

Abstract
Hemophagocytic lymphohistiocytosis is a hyperinflammatory syndrome defined by clinical and laboratory criteria. Current criteria were created to identify patients with familial hemophagocytic lmyphohistiocytosis in immediate need of immunosuppressive therapy. However, these criteria also identify patients with infection-associated hemophagocytic inflammatory states lacking genetic defects typically predisposing to hemophagocytic lymphohistiocytosis. These patients include those with primary immunodeficiencies, in whom the pathogenesis of the inflammatory syndrome may be distinctive and aggressive immunosuppression is contraindicated. To better characterize hemophagocytic inflammation associated with immunodeficiencies, we combined an international survey with a literature search and identified 63 patients with primary immunodeficiencies other than cytotoxicity defects or X-linked lymphoproliferative disorders, presenting with conditions fulfilling current criteria for hemophagocytic lymphohistiocytosis. Twelve patients had severe combined immunodeficiency with <100/μL T cells, 18 had partial T-cell deficiencies; episodes of hemophagocytic lymphohistiocytosis were mostly associated with viral infections. Twenty-two patients had chronic granulomatous disease with hemophagocytic episodes mainly associated with bacterial infections. Compared to patients with cytotoxicity defects, patients with T-cell deficiencies had lower levels of soluble CD25 and higher ferritin concentrations. Other criteria for hemophagocytoc lymphohistiocytosis were not discriminative. Thus: (i) a hemophagocytic inflammatory syndrome fulfilling criteria for hemophagocytic lymphohistiocytosis can be the initial manifestation of primary immunodeficiencies; (ii) this syndrome can develop despite severe deficiency of T and NK cells, implying that the pathophysiology is distinct and not appropriately described as “lympho”-histiocytosis in these patients; and (iii) current criteria for hemophagocytoc lymphohistiocytosis are insufficient to differentiate hemophagocytic inflammatory syndromes with different pathogeneses. This is important because of implications for therapy, in particular for protocols targeting T cells.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome. The term was initially coined based on histomorphological features.1 The hereditary disorders, of which the HLH syndrome is the defining clinical manifestation, have been associated with autosomal recessive mutations in genes encoding perforin (familial hemophagocytic lymphohistiocytosis, FHL-2) and a group of proteins required for secretion of perforin-containing cytotoxic granules (FHL 3–5, Griscelli syndrome type 2 and Chediak-Higashi syndrome).1 HLH is also a frequent manifestation of some defined genetic disorders of Epstein-Barr virus (EBV) susceptibility, e.g. X-linked lymphoproliferative syndromes (XLP1 and XLP2).2 Impaired lymphocyte cytotoxicity with highly activated, but inefficient T cells are the main pathogenic factors in the former group of disorders,3 while the pathophysiological basis of HLH in XLP and other syndromes of EBV susceptibility is less well understood.
As defined by the Histiocyte Society, the diagnosis of HLH syndrome is based on fulfillment of five out of eight clinical and laboratory parameters or a molecular diagnosis of a disease conferring a high risk of developing HLH. These criteria have been useful for the identification of patients with genetic defects in lymphocyte cytotoxicity. However, they are also fulfilled by a range of other patients presenting with hemophagocytic inflammatory disease, but normal cytotoxicity. Thus, the HLH syndrome can manifest in the context of severe infections including viral infections or sepsis/systemic inflammatory response syndrome, autoimmune and autoinflammatory diseases or malignancies such as lymphomas.4–6 These disease states are frequently summarized as “secondary HLH”, “acquired HLH” or “macrophage activation syndrome”. Affected patients usually present with clinical and laboratory manifestations that cannot be readily distinguished from those observed in patients with defects in cytotoxicity.4–6 However, the fact that defective cytotoxicity cannot be consistently found in such patients suggests that the pathophysiological pathways leading to HLH syndrome may differ between different groups of patients.5
Current treatment guidelines based on the HLH-2004 study of the Histiocyte Society recommend that HLH-directed therapy should be strongly considered if five out of eight diagnostic criteria are fulfilled, irrespective of whether they occur in the presence of defects in lymphocyte cytotoxicity or in other forms of the disease.7 While there is no doubt that this therapy can be life-saving in patients with FHL and many instances of infection-associated HLH,8 less intensive anti-inflammatory treatment is frequently sufficient for patients with other forms of hemophagocytic inflammatory disease and aggressive immunosuppression may even be contraindicated.1,9 Moreover, more specific therapies for HLH targeting T cells9 or interferon-γ3,10 are undergoing prospective evaluation. Potential differences in the pathogenic events leading to HLH syndrome are, therefore, becoming increasingly relevant.
One well-defined group of patients in whom the HLH syndrome has been described, are patients with primary immunodeficiencies (PID) other than FHL or XLP. Single cases or small case series of HLH syndrome have been reported in a variety of PID, and the clinical presentation of some of these cases has recently been summarized.11 However, a multicenter systematic analysis of the clinical and laboratory features of HLH syndrome in these patients in comparison to HLH associated with defects in lymphocyte cytotoxicity has not been performed. We reasoned that such an analysis might offer the opportunity to identify parameters for differential diagnosis, facilitating early identification of patients with hemophagocytic inflammatory disease in whom aggressive immunosuppressive therapy may be contraindicated. Furthermore, since PID provide an excellent possibility to study the role of constitutional immunological abnormalities in immunopathological conditions, we expected to gain some insights relevant to the pathogenesis of HLH syndrome.
