

Abstract
In a 28-year-old male with a mild mitochondrial myopathy manifesting as exercise intolerance and early signs of cardiomyopathy without muscle weakness or ophthalmoplegia, we identified two novel mutations in the SLC25A4 gene: c.707G>C in exon 3 (p.(R236P)) and c.116_137del in exon 2 (p.(Q39Lfs*14)). Serum lactate levels at rest were elevated (12.7 mM). Both the patient’s father and brother were heterozygous carriers of the c.707G>C mutation and were asymptomatic. The second mutation causes a 22 bp deletion leading to a frame shift likely giving rise to a premature stop codon and nonsense-mediated decay (NMD). The segregation of the mutations could not be tested directly as the mother had died before. However, indirect evidence from NMD experiments showed that the two mutations were situated on two different alleles in the patient. This case is unique compared to other previously reported patients with either progressive external ophthalmoplegia (PEO) or clear hypertrophic cardiomyopathy with exercise intolerance and/or muscle weakness carrying recessive mutations leading to a complete absence of the SLC25A4 protein. Most likely in our patient, although severely reduced, SLC25A4 is still partially present and functional.
Keywords: Hypertrophic cardiomyopathy, Mitochondrial myopathy, Novel mutations, SLC25A4
Introduction
ANT (adenine nucleotide transporter) is the most abundant protein located in the inner membrane of the mitochondria. It plays a crucial role in the exchange of ADP and ATP between the cytosolic and mitochondrial compartments and is therefore vital for the function of the oxidative phosphorylation (OXPHOS) system. Four isoforms of human ANT can be distinguished (Cozens et al. 1989; Dolce et al. 2005; Li et al. 1989; Ku et al. 1990; Houldsworth and Attardi 1988; Stepien et al. 1992). SLC25A4 (ANT1) is expressed in the heart and skeletal muscle. It has a 91–93% amino acid homology with SLC25A5 (ANT2), SLC25A6 (ANT3) and SLC25A31 (ANT4) of which the first two are expressed in almost all body tissues and the last in the liver, testis, and brain (Dolce et al. 2005; Neckelmann et al. 1987) that characterized the cDNA of SLC25A4, which consists of 1,400 nucleotides. The SLC25A4 gene is located on chromosome 4, spans 5.8 kb, and consists of 4 exons.
Mutations in SLC25A4 are associated with the presence of multiple mtDNA deletions in different tissues (skeletal and heart muscle, brain, kidney, and liver) (Palmieri et al. 2005). Until now, five different autosomal dominant SLC25A4 mutations have been described primarily associated with adPEO (autosomal dominant progressive external ophthalmoplegia) (Deschauer et al. 2005; Napoli et al. 2001; Komaki et al. 2002; Kaukonen et al. 2000). An autosomal recessively inherited mitochondrial myopathy and hypertrophic cardiomyopathy was described in two cases (Palmieri et al. 2005; Echaniz-Laguna et al. 2012).
Here, we describe two novel mutations in the SLC25A4 gene in a patient first reported by Bakker et al. (1993a, b). In the patient (1) immunostaining of Western blots revealed a 4-fold decrease in the concentration of the adenine nucleotide translocator; (2) the activities of complexes I to V catalyzing oxidative phosphorylation and the pyruvate and the 2-oxoglutarate dehydrogenase complexes showed a 2- to 20-fold increase, (3) in the serum, lactate levels up to 12.7 mM at rest were reported; and (4) 31P-nuclear magnetic resonance spectroscopy in the resting muscle showed half the creatine phosphate level as compared to controls. No ragged red fibers were observed in muscle, but electron microscopy revealed abnormal mitochondria. At the age of 28, he was still suffering from only mild exercise intolerance and was using a wheelchair for outdoor transportation. There were no signs of ophthalmoplegia and he only showed mild signs of hypertrophic cardiomyopathy.
