Australia) and Dr. Andrew Roberts (Royal Melbourne Hospital, Melbourne, Australia) for provision of patients’ biopsy samples; Soo Min Heng, Dean Inwood, and Simon Downes (Radiation Oncology Department, Prince of Wales Hospital, Sydney, Australia) for mouse irradiation. The EBV transformed B lymphoblastoid cell line (SMI-LCL) was a kind gift from Capt. Richard Childs (National Institutes of Health), and K562-mIL15-41BBL was generously provided by Dr. Dario Campana (St. Jude Children’s Research Hospital, Memphis, TN, USA).
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by the Children’s Cancer Institute Australia for Medical Research and by CSL Limited. The Children’s Cancer Institute Australia for Medical Research is affiliated with the University of New South Wales and the Sydney Children’s Hospitals Network. HSR is supported by The Peter Nelson Leukaemia Research Fund. AFL is supported by the National Health and Medical Research Council of Australia and Cancer Australia. RBL is supported by a fellowship from the National Health and Medical Research Council of Australia.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113(18):4179–4187. [DOI] [PubMed] [Google Scholar]
- 2.Farag SS, Archer KJ, Mrozek K, et al. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: results from Cancer and Leukemia Group B 8461. Blood. 2006;108(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ossenkoppele GJ, Graveland WJ, Sonneveld P, et al. The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood. 2004;103(8):2908–2913. [DOI] [PubMed] [Google Scholar]
- 4.List AF, Kopecky KJ, Willman CL, et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2001;98(12):3212–3220. [DOI] [PubMed] [Google Scholar]
- 5.Becton D, Dahl GV, Ravindranath Y, et al. Randomized use of cyclosporin A (CsA) to modulate P-glycoprotein in children with AML in remission: Pediatric Oncology Group Study 9421. Blood. 2006;107(4):1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cripe LD, Uno H, Paietta EM, et al. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116(20): 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauswirth AW, Florian S, Printz D, et al. Expression of the target receptor CD33 in CD34+/CD38−/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73–82. [DOI] [PubMed] [Google Scholar]
- 8.Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. [DOI] [PubMed] [Google Scholar]
- 9.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–1784. [DOI] [PubMed] [Google Scholar]
- 10.Munoz L, Nomdedeu JF, Lopez O, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86(12):1261–1269. [PubMed] [Google Scholar]
- 11.Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–2988. [DOI] [PubMed] [Google Scholar]
- 12.Sun Q, Woodcock JM, Rapoport A, et al. Monoclonal antibody 7G3 recognizes the N-terminal domain of the human interleukin-3 (IL-3) receptor alpha-chain and functions as a specific IL-3 receptor antagonist. Blood. 1996;87(1):83–92. [PubMed] [Google Scholar]
- 13.Roberts AW, He S, Bradstock KF, et al. A Phase 1 and correlative biological study of CSL360 (anti-CD123 mAb) in AML. ASH Annual Meeting Abstracts, 2008:2956. [Google Scholar]
- 14.Lazar GA, Dang W, Karki S, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. 2006;103(11):4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213–2221. [DOI] [PubMed] [Google Scholar]
- 16.Lanier LL, Le AM, Phillips JH, Warner NL, Babcock GF. Subpopulations of human natural killer cells defined by expression of the Leu-7 (HNK-1) and Leu-11 (NK-15) antigens. J Immunol. 1983;131(4):1789–1796. [PubMed] [Google Scholar]
- 17.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–393. [DOI] [PubMed] [Google Scholar]
- 18.Hsu KC, Gooley T, Malkki M, et al. KIR ligands and prediction of relapse after unrelated donor hematopoietic cell transplantation for hematologic malignancy. Biol Blood Marrow Transplant. 2006;12(8):828–836. [DOI] [PubMed] [Google Scholar]
- 19.Scquizzato E, Zambello R, Teramo A, et al. KIR/HLA-I mismatching and risk of relapse in paediatric patients undergoing non-haploidentical allogeneic haematopoietic stem cell transplantation. Pediatr Transplant. 2011;15(2):198–204. [DOI] [PubMed] [Google Scholar]
- 20.Giebel S, Locatelli F, Lamparelli T, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood. 2003;102(3):814–819. [DOI] [PubMed] [Google Scholar]