Methods
Recruitment of patients
To identify patients with PID other than disorders of cytotoxicity or XLP and a clinical presentation with a disease state fulfilling the HLH-2004 criteria for HLH, we performed a survey among centers involved in the diagnosis and treatment of PID and/or HLH through the Histiocyte Society, the European Society of Blood and Bone Marrow Transplantation’s Inborn Errors Working Party and the German Society for Pediatric Oncology and Hematology. These data were supplemented by a literature review based on a PubMed search for “hemophagocytic lymphohistiocytosis”, “macrophage activation syndrome”, and “immunodeficiency” or individual immunodeficiencies such as “chronic granulomatous disease” (CGD) up to December 31st, 2014. All cases were reviewed by SFNB and SE. Patients were included if they fulfilled the following criteria: (i) genetic or clinical diagnosis of a defined PID as classified by the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency (IUIS) except for genetic defects in cytotoxicity, XLP1 or XLP2;12 and (ii) presence of at least five out of eight diagnostic criteria for HLH according to the HLH-2004 criteria.7 These eight criteria are: (i) fever; (ii) splenomegaly; (iii) cytopenia of two or more cell lines (hemoglobin ≤90 g/L, platelets ≤100×109/L, neutrophils ≤1×109/L); (iv) hypofibrinogenemia (≤1.5 g/L) or hypertriglyceridemia (≥265 mg/dL); (v) hyperferritinemia (≥500 ng/mL); (vi) increased level of soluble CD25 (sCD25, ≥2400 U/mL); (vii) evidence of hemophagocytosis; and (viii) decreased or absent NK cell cytotoxicity. From all patients, we recorded the underlying immunodeficiency diagnosis, the individual diagnostic criteria for HLH, associated infections, treatment and outcome of the HLH episode and whether or not HLH developed at/before or after the diagnosis of PID.
Control groups
Laboratory values of these patients were compared to those observed in a cohort of patients with active “primary” HLH (FHL 2–5; n=90) and a cohort of patients with infection-associated “secondary” HLH (I-HLH; n=40) without another known underlying disease such as malignancy, rheumatological or metabolic disease from the German HLH registry. This registry collects clinical, immunological and genetic information on patients referred for evaluation for HLH from German speaking countries. Around 100 patients are referred each year and around 60 of these fulfill the diagnostic criteria for HLH. The group of patients with I-HLH consisted of patients who: (i) fulfilled at least five of the eight diagnostic criteria for HLH; (ii) had an infection proven by positive polymerase chain reaction or unequivocal serology at the time of manifesting HLH; (iii) did not have either a mutation in HLH-related genes (if investigated) or another obvious underlying genetic disease or a positive family history for HLH; and (iv) had not had a disease relapse within at least 12 months after the HLH manifestation.13 About 20% of the patients in this group were thoroughly evaluated for mutations in HLH-causing genes because of ambiguous or abnormal results in the functional analysis.
Statistical analysis
Statistical analysis was performed with GraphPad InStat software version 6.05. First we calculated the natural logarithms of the means of sCD25, ferritin, fibrinogen, and triglyceride levels. Differences between group means were then evaluated with one-way analysis of variance with a post-hoc Tukey test. Differences were considered statistically significant at P values <0.05.
Ethics