Material and Methods
Molecular Studies
DNA Analysis
Total DNA was extracted from blood using the wizard genomic DNA purification kit (Promega, Leiden, The Netherlands) and from fibroblasts using the Puregene kit (Gentra, Minneapolis, Minnesota, USA). Specific intronic primers were designed to amplify the exons and flanking introns of SLC25A4 (Table 1). All primers contained an additional M13 sequence. Exon 1 was amplified in a volume of 25 μl using 5 ng genomic DNA as template with 0.5 U KAPA2G Fast HotStart DNA polymerase, KAPA2G HotStart Buffer A, 5 mM dNTP-mix (KapaBiosystems, Massachusetts,USA), 3.75 pmol each primer, and 10% DMSO (Merck, Darmstadt, Germany). The reactions for exon 2, 3, and 4 were performed in a 10 μl volume using 5 ng genomic DNA as template, AmpliTaq Gold Master Mix (Applied Biosystems, Foster City, USA), 2 pmol of both primers, and 8% glycerol (Invitrogen, Carlsbad, USA). PCR conditions for all the exons were as follows: first one cycle of 96°C for 5 min, followed by 40 cycles of 94°C for 30 s, 60°C for 45 s, and 72°C for 45 s; and finally one cycle 72°C for 10 min. PCR products were directly sequenced with the PRISM Ready Reaction Sequencing Kit (Perkin-Elmer Life Sciences) on an ABI3100 DNA Analyzer (Applied Biosystems).
Table 1.
Primer sequences for amplification of SLC25A4
| Exon | Length (bp) | Sequence forward primer | Sequence reverse primer |
|---|---|---|---|
| 1 | 419 | 5′-CCTCCTCTCGCGAGAGC-3′ | 5′-GCCTGGCGCAGATTTTC-3′ |
| 2A | 393 | 5′-GTCCTCTTCCCTTCTCTCTA-3′ | 5′-CCAACCTGGTCCTAGCAAAG-3′ |
| 2B | 452 | 5′-GGCGCTACTTTGCTGGTAAC-3′ | 5′-GCACATCACCTCCTCATTCA-3′ |
| 3 | 445 | 5′-GCAAGGTCAGAGCATGGAG-3′ | 5′-GTTGAGAACGTTAGGGGGAAT-3′ |
| 4 | 485 | 5′-GTTGCATGGAGCTGGGACT-3′ | 5′-CTCAATGAAGCATCTCTTCTGA-3′ |
| cDNA (c.25F-c.270R) | 246 | 5′-CTAAAGGACTTCCTGGCCG-3′ | 5′-GGCGAAGTTGAGAGCTTG-3′ |
| cDNA (c.533F-c.827R) | 295 | 5′-ACGTCTCTGTCCAAGGCATC-3′ | 5′-GACCAGGCACCTTTGAAGAA-3′ |
| M13 sequence | 18 | 5′-TGTAAAACGACGGCCAGT-3′ | 5′-CAGGAAACAGCTATGACC-3′ |
cDNA Analysis
The Tripure isolation reagent kit (Roche Applied Science, Mannheim, Germany) was used to extract total RNA from fibroblasts. All extractions were performed according to the instruction manuals. cDNA synthesis was performed by the SuperScript First-Strand Synthesis System for RT-PCR, using oligo(dT) (Invitrogen, Carlsbad, USA). The synthesis was performed according the instruction manual, except for the RNase H step, which was not performed. The PCR was performed in a 10 μl volume with 40 ng cDNA as template, using Phire Hot Start II PCR Master Mix (Finnzymes, Espoo, Finland), 3 pmol each primer, and 3% DMSO (Merck, Darmstadt, Germany) with PCR conditions according to the manual, with the exception of the number of PCR cycles and the annealing temperature, which were respectively 40 cycles and 60°.
Fibroblast Culture
Fibroblasts were grown in Dulbecco’s modified Eagle’s medium (DMEM), high glucose, pyruvate, and glutamine (Invitrogen, Carlsbad, USA); 10% fetal bovine serum, 10 U/ml penicillin and 10 U/ml streptomycin; and 0.2 mM uridine in a humidified atmosphere of 95% air and 5% CO2 at 37°C. To investigate the process of nonsense-mediated decay (NMD) we cultured the cells in duplicate and added 200 μg/ml puromycin to the medium 5 h before harvesting the cells.
