

Diffuse large B-cell lymphoma (DLBCL) is the most common form of lymphoma, accounting for 30–40% of newly diagnosed cases of non-Hodgkin lymphomas. The molecular heterogeneity of DLBCL has been deciphered by gene expression profiling, and DLBCL have been divided into three main molecular subtypes: the germinal center B-cell-like (GCB) subtype, the activated B-cell-like (ABC) subtype, and the primary mediastinal B-cell lymphoma subtype with distinct clinical outcomes and responses to immunochemotherapy. Next-generation sequencing (NGS) technologies, which allow for massive, parallel, high-throughput DNA sequencing, have emerged over the past decade and have provided new insights into the genomic characterization of DLBCL. Recurrent single nucleotide variants (SNV) are now well defined and provide new therapeutic opportunities for the three molecular subtypes. The SNV target genes play a crucial role in several pathways, including B-cell receptor signaling (CD79A/CD79B), NFκ-B (CARD11), Toll-like receptor signaling (MYD88), immunity (CD58, TNFSRF14, B2M), cell cycle/apoptosis (TP53, BCL2) and epigenetic regulation (EZH2,CREBBP, MLL2).1,2
Recently, whole exome sequencing in breast cancer has shown that mutations observed in the tumor could also be detected in circulating, cell-free DNA (cfDNA) and could be used to detect genetic changes during treatments and relapse, defining the concept of “liquid biopsy”.3 In DLBCL, whereas tumor circulating cells or leukemic phase are not usually detectable, clonotypic sequences have been constantly detected in cfDNA extracted from serum/plasma or peripheral blood mononuclear cells.4–8
In this study we sought to determine, by routinely applicable NGS technology, whether the pattern of acquired SNV observed in tumor DNA could also be detected in cfDNA in DLBCL patients at the time of diagnosis. For this purpose, we analyzed 12 DLBCL cases with available matched tumor DNA and plasma collected at the time of diagnosis. Patients harboring typical GCB/ABC-related mutations targeting CD79A/B, EZH2, CARD11 or MYD88 genes, previously identified by the Sanger method, were selected.9 This study was approved by the regional ethical committee (numbered as CPP N°01/006/2014).
The main clinical features of the patients are summarized in Table 1. None of the selected cases harbored detectable circulating lymphoma cells by routine blood smear examination. Of note, no peripheral blood cytometry was performed, in accordance with our center’s initial staging procedures for DLBCL patients. Tumor DNA was extracted from frozen lymph node samples by standard methods. cfDNA was extracted from archived EDTA-anticoagulated plasma aliquots (1 mL) stored at −80°C using the QIAamp® Circulating Nucleic Acid Kit (Qiagen) (with the QIAvac 24 Plus vacuum manifold, following the manufacturer’s instructions), and concentrations were measured using a fluorometric assay (Qubit® dsDNA HS Assay Kit, Life Technologies). The mean cfDNA concentration in plasma was 1.65 ng/μL (range, 0.46–11.2 ng/μL) (Table 1). The cell-of-origin signature was determined by cDNA-mediated annealing, selection, extension, and ligation technology based on the expression of 19 genes, as previously reported.10 Among the 12 cases analyzed, five belonged to the ABC subgroup, six to the GCB subgroup and one case was unclassified (Table 1).
Table 1.
Clinical characteristics and list of somatic variants (insertion/deletion/single nucleotide variant) detected by sequencing in tumor DNA and cell-free, plasma circulating DNA. Details of the locations of the mutations are indicated in Online Supplementary Table S1.
Tumor DNA was sequenced using an Ion Torrent Personal Genome Machine (Life Technologies). Ten nanograms of genomic DNA were submitted to NGS using a laboratory-developed Lymphopanel set, designed to identify mutations in 34 genes relevant to lymphomagenesis (Online Supplementary File S1A). This design covers 87,703 bases and generates 872 amplicons. Amplified libraries (Ion AmpliSeq™ Library Kit 2.0) were submitted to emulsion polymerase chain reaction with the Ion OneTouch™ 200 Template Kit (Life Technologies) using the Ion OneTouch™ System (Life Technologies) according to the manufacturer’s instructions. The templated Ion Sphere™ Particles were enriched with the Ion OneTouch™ Enrichment System and loaded and sequenced on an Ion 316™ v2 Chip (Life Technologies).