The study was approved by the ethics committee of the Albert-Ludwigs-Universität Freiburg (AK28/14) and conducted according to the Declaration of Helsinki.
Results
Patients and sequence of diagnosis
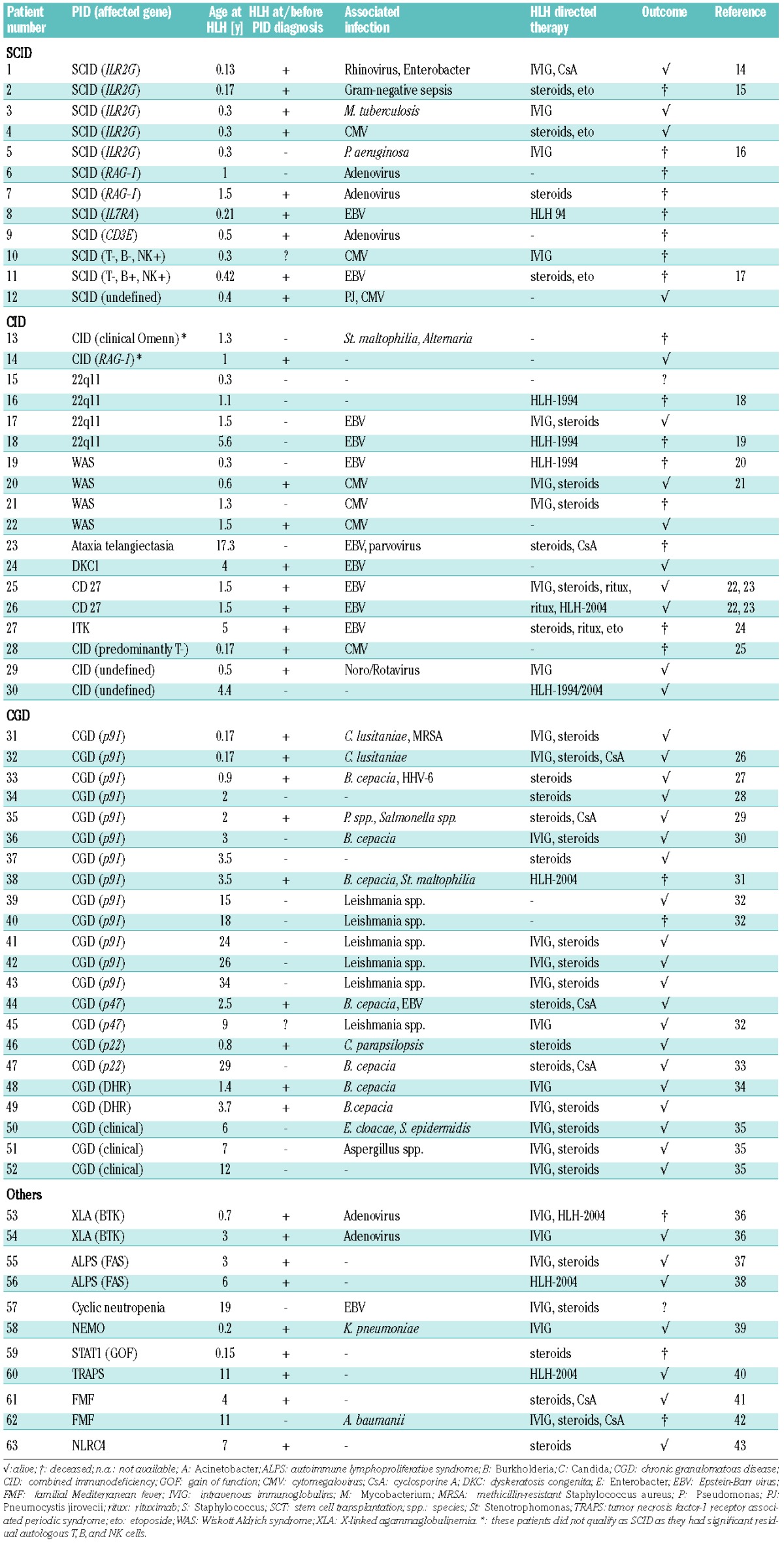
We identified 28 patients with PID other than defects in cytotoxicity or XLP fulfilling the HLH-2004 diagnostic criteria through the survey and found 30 publications describing another 35 cases (Table 1).14–43 Six of the 18 cases recently summarized by Faitelson11 were not included in this analysis because of incomplete information on our inclusion criteria as were ten cases identified by either the survey or the literature review. In total, we identified 63 patients with PID and an HLH syndrome. The median age at diagnosis of the HLH syndrome was 1.5 years (range 0.13–34 years). Notably, an HLH syndrome was the initial presentation leading to diagnosis of the underlying PID in 36 patients, whereas 25 patients developed HLH after the diagnosis of PID had been established; for two patients this information was not available.
Table 1.
Cohort of patients with HLH syndrome in the context of primary immunodeficiencies.
Spectrum of primary immunodeficiencies presenting with hemophagocytic lymphohistiocytosis syndrome
More than 80% of patients had two main groups of PID. Thirty patients had combined immunodeficiencies (CID). More specifically, 12 patients had SCID with a variety of molecular causes (Table 1). Interestingly, in nine of these 12 patients, the HLH syndrome developed in the first months of life, at or before the diagnosis of the immunodeficiency. Eighteen patients had other well-characterized CID affecting T-cell development or function, including “atypical” SCID44 with significant numbers of autologous T cells (n=2), 22q11 deletion syndrome (n=4), Wiskott-Aldrich syndrome (n=4), ataxia telangiectasia (n=1) and dyskeratosis congenita (n=1). Two patients had CD27 deficiency and one had ITK deficiency, conditions that have previously been associated with poor control of EBV infection (Table 1).45,46 The second main group consisted of 22 patients with chronic granulomatous disease (CGD) (Table 1). Most of these patients presented with HLH syndrome beyond the first year of life, some in adulthood. In nine of the younger patients (<4 years), the HLH syndrome developed at or before the diagnosis of CGD. Only 11 patients had other PID, including antibody deficiencies (n=2), diseases of immune dysregulation (n=2), other congenital defects of phagocytes (n=1), defects in innate immunity (n=2), or autoinflammatory disorders (n=4) (Table 1).
Infections associated with hemophagocytic lymphohistiocytosis syndrome in patients with primary immunodeficiencies
In 50/63 patients (79%), the HLH syndrome was associated with an infection. In SCID and CID patients, it was mainly associated with viral infections. Ten of 30 (33%) patients had EBV infection, seven had cytomegalovirus infection, three had infections with adenovirus and two had infections with other gastrointestinal or respiratory viruses. In contrast, in only two of the 22 (9%) CGD patients was the HLH syndrome associated with a viral infection and in both cases an additional bacterial infection was diagnosed. The main infectious agents in the group of CGD patients were Burkholderia cepacia (n=7), Leishmania spp. (n=6), and fungi (n=4). In the group of 11 patients with other PID, the HLH syndrome was associated with a viral infection in three and a bacterial infection in two patients (Table 1).