Results
Mutation Analysis
In our patient two novel SLC25A4 mutations were identified. The first mutation is a heterozygous deletion c.116_137del in exon 2 (p.(Q39Lfs*14)), whereas the second mutation is a missense mutation c.707G>C (heterozygous) in exon 3 leading to the substitution of arginine at position 236 into a proline (p.(R236P)) as displayed in Fig. 1. Neither of the mutations were found in a control panel of the same ethnicity (n = 114), present in the HGMD® or in SNP databases like dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) and the 1000 Genomes Project (Altshuler et al. 2010). In addition, they were not present in the exomes of 61,486 unrelated individuals (Exome Aggregation Consortium, ExAC; http://exac.broadinstitute.org). P.Arg236 is highly conserved among different species including S. cerevisiae (baker’s yeast). The sequence which harbors the deletion c.116_137del in exon 2 (p.(Q39Lfs*14)) is also highly conserved (up to Drosophila melanogaster). Aminoacid alignments are also shown in Fig. 1. Both the patient’s father and brother are heterozygous carriers of the c.707G>C mutation and are asymptomatic. We were unable to perform DNA analysis on the patient’s mother since she died at 59 years of age from leukaemia.
Fig. 1.
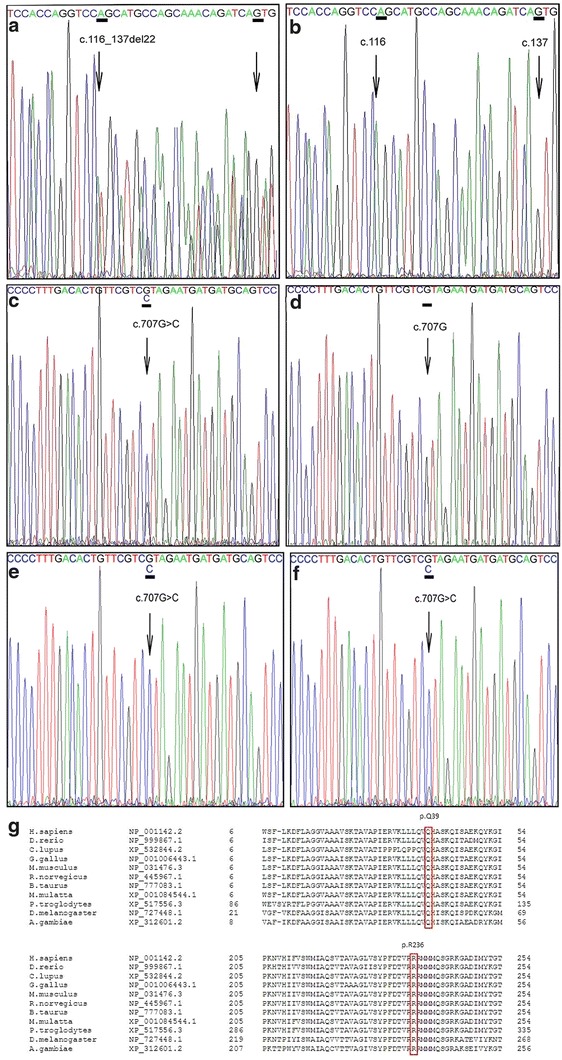
The c.116_137del p.(Q39Lfs*14) and the c.707G>C p.(R236P) mutation. DNA from fibroblasts of the proband and DNA from blood of the control showing the c.116_137del mutation (A and B) and the c.707G>C mutation (C en D). cDNA from fibroblasts of the proband cultured with puromycin, which inhibits NMD (F), shows that the signal of the wild-type nucleotide c.707G is relatively higher compared to cDNA from fibroblasts cultured without puromycin (E). Amino acid alignment of the human SLC25A4 reference sequence versus various species for position 6 to 54 and position 205 to 254. Indicated is the position of the p.(Q39Lfs*14) mutation and the p.(R236P) mutation. The arginine at position 236 is conserved in all species shown (G)
Q39Lfs*14 Mutation Leads to Partial Nonsense-Mediated Decay in Fibroblasts
The p.(Q39Lfs*14) mutation is caused by a 22 bp deletion, predicted to lead to a frameshift with a premature stop codon. cDNA analysis in fibroblasts with and without puromycin, which suppresses NMD, was performed to test if this mutation would lead to NMD. The results confirm that NMD is at least partially involved.
Discussion
In this paper we report a patient with a severe reduction in SLC25A4 gene expression with a fourfold decreased expression of SLC25A4 protein (Bakker et al. 1993a). We detected two novel mutations in the SLC25A4 gene, a c.707G>C in exon 3 leading to the substitution of arginine at position 236 into a proline (p.(R236P)) and a c.116_137del in exon 2 (p.(Q39Lfs*14)).