After alignment to a reference genome sequence (hg19) and a variant calling procedure, variants were filtered through a dedicated bioinformatic pipeline that eliminated synonymous variants and variants with a variant allele frequency (VAF) greater than 1% in the 1000 genome database (considered as polymorphisms). Only non-synonymous SNV/In/Del with a quality score >22 and/or confirmed by a Sanger experiment were retained as acquired somatic mutations in lymphoma cases and were used for the subsequent sequencing of the matched cfDNA (for the detailed pipeline see Online Supplementary File S1B). Somatic mutations identified in tumor DNA were used to build a hot-spot file enabling higher sensitivity tracking of somatic mutations in circulating DNA.
In order to increase the sensitivity and specificity of variant detection in cfDNA, we exclusively amplified amplicons targeting mutations detected in the corresponding tumor DNA by the complete Lymphopanel by performing a dedicated sequencing procedure with a pool of oligonucleotide primers selected among the 872 pairs provided by Life Technologies. The procedure to make libraries and sequence amplicons was the same as that for tumor DNA but used a 314™ v2 Chip. When possible, circulating DNA was extracted and sequenced from two different aliquots of plasma.
The SNV/In/Del detected in both tumor and circulating DNA are indicated in Table 1 with their corresponding VAF. As expected, we identified a typical SNV pattern in the five ABC DLBCL cases, including mutations targeting MYD88, CD79A/B, PIM1, PRDM1, CARD11 or IRF4, whereas EZH2, BCL2, GNA13, or TNFSRF14 were mutated in the unclassified and GCB DLBCL cases. MLL2 (KMT2D), CREBBP or ITPKB were targeted by somatic mutations shared in the two cell-of-origin subtypes. The sequencing depths obtained for each sample and targets are indicated in Table 1.
The mean number of reads targeting the mutated regions for tumor DNA was 241 (range, 23–741) as compared to 5,987 (range, 6–22,541) for plasma DNA, indicating a 24-fold mean depth sequencing increase. The mean VAF in the tumor DNA was 35% (range, 17–64%) as compared to a mean of 11% for plasma DNA (range, 2–89%) (Table 1).
In 11/12 DLBCL cases, we observed somatic mutations in cfDNA, similar or partially similar to those observed in the tumor (Figure 1, Table 1 and Online Supplementary Figure S1). We defined the concordance rate as the ratio of the number of mutated cfDNA genes and the number of mutated tumor DNA genes. This rate ranged from 33% to 100% (5 cases) in the 11/12 cases with detected mutated cfDNA. Overall, the median concordance rate between tumor DNA and cfDNA was 85%.