Lymphocyte compartment in patients with severe combined immunodeficiency
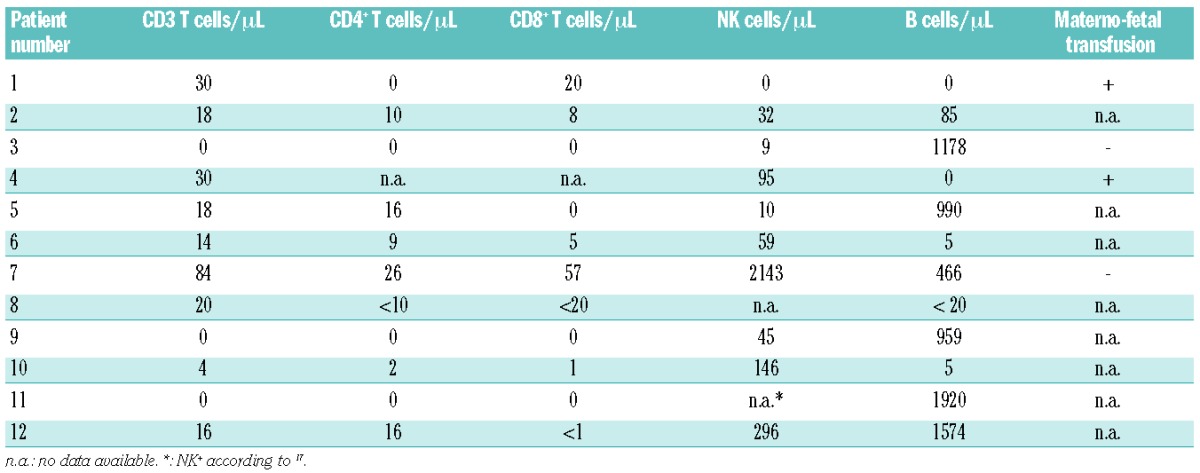
Since activated T cells and possibly NK cells are regarded as major effectors in the pathogenesis of FHL,1,3 we had a closer look at the lymphocyte compartment in SCID patients. Interestingly, all SCID patients had fewer than 100 T cells/μL (including 9 patients with fewer than 20 T cells/μL) (Table 2). Four patients had been analyzed for maternal T cells, which were detected at low numbers in two of them. Three patients also had 10 or fewer NK cells/μL. The number of B cells was more variable.
Table 2.
Lymphocyte subsets in patients with SCID and hemophagocytic inflammatory syndrome.
Diagnostic parameters in patients with primary immunodeficiencies presenting with hemophagocytic lymphohistiocytosis syndrome
Information on all eight HLH-2004 diagnostic criteria was only available for seven patients, while seven criteria were reported in an additional 15 patients. Information was usually lacking on sCD25 (reported in 23/63 patients, 36%) or NK cell cytotoxicity (reported in 19/63 patients, 30%). On the other hand, all patients fulfilled at least five diagnostic criteria. In 36 patients (57%), all of the five to eight reported criteria were positive. When looking at individual criteria, no particular pattern emerged that was characteristic for PID or PID subgroups. Of the patients for whom information on these criteria was available, 98% had fever, 91% had splenomegaly, 91% had cytopenias, 80% had elevated triglycerides, 75% had low fibrinogen, 100% had elevated ferritin, 82% had elevated sCD25 and 63% had low or absent NK cell cytotoxicity.
We then quantitatively compared the laboratory values of patients with T-cell deficiencies and CGD to those obtained in a cohort of patients with FHL (n=90) and a cohort of patients with infection-triggered “secondary” HLH (I-HLH) (n=40). The most significant differences were observed in the serum concentrations of soluble interleukin-2 receptor (sCD25) (Figure 1A, Table 3). Patients with T-cell deficiencies had significantly lower values than those observed in patients with FHL (mean: T-cell deficiencies: 5290 U/mL, FHL: 23074 U/mL; P<0.0001). Notably, levels were also lower in patients with I-HLH than in patients with FHL. In contrast, the concentrations of ferritin were higher in the patients with T-cell deficiencies than in the other three groups of patients, but only significantly higher than those in the FHL patients (P<0.05; mean: T-cell deficiencies: 64808 ng/mL, FHL: 10154 ng/mL, I-HLH: 8359 ng/mL, CGD 8819 ng/mL; Figure 1B). These differences were even more obvious, when the ratio of ferritin/sCD25 was determined, which was significantly higher in patients with T-cell deficiencies than in FHL, I-HLH, and CGD patients (mean: T-cell deficiencies: 14.18, FHL: 0.37, I-HLH: 0.48, CGD: 0.90; Figure 1C; see figure for P-values). Apart from a significant difference in fibrinogen levels between FHL and I-HLH patients, no relevant differences were observed in the other HLH-defining parameters (fibrinogen: mean FHL: 1.218 g/L, I-HLH: 1.869 g/L; P<0.05; Figure 2).
Figure 1.
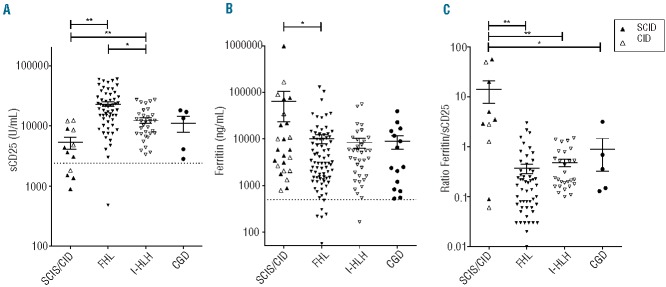
(A) Serum sCD25 levels in patients with SCID/CID, FHL, I-HLH (infection triggered “secondary” HLH), and CGD. (B) Serum ferritin levels in SCID/CID, FHL, I-HLH, and CGD patients. (C) Ratio of ferritin/sCD25 in SCID/CID, FHL, I-HLH, and CGD patients. *P<0.05. **P<0.01. The dotted line indicates the cut-off value according to the HLH diagnostic criteria. The bars indicate means ± standard deviation.
Table 3.
HLH diagnostic criteria in patients with PID and hemophagocytic inflammatory syndrome.