The p.Arg236 is a highly conserved amino acid, located in the predicted transmembrane region (helix) of the protein involved in maintaining the conformation of the protein. The p.(R236P) mutation is likely to affect the folding of SLC25A4 and its function because proline is known to interfere with alpha-helix formation (Fig. 2). Moreover, previous experiments in yeast, in which position R253 (human SLC25A4 position R236) was substituted for isoleucine, transport function was disrupted (Nelson et al. 1993; Heidkämper et al. 1996). Also, studies in bovine showed that R254 (human SLC25A4 position R236) is the second arginine in the RRRMMM motif, which appears to take part in nucleotide binding (Pebay-Peyroula et al. 2003).
Fig. 2.
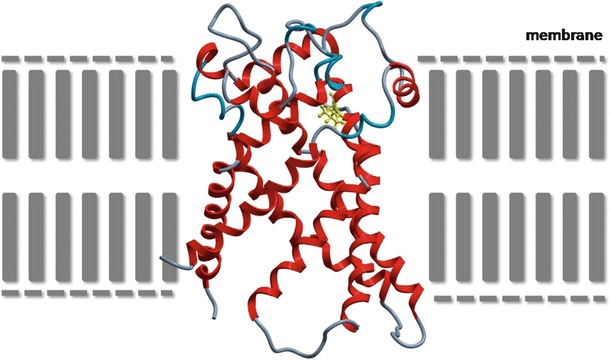
Model for human SLC25A4. A homology model was built, employing the crystal structure of the mitochondrial ADP/ATP carrier (PDB accession file 1OKC.pdb, sequence identity 96%) with the molecular modeling package ICM Pro (Molsoft, Inc, La Jolla, USA) and was next optimized and minimized following standard parameters. Alpha-helices are depicted in red, other areas in gray. Location of mutated proline (yellow) in SLC25A4 protein is situated in the membrane
From the asymptomatic patient’s father and brother who were carriers of this pathogenic mutation one may infer that the expression of functional SLC25A4 was still sufficient for a normal function (autosomal recessive inheritance). The 22 base pair deletion in exon 2, c.116_137del p.(Q39Lfs*14) leads to a frameshift with a new reading frame which introduces a stop codon at position 13. Both mutations were not found in controls. Our patient is most likely compound heterozygous resulting in little SLC25A4 functional rest activity as the explanation for the clinical signs of exercise intolerance and lactic acidosis. We demonstrated by NMD experiments (the level of mutated vs wild-type RNA levels in presence and absence of puromycin; data not shown) that both mutations are situated on different alleles (trans position) indicating that mutations had different origins (paternal/de novo or paternal/maternal) and suggests compound heterozygosity for the mutations. The deletion is most likely inherited from the mother, although this could not be tested as she had died before. Although it would have been interesting to study the presence of mtDNA deletions in muscle, as could be expected based on previous reports of SLC25A4 mutations (Komaki et al. 2002), this has not been performed at the time (Bakker et al. 1993a), and unfortunately no muscle tissue is available anymore. Unfortunately it is also not possible to re-biopsy the patient at this age.
Humans have different tissue-specific isoforms of ADP/ATP translocase (also known as adenine nucleotide translocator or ANT). SLC25A4 is a 60 kD protein encoded by the SLC25A4 gene and is highly expressed in the heart and skeletal muscle, whereas ANT3 is encoded by the SL2C5A6 and is ubiquitously expressed. The main function of both SLC25A4 and ANT3 is the exchange of ADP and ATP across the inner mitochondrial membrane in order to maintain the cellular bioenergetic state. ANT therefore plays a central role in OXPHOS (Adrian et al. 1986; Klingenberg 1981, 1989; Fiore et al. 1998).
Dysfunction of the ATP/ADP transporter is expected to result in severe defects in OXPHOS, giving rise to considerable energy deficiency primarily in the cytosol and a compensatory induction in mitochondrial proliferation. In a knockout (KO) mouse model (Graham et al. 1997), it was demonstrated that mice lacking SLC25A4 did indeed show progressive hypertrophy of the heart, severe exercise intolerance, a fourfold resting lactate as compared to the wild-type mice, and mitochondrial abnormalities in skeletal muscle.