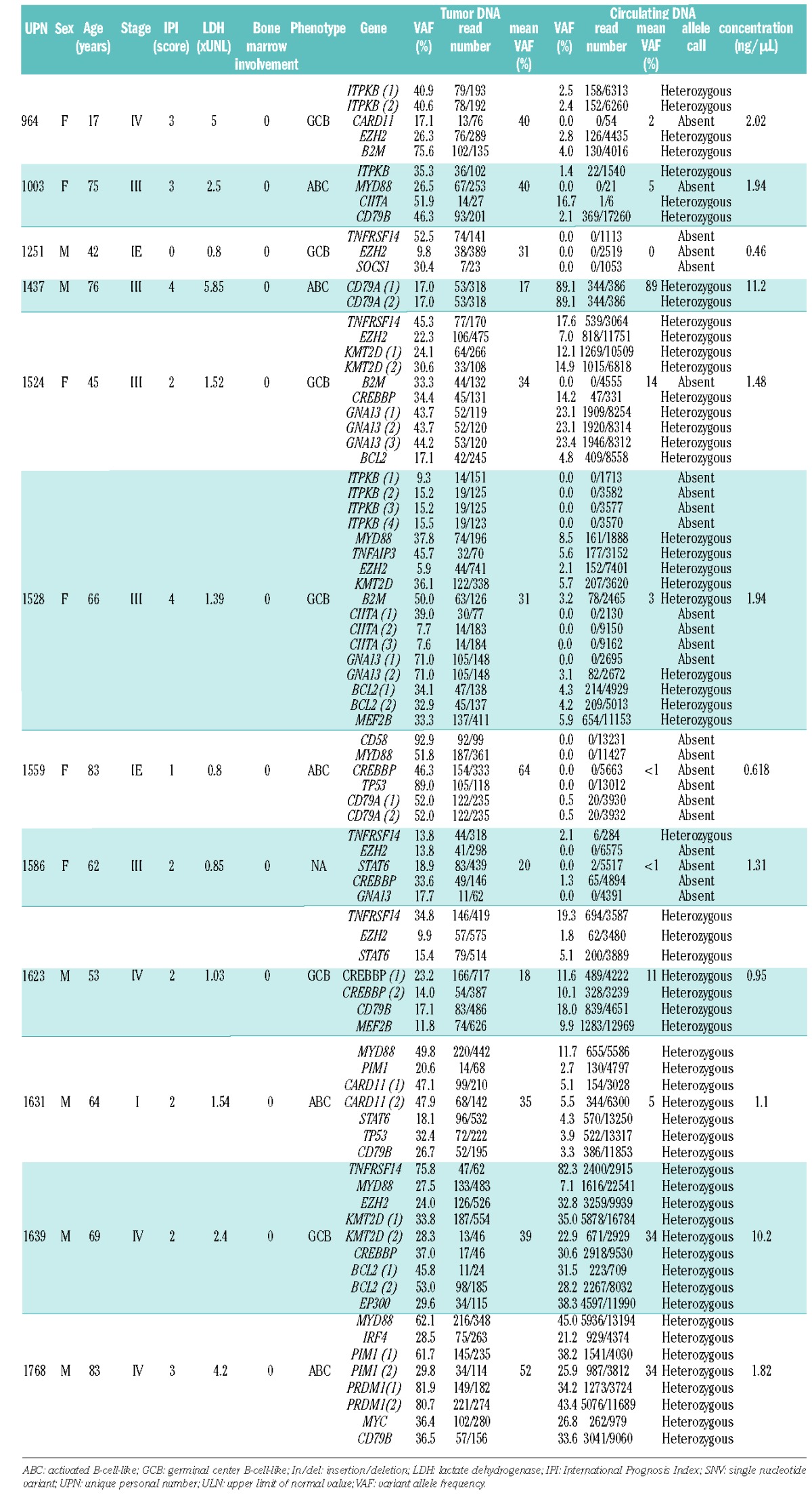
Figure 1.
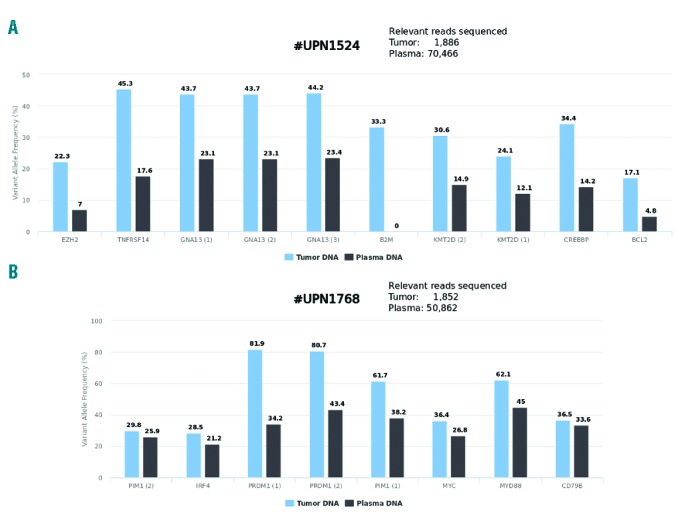
Representative examples of variant allele frequencies observed in tumor DNA and matched circulating cell-free DNA at the time of diagnosis in two different patients (A and B).