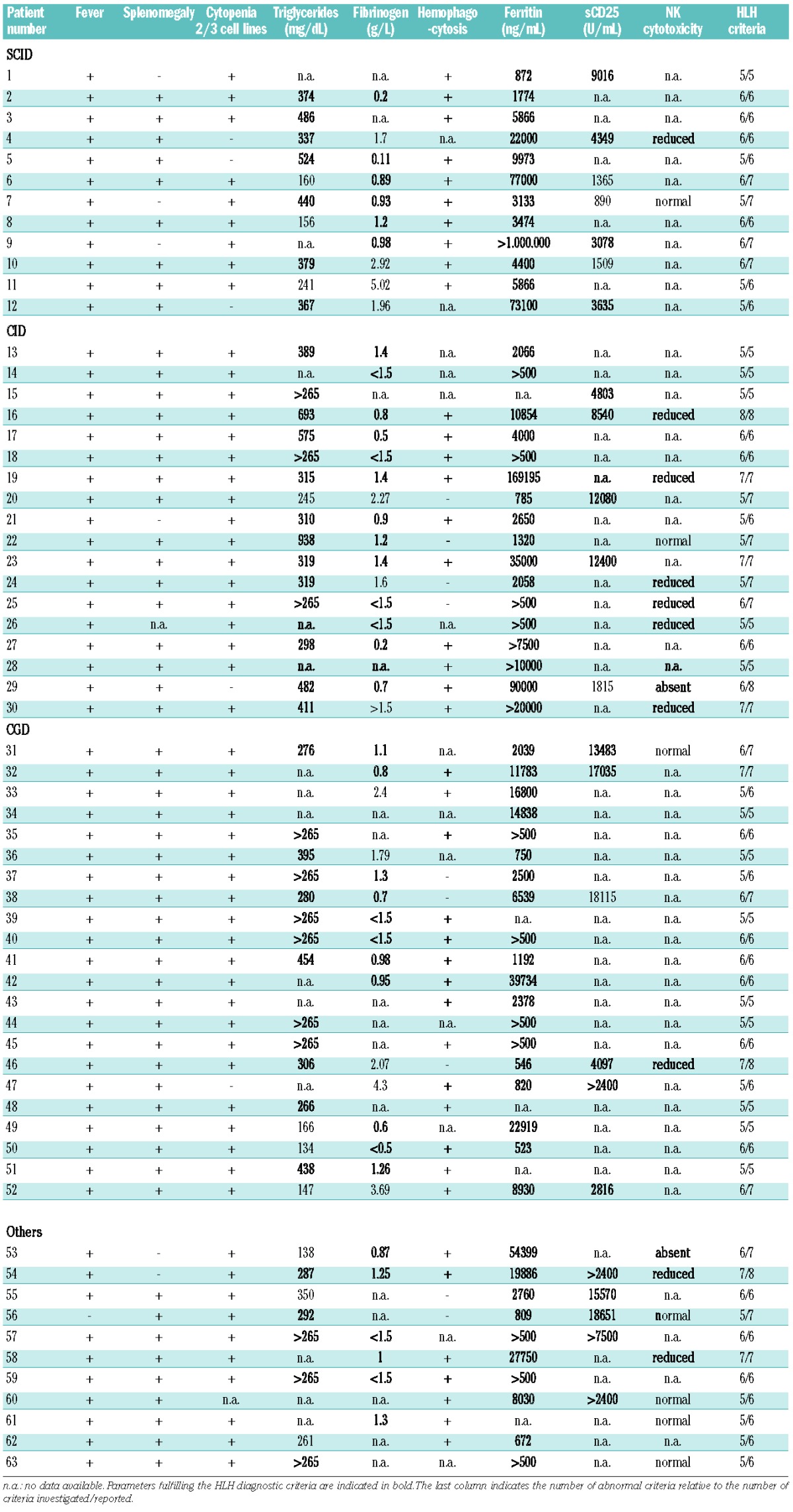
Figure 2.
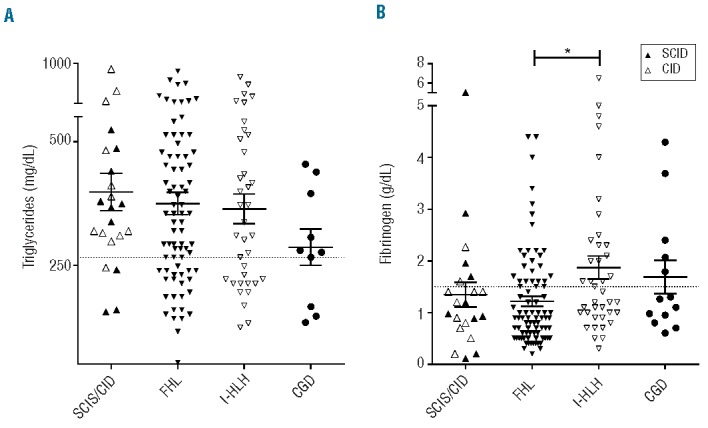
(A) Serum triglyceride levels in SCID/CID, FHL, I-HLH, and CGD patients. (B) Serum fibrinogen levels in SCID/CID, FHL, I-HLH, and CGD patients. *P<0.05. The dotted line indicates the cut-off value according to the HLH diagnostic criteria. The bars indicate means ± standard deviation.
Treatment and outcome
The patients received a wide range of HLH-directed treatment regimens, including corticosteroids alone or in combination with intravenous immunoglobulins, cyclosporine or rituximab (Table 1). Etoposide-based therapy was given to 14 patients, including ten patients with T-cell deficiencies, six of whom received their PID diagnosis during or after the episode of HLH syndrome. Only one patient with CGD received etoposide-based therapy. Thirty-nine (65%) of 60 patients with reported outcome survived the episode of HLH syndrome. Of note, only four of 12 SCID patients and nine of 18 patients with CID and reported outcome survived, which contrasted with 20 of 22 CGD patients.
Discussion
This study identified 63 patients with PID other than genetic disorders of cytotoxicity or XLP, who fulfilled the current clinical criteria for HLH. In 36 of them, this HLH syndrome was the initial manifestation before or at the diagnosis of PID. PID other than disorders of cytotoxicity or XLP are, therefore, a relevant differential diagnosis in patients presenting with the clinical picture of HLH syndrome.