- 21.Multhoff G, Pfister K, Botzler C, et al. Adoptive transfer of human natural killer cells in mice with severe combined immunodeficiency inhibits growth of Hsp70-expressing tumors. Int J Cancer. 2000;88(5):791–797. [DOI] [PubMed] [Google Scholar]
- 22.Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegler U, Kalberer CP, Nowbakht P, Sendelov S, Meyer-Monard S, Wodnar-Filipowicz A. Activated natural killer cells from patients with acute myeloid leukemia are cytotoxic against autologous leukemic blasts in NOD/SCID mice. Leukemia. 2005;19(12):2215–2222. [DOI] [PubMed] [Google Scholar]
- 24.Berg M, Lundqvist A, McCoy P, Jr, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11(3):341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lock RB, Liem N, Farnsworth ML, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood. 2002;99(11):4100–4108. [DOI] [PubMed] [Google Scholar]
- 26.Murphy KM, Levis M, Hafez MJ, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5(2):96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Q, Chen W, Gaal KK, Slovak ML, Stein A, Weiss LM. A rapid, one step assay for simultaneous detection of FLT3/ITD and NPM1 mutations in AML with normal cytogenetics. Br J Haematol. 2008;142(3):489–492. [DOI] [PubMed] [Google Scholar]
- 28.Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009;Chapter 2:Unit 2 12. [DOI] [PubMed] [Google Scholar]
- 29.Warren HS, Rana PM. An economical adaptation of the RosetteSep procedure for NK cell enrichment from whole blood, and its use with liquid nitrogen stored peripheral blood mononuclear cells. J Immunol Methods. 2003;280(1–2):135–138. [DOI] [PubMed] [Google Scholar]
- 30.Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9): 3175–3182. [DOI] [PubMed] [Google Scholar]
- 31.Shultz LD, Schweitzer PA, Christianson SW, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154(1):180–191. [PubMed] [Google Scholar]
- 32.Sanchez PV, Perry RL, Sarry JE, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23(11): 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111(5):2776–2784. [DOI] [PubMed] [Google Scholar]
- 34.Shih LY, Huang CF, Wu JH, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100(7):2387–2392. [DOI] [PubMed] [Google Scholar]
- 35.Stirewalt DL, Kopecky KJ, Meshinchi S, et al. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood. 2006;107(9): 3724–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104(4):1145–1150. [DOI] [PubMed] [Google Scholar]
- 37.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368(9550):1894–1907. [DOI] [PubMed] [Google Scholar]
- 38.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2008;22(3):659–661. [DOI] [PubMed] [Google Scholar]
- 39.Wunderlich M, Mizukawa B, Chou FS, et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121(12):e90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuber J, Radtke I, Pardee TS, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23(7):877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–6489. [DOI] [PubMed] [Google Scholar]
- 42.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autolo-gous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17(19):6287–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Covassin L, Jangalwe S, Jouvet N, et al. Human immune system development and survival of non-obese diabetic (NOD)-scid IL2rgamma(null) (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin Exp Immunol. 2013;174(3):372–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Qu YH, Wu YF, et al. Bone marrow mesenchymal stem cells reduce the antitumor activity of cytokine-induced killer/natural killer cells in K562 NOD/SCID mice. Ann Hematol. 2011;90(8):873–885. [DOI] [PubMed] [Google Scholar]
- 45.Ek T, Jarfelt M, Mellander L, Abrahamsson J. Proinflammatory cytokines mediate the systemic inflammatory response associated with high-dose cytarabine treatment in children. Med Pediatr Oncol. 2001;37(5):459–464. [DOI] [PubMed] [Google Scholar]
- 46.Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11(12):1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stern M, Passweg JR, Meyer-Monard S, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant. 2013;48(3):433–438. [DOI] [PubMed] [Google Scholar]
- 48.Chan WK, Kung Sutherland M, Li Y, Zalevsky J, Schell S, Leung W. Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin Cancer Res. 2012;18(22):6296–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. 2007;7(5): 329–339. [DOI] [PubMed] [Google Scholar]
- 50.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]