In one case (#1251), SNV observed in the tumor DNA were not detected in plasma DNA. In another case (#1559), SNV were barely detectable (VAF of 0.5% for two variants). Of note, both cases displayed limited disease (stage I or II) and normal lactate dehydrogenase levels, indicating that the amount of tumor-specific circulating cfDNA is at least partially related to tumor burden. Despite a low amount of circulating DNA extracted from plasma for cases #1251 and #1559, we obtained adequate sequencing quality and depth (the overall number of reads sequenced with mutated targets was 4,685 and 51,195 respectively; Table 1), indicating that in some rare cases, tumor-specific cfDNA is absent or beneath the level of sensitivity of the NGS method used. Of note, in case #1631 characterized by limited stage I disease, SNV were detected with a mean VAF of 5.2% in plasma DNA, as compared to a mean VAF of 34.6% in the tumor DNA (Table 1). In contrast, cases #1639 and #1768 (both with stage IV disease and elevated levels of lactate dehydrogenase) displayed a high proportion of tumor-specific circulating DNA, as indicated by the high VAF observed (Table 1). Interestingly, in these two cases, the sub-clonal distribution of certain mutations, as indicated by the VAF distribution of each individual variant, was also observed in cfDNA, suggesting that sequencing cfDNA can reflect the SNV pattern observed in tumor cells in some instances (Figure 1 and Online Supplementary Figure S1). In case #1524, despite a sufficient number of relevant reads (> 4,000) we failed to detect the B2M SNV present in the lymph node biopsy. This result was confirmed by manual Integrative Genomics Viewer checking, suggesting that the B2M SNV is present only in a subclone caught in the biopsy sample but not highlighted by the cfDNA that reflects the entire tumor burden.
By contrast, in some cases, the number of target reads was clearly insufficient for adequate SNV detection (case #964, CARD11; case #1003, MYD88; Table 1), most likely reflecting the low amount of cfDNA available rather than a true clonal divergence between tumor DNA and cfDNA. This was not observed in cases with higher amounts of cfDNA.
Failure to detect SNV in cfDNA appears related more to the proportion of tumor-specific DNA than to the total amount or quality of total cfDNA. Of note, we failed to find any tumor-related SNV in DNA extracted from 11/12 samples of peripheral blood mononuclear cells using this approach (data not shown), indicating that serum or plasma is preferable for the detection of mutated circulating DNA. In a large cohort of cases of Hodgkin lymphoma, mantle cell lymphoma and DLBCL, increased levels of plasma DNA (determined using quantitative polymerase chain reaction for the β-globin gene) were associated with advanced stage disease, presence of B-symptoms, elevated lactate dehydrogenase levels, and age >60 years also indicating that the amount of circulating DNA is partially related to tumor burden.8 Furthermore, it was shown in a cohort of patients with Epstein-Barr virus-positive lymphoma that serum and plasma were equivalent for detecting lymphoma-specific DNA but that only the lymphoma-specific DNA could be used to monitor disease response in lymphoma.7
To our knowledge this is the first report of the detection of non-immunoglobulin somatic mutations in DLBCL from circulating DNA by routine NGS, enabling the identification of lymphoma-specific cfDNA. Other quantitative approaches, including digital polymerase chain reaction, are also suitable and could be used in this setting for detecting recurrent translocations or mutations.11 More recently LymphoSIGHT®, a high-throughput DNA sequencing method, was developed to detect and quantify circulating tumor DNA as minimal residual disease and was able to predict both early treatment failure and relapse in patients with newly diagnosed DLBCL, chronic lymphocytic leukemia or acute lymphoblastic leukemia.12–15 This approach is based on tumor DNA amplification using locus-specific primer sets for the immunoglobulin heavy/light-chain which failed in a substantial number of cases of DLBCL. Importantly the Lymphopanel used in this study is able to detect at least one acquired SNV in 95% of DLBCL cases at initial diagnosis (manuscript in preparation) and may, therefore, constitute a simple, routinely applicable test to provide the cell-of-origin subtype or to detect targetable mutations at the time of diagnosis or relapse. However, its capacity to detect minimal residual disease with a high level of sensitivity remains to be determined and we can hypothesize that at least 5–10% of DLBCL cases will not display any SNV detectable by our Lymphopanel. Furthermore, cfDNA sequencing was successfully performed using the entire Lymphopanel (including the 34 targeted genes) in one case (data not shown), indicating that this approach is feasible without the knowledge of the tumor variant calling. However this requires an increase in the sequencing depth capacity and entails a substantial increase of costs.