While many PID may predispose to an infection-triggered HLH syndrome, this was largely restricted to two groups in this cohort: patients with T-cell deficiencies, who are susceptible to a broad range of infections, viral infections in particular, and patients with CGD, who are susceptible to a narrower spectrum of bacterial and opportunistic, but not viral, infections. Infections associated with the HLH syndrome in PID patients reflected this specific susceptibility, compatible with the notion that uncontrolled pathogen replication is a major risk factor for this hyperinflammatory syndrome. However, while HLH syndrome was observed in a relevant number of CGD patients, it was not reported in other congenital defects in phagocytes and only rarely in other defects in innate immunity. It is, therefore, tempting to hypothesize that HLH syndrome in CGD may not only reflect impaired infection control, but also a genetic predisposition to hyperinflammation. Colitis and other inflammatory organ pathologies associated with granuloma formation are well-known complications in this disease,47 which were recently linked to autophagic dysfunction.48 This apparent association of impaired autophagy with HLH syndrome warrants future investigation. It should be noted that the design of our study did not allow us to exclude a potential contribution of monoallelic mutations or polymorphisms in FHL-associated genes to the manifestation of HLH in all patients. Nevertheless, at least seven patients were tested for all known genes associated with FHL and in a number of other patients individual genes were sequenced without detection of relevant mutations or polymorphisms such as perforin A91V.
An imbalance between viral control and immune activation is thought to be an important determinant of the HLH syndrome associated with viral infections (such as EBV) and in a proportion of patients with FHL. This is supported by evidence from mouse models.3,49 In these models, hyperactive T cells are a key factor in disease pathogenesis.2,50 It was, therefore, surprising that, paradoxically, a significant number of patients with T-cell deficiencies, including several patients without detectable circulating T cells, developed the HLH syndrome. Notably, at least three T-cell deficient X-SCID patients also had ≤10/μL NK cells, rendering it unlikely that hyperactivated NK cells replaced T cells as a pathogenic factor in these cases. This is further supported by a significantly lower level of sCD25 in this group of patients compared to patients with FHL or I-HLH. These observations suggest that the HLH syndrome can develop despite severe deficiency of T and NK cells, two main effectors implicated in “primary” HLH. Due to this absence of T and NK cells, the term hemophagocytic “lympho”-histiocytosis appears inappropriate to describe the “HLH-like” disease in SCID patients. It seems likely that in these patients activation of macrophages and the associated cytokine release proceed independently of lymphocytes. The term “hemophagocytic inflammatory syndrome” (HIS) may, therefore, better describe this condition. Although not addressed in this study, it is possible that such a distinction may also be relevant for some other disease states currently classified as “secondary” HLH.
The current HLH-2004 criteria were insufficient for this discrimination. Neither the number nor the combination of HLH-2004 criteria fulfilled in patients with PID and HIS was different from what is usually observed in “primary” or infection-associated “secondary” HLH. It should be stated that in most PID patients summarized in this study, not all eight criteria were documented. However, sCD25 is frequently also not determined in many patients with familial HLH and in most centers NK cell cytotoxicity testing has been replaced by intracellular protein and degranulation studies.51 The only notable differences between the two groups were the relatively low sCD25 levels and high ferritin levels in most patients with T-cell deficiencies. This most likely reflects the lack of T cells in SCID and the low T-cell counts and/or impaired T-cell function in CID patients. Furthermore, the ratio of ferritin to sCD25 heightened these differences and appears to be a useful measure, as has previously been suggested in lymphoma-associated HLH.52 Our (admittedly limited) data suggest that a ferritin:sCD25 ratio of ≥3 should be viewed as suggestive of SCID/CID in an infant with the HLH syndrome. Because current understanding of HLH pathogenesis suggests that “overactivated” T cells exuberantly recruit innate effectors (macrophages) to drive HLH disease manifestations,3,53 these findings suggest that uncon trolled viral infections in the context of SCID/CID may circumvent the typical HLH pathogenesis. It remains to be shown whether such an alternative pathophysiology may also be relevant for some cases of infection-associated or rheumatologic “secondary” HLH in apparently immunocompetent individuals. In this context it is noteworthy that sCD25 levels were also significantly lower in patients with infection-associated “secondary” HLH than in patients with FHL, though this may simply illustrate the propensity towards T-cell ‘overactivation’ in individuals with defective cytotoxic function.
Disease classifications should be based on pathophysiological knowledge with the goal of facilitating the best treatment decisions for affected patients. The observations of this study further illustrate that the current diagnostic HLH criteria defined by the HLH-2004 protocol describe a common clinical and biological endpoint resulting from different conditions and pathways of pathogenesis. We think that there are good arguments to reserve the term hemophagocytic “lympho”-histiocytosis (HLH) to disease states that are associated with a predominantly T-cell-mediated immunopathogenesis, including “primary” HLH and some cases of infection-associated “secondary” HLH. In these cases, etoposide-based protocols or T-cell-directed immune therapy, e.g. based on antithymocyte globulin9 or alemtuzumab,54 are indicated and can be highly effective. In contrast, strong immunosuppressive therapy may not always be appropriate and may even be harmful for other groups of patients, especially those with PID other than FHL or XLP. In this study, we propose the provisional term “hemophagocytic inflammatory syndrome” (HIS) to classify these patients, in whom the intensity of immunosuppressive treatment needs to be carefully adapted to the underlying immunodeficiency and ongoing infections.