A patient with a homozygous recessive null mutation p.(A123M) in SLC25A4 had a similar clinical manifestation, including hypertrophic cardiomyopathy, easy fatigability and exercise-related muscle pain, and episodes of headache associated with vomiting.
Muscle weakness and external ophthalmoplegia were conspicuously absent. Biochemically, lactic acidosis was present and the muscle biopsy showed ragged red fibers (Palmieri et al. 2005). The activities of mtDNA-dependent respiratory chain enzymes were partially reduced.
The other patient with a complete loss of expression of the SLC25A4 gene due to homozygosity for a new nucleotide variation, c.111+1G>A p.(A123D), was found to have muscle weakness in the arms and legs when examined at age 21 years. Also, the patient showed hypertrophic cardiomyopathy and congenital cataracts and slight mental retardation (Echaniz-Laguna et al 2012). There was no ophthalmoplegia present. Biochemically she had lactic acidosis, ragged red fibers in muscle biopsy and normal activities of mitochondrial respiratory chain complexes.
The patient with a severe SLC25A4 deficiency described in this paper had increased OXPHOS complex activities in muscle as described by Bakker et al. (1993a). This increase was explained as a secondary effect due to the hyperproliferation of mitochondria or an enhanced synthesis of mitochondrial enzymes, which can be a common phenomenon in patients with mitochondrial myopathies and might be an attempt to compensate for the decreased function of individual mitochondria (Bakker et al. 1993a). From the great difference between the coupled and uncoupled respiration activities (in controls these activities were similar), it could be concluded that the patient has a severe defect in the mitochondrial phosphorylation, despite the increased OXPHOS activities. In addition, energy production from pyruvate plus malate was barely normal, further supporting the presence of a phosphorylation defect (Bakker et al. 1993a).
The major effect of SLC25A4 deficiency as demonstrated in SLC25A4-KO mice (with characteristics of a cardiomyopathy and mitochondrial myopathy) is a deficiency of ADP-stimulated but not uncoupled respiration defect resulting from the blockade of ADP/ATP exchange. This defect was most pronounced in muscular and less in heart tissue. In SLC25A4-KO mice, ANT2 could replace SLC25A4 and thus prevent premature heart failure.
At the time of measurement of SLC25A4 activity, no specific isoforms were distinguished during protein analysis (Bakker et al. 1993a). However, it was reported that preliminary results of a transcript analysis of isoform-specific probes showed a specific reduction of the muscle isoform in the patient versus controls (Bakker et al. 1993a). Although it seems unlikely that other isoforms are responsible for residual ANT activity based on these results, we cannot entirely exclude this possibility. We speculate that the patient had still sufficient residual SLC25A4 activity to sustain ATP supply and together with the apparent enhanced mitochondrial function was able to compensate for the defect and prevent transition to a more severe phenotype.
Mitochondria-generated reactive oxygen species which clearly contribute to worsening of the cardiomyopathy was demonstrated in SLC25A4-KO mice (Narula et al. 2011). The residual SLC25A4 activity in combination with the ANT3 backup function might have prevented an excessive ROS production; in addition, the patient described by Bakker et al. (1993b) benefitted from supplementation with the antioxidant vitamin E. Currently, however, the patient is not treated with antioxidants anymore.
In summary, our index patient with only a mild mitochondrial myopathy manifesting as exercise intolerance and early signs of hypertrophic cardiomyopathy did not develop muscle weakness, ophthalmoplegia or cerebellar symptoms over a period of 20 years. In this respect our case is unique as compared to those previously described. In light of this, the absence of ophthalmoplegia seems to contrast with dominant mitochondrial disease due to SLC25A4 mutations, which is also supported by the absence of opthalmoplegia in the patients described by Palmieri et al. (2005) and Echaniz-Laguna et al. (2012). The milder phenotype in our patient is likely correlated with the nature of the two novel recessive mutations which lead to a severely reduced yet still present and partially functioning SLC25A4 protein.
Synopsis
Two novel recessive SLC25A4 mutations leading to a relatively mild phenotype without progressive external ophthalmoplegia.
Details of the Contributions of Individual Authors
IKK set up the SLC25A4 sequencing and wrote the paper. She is the guarantor. MdV provided medical information of the patient and wrote part of the paper. HB, HS, LD, LS, and RW were involved in the