To conclude, our results indicate that cfDNA can also be used in DLBCL to detect somatic variants, validating the concept of “liquid biopsy” in this type of tumor.3 These preliminary results have prompted us to start a prospective study with the aim of serially sequencing cfDNA during DLBCL treatment and follow-up (registered at clinicaltrials.gov as NCT02339805). If these preliminary results are confirmed by a prospective study, new strategies should be proposed for both diagnosis and treatment tailoring based on the simple detection and quantification of SNV in plasma.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Morin RD, Gascoyne RD. Newly identified mechanisms in B-cell non-Hodgkin lymphomas uncovered by next-generation sequencing. Semin Hematol. 2013;50(4):303–313. [DOI] [PubMed] [Google Scholar]
- 2.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He J, Wu J, Jiao Y, et al. IgH gene rearrangements as plasma biomarkers in Non- Hodgkin’s lymphoma patients. Oncotarget. 2011; 2(3):178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosny G, Farahat N, Hainaut P. TP53 mutations in circulating free DNA from Egyptian patients with non-Hodgkin’s lymphoma. Cancer Lett. 2009;275(2):234–239. [DOI] [PubMed] [Google Scholar]
- 6.Mussolin L, Burnelli R, Pillon M, et al. Plasma cell-free DNA in paediatric lymphomas. J Cancer. 2013;4(4):323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones K, Nourse JP, Keane C, et al. Tumor-specific but not nonspecific cell-free circulating DNA can be used to monitor disease response in lymphoma. Am J Hematol. 2012;87(3):258–265. [DOI] [PubMed] [Google Scholar]
- 8.Hohaus S, Giachelia M, Massini G, et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Ann Oncol. 2009; 20(8):1408–1413. [DOI] [PubMed] [Google Scholar]
- 9.Bohers E, Mareschal S, Bouzelfen A, et al. Targetable activating mutations are very frequent in GCB and ABC diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2014;53(2):144–153. [DOI] [PubMed] [Google Scholar]
- 10.Lanic H, Mareschal S, Mechken F, et al. Interim positron emission tomography scan associated with international prognostic index and germinal center B cell-like signature as prognostic index in diffuse large B-cell lymphoma. Leuk Lymphoma. 2012;53(1):34–42. [DOI] [PubMed] [Google Scholar]
- 11.Shuga J, Zeng Y, Novak R, et al. Single molecule quantitation and sequencing of rare translocations using microfluidic nested digital PCR. Nucleic Acids Res. 2013;41(16):e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armand P, Oki Y, Neuberg DS, et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br J Haematol. 2013;163(1):123–126. [DOI] [PubMed] [Google Scholar]
- 13.Logan AC, Vashi N, Faham M, et al. Immunoglobulin and T cell receptor gene high-throughput sequencing quantifies minimal residual disease in acute lymphoblastic leukemia and predicts post-transplantation relapse and survival. Biol Blood Marrow Transplant. 2014;20(9):1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logan AC, Zhang B, Narasimhan B, et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia. 2013;27(8):1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roschewski M, Dunleavy K, Pittaluga S, et al. Monitoring of circulating tumor DNA as minimal residual disease in diffuse large B-cell lymphoma. Blood. 2014;124(21):139 Abstr. [Google Scholar]