HIS could be an umbrella term for different diseases currently referred to as “primary” or “secondary” forms of HLH or macrophage activation syndrome. According to this concept, HLH would be one form of HIS. We propose maintaining the term “hemophagocytic” despite the fact that not all patients fulfill this criterion (41/50 in our study) for the following reasons: (i) the probability of detecting hemophagocytosis depends on the time point of the analysis, which in many cases is performed only once; (ii) the element of activated phagocytes producing cytokines is likely to be essential for all forms of HIS; (iii) keeping this element will keep the term searchable in the literature and maintain awareness of physicians that this is a serious condition; and (iv) the term hyperinflammatory alone is poorly discriminative with regards to other inflammatory conditions. It is important to note that these considerations are suggestions and any change in nomenclature should be legitimated by the Histiocyte Society, which is currently engaged in revising the terminology for this group of diseases.
It is an obvious difficulty of current nomenclature that a single clinical scenario (e.g. EBV-associated HLH in an infant) could exemplify HLH associated with a defect in lymphocyte cytotoxicity, a T-cell-driven “secondary” HLH, or a T/NK-cell-independent HIS in a SCID patient, each with a potentially different pathophysiology and a common phenotypic endpoint. It is thus important to remember that the HLH syndrome itself never represents a complete diagnosis. It will be an important challenge to produce diagnostic criteria that can help to separate etiologically different conditions. The ratio of ferritin to sCD25 may represent one such criterion, but new bio-markers specifically reflecting the individual pathophysiological pathway leading to the inflammatory disease state of the HLH syndrome are needed. It will be of particular interest to evaluate whether markers of immune cell activation in addition to cytokine patterns may more specifically reflect disease pathogenesis.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Janka GE, Lehmberg K. Hemophagocytic syndromes - an update. Blood Rev. 2014;28(4):135–142. [DOI] [PubMed] [Google Scholar]
- 2.Pachlopnik Schmid J, Cote M, Menager MM, et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev. 2010;235(1):10–23. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–743. [DOI] [PubMed] [Google Scholar]
- 4.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9–15. [DOI] [PubMed] [Google Scholar]
- 6.Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10(3):387–392. [DOI] [PubMed] [Google Scholar]
- 7.Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. [DOI] [PubMed] [Google Scholar]
- 8.Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. [DOI] [PubMed] [Google Scholar]
- 9.Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–628. [DOI] [PubMed] [Google Scholar]
- 10.Pachlopnik Schmid J, Ho CH, Chretien F, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1(2):112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014; 155(1):118–125. [DOI] [PubMed] [Google Scholar]
- 12.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmberg K, Pink I, Eulenburg C, Beutel K, Maul-Pavicic A, Janka G. Differentiating macrophage activation syndrome in systemic juvenile idiopathic arthritis from other forms of hemophagocytic lymphohistiocytosis. J Pediatr. 2013;162(6):1245–1251. [DOI] [PubMed] [Google Scholar]
- 14.Dvorak CC, Sandford A, Fong A, Cowan MJ, George TI, Lewis DB. Maternal T-cell engraftment associated with severe hemophagocytosis of the bone marrow in untreated X-linked severe combined immunodeficiency. J Pediatr Hematol Oncol. 2008;30(5):396–400. [DOI] [PubMed] [Google Scholar]
- 15.Grunebaum E, Zhang J, Dadi H, Roifman CM. Haemophagocytic lymphohistiocytosis in X-linked severe combined immunodeficiency. Br J Haematol. 2000;108(4):834–837. [DOI] [PubMed] [Google Scholar]
- 16.Patiroglu T, Haluk Akar H, van den Burg M, et al. X-linked severe combined immunodeficiency due to a novel mutation complicated with hemophagocytic lymphohistiocytosis and presented with invagination: a case report. Eur J Microbiol Immunol (Bp). 2014;4(3):174–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid I, Reiter K, Schuster F, et al. Allogeneic bone marrow transplantation for active Epstein-Barr virus-related lymphoproliferative disease and hemophagocytic lymphohistiocytosis in an infant with severe combined immunodeficiency syndrome. Bone Marrow Transplant. 2002;29(6):519–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cesaro S, Messina C, Sainati L, Danesino C, Arico M. Del 22Q11.2 and hemophagocytic lymphohistiocytosis: a non-random association. Am J Med Genet A. 2003;116A(2):208–209. [DOI] [PubMed] [Google Scholar]
- 19.Arico M, Bettinelli A, Maccario R, Clementi R, Bossi G, Danesino C. Hemophagocytic lymphohistiocytosis in a patient with deletion of 22q11.2. Am J Med Genet. 1999;87(4):329–330. [PubMed] [Google Scholar]
- 20.Pasic S, Micic D, Kuzmanovic M. Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott-Aldrich syndrome. Acta Paediatr. 2003;92(7):859–861. [DOI] [PubMed] [Google Scholar]
- 21.Almagor Y, Revel-Vilk S, Averbuch D, et al. Congenital cytomegalovirus infection and Wiskott-Aldrich syndrome successfully treated with unrelated cord blood transplantation. Pediatr Blood Cancer. 2011;57(4):681–683. [DOI] [PubMed] [Google Scholar]
- 22.Seidel MG. CD27: a new player in the field of common variable immunodeficiency and EBV-associated lymphoproliferative disorder? J Allergy Clin Immunol 2012;129(4):1175; author reply 1175–6. [DOI] [PubMed] [Google Scholar]
- 23.Salzer E, Daschkey S, Choo S, Gombert, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. 2013;98(3):473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stepensky P, Weintraub M, Yanir A, et al. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica. 2011;96(3):472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kashiwagi Y, Kawashima H, Sato S, et al. Virological and immunological characteristics of fatal virus-associated haemophagocytic syndrome (VAHS). Microbiol Immunol. 2007;51(1):53–62. [DOI] [PubMed] [Google Scholar]
- 26.Valentine G, Thomas TA, Nguyen T, Lai YC. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: a case report. Pediatrics. 2014;134(6):e1727–1730. [DOI] [PubMed] [Google Scholar]
- 27.Araujo A, Pagnier A, Frange P, et al. [Lymphohistiocytic activation syndrome and Burkholderia cepacia complex infection in a child revealing chronic granulomatous disease and chromosomal integration of the HHV-6 genome]. Arch Pediatr. 2011;18(4): 416–419. [DOI] [PubMed] [Google Scholar]
- 28.Akagi K, Kawai T, Watanabe N, et al. A case of macrophage activation syndrome developing in a patient with chronic granulomatous disease-associated colitis. J Pediatr Hematol Oncol. 2014;36(3):e169–172. [DOI] [PubMed] [Google Scholar]
- 29.Scheffler-Mendoza SC, Yamazaki-Nakashimada MA, Olaya-Vargas A, et al. Successful stem cell transplantation in a child with chronic granulomatous disease associated with contiguous gene deletion syndrome and complicated by macrophage activation syndrome. Clin Immunol. 2014; 154(2):112–115. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez-Cardona A, Rodriguez-Lozano AL, Blancas-Galicia L, Rivas-Larrauri FE, Yamazaki-Nakashimada MA. Intravenous immunoglobulin treatment for macrophage activation syndrome complicating chronic granulomatous disease. J Clin Immunol. 2011;32(2):207–211. [DOI] [PubMed] [Google Scholar]
- 31.van Montfrans JM, Rudd E, van de Corput L, et al. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr Blood Cancer. 2009;52(4):527–529. [DOI] [PubMed] [Google Scholar]
- 32.Martin A, Marques L, Soler-Palacin P, et al. Visceral leishmaniasis associated hemophagocytic syndrome in patients with chronic granulomatous disease. Pediatr Infect Dis J. 2009;28(8):753–754. [DOI] [PubMed] [Google Scholar]
- 33.Hisano M, Sugawara K, Tatsuzawa O, Kitagawa M, Murashima A, Yamaguchi K. Bacteria-associated haemophagocytic syndrome and septic pulmonary embolism caused by Burkholderia cepacia complex in a woman with chronic granulomatous disease. J Med Microbiol. 2007;56(Pt 5):702–705. [DOI] [PubMed] [Google Scholar]
- 34.Sirinavin S, Techasaensiri C, Pakakasama S, Vorachit M, Pornkul R, Wacharasin R. Hemophagocytic syndrome and Burkholderia cepacia splenic microabscesses in a child with chronic granulomatous disease. Pediatr Infect Dis J. 2004;23(9):882–884. [DOI] [PubMed] [Google Scholar]
- 35.Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer. 2011;56(3):460–462. [DOI] [PubMed] [Google Scholar]
- 36.Schultz KA, Neglia JP, Smith AR, Ochs HD, Torgerson TR, Kumar A. Familial hemophagocytic lymphohistiocytosis in two brothers with X-linked agammaglobulinemia. Pediatr Blood Cancer. 2008;51(2):293–295. [DOI] [PubMed] [Google Scholar]
- 37.Kuijpers TW, Baars PA, vaan de Kerk DJ, et al. Common variable immunodeficiency and hemophagocytic features associated with a FAS gene mutation. J Allergy Clin Immunol. 2011;127(6):1411–1414e2. [DOI] [PubMed] [Google Scholar]
- 38.Rudman Spergel A, Walkovich K, Price S, et al. Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics. 2013;132(5):e1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pachlopnik Schmid JM, Junge SA, Hossle JP, et al. Transient hemophagocytosis with deficient cellular cytotoxicity, monoclonal immunoglobulin M gammopathy, increased T-cell numbers, and hypomorphic NEMO mutation. Pediatrics. 2006;117(5):e1049–1056. [DOI] [PubMed] [Google Scholar]
- 40.Horneff G, Rhouma A, Weber C, Lohse P. Macrophage activation syndrome as the initial manifestation of tumour necrosis factor receptor 1-associated periodic syndrome (TRAPS). Clin Exp Rheumatol. 2013;31(3 Suppl 77):99–102. [PubMed] [Google Scholar]
- 41.Rossi-Semerano L, Hermeziu B, Fabre M, Kone-Paut I. Macrophage activation syndrome revealing familial Mediterranean fever. Arthritis Care Res. 2011;63(5):780–783. [DOI] [PubMed] [Google Scholar]
- 42.Uslu N, Demir H, Balta G, et al. Hemophagocytic syndrome in a child with severe Crohn’s disease and familial Mediterranean fever. J Crohns Colitis. 2010;4(3):341–344. [DOI] [PubMed] [Google Scholar]
- 43.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Felgentreff K, Perez-Becker R, Speckmann C, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141(1):73–82. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-inducible T-cell kinase (ITK) deficiency - clinical and molecular aspects. J Clin Immunol. 2014;34(8): 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Montfrans JM, Hoepelman AI, Otto S, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129(3):787–793 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magnani A, Brosselin P, Beaute J, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. 2014;134(3):655–662 e8. [DOI] [PubMed] [Google Scholar]
- 48.de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. 2014;111(9):3526–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jessen B, Kogl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. 2013;4:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118(3):618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryceson YT, Pende D, Maul-Pavicic A, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012; 119(12):2754–2763. [DOI] [PubMed] [Google Scholar]
- 52.Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol 2012;91:63–71. [DOI] [PubMed] [Google Scholar]
- 53.Zoller EE, Lykens JE, Terrell CE, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med. 2011; 208(6):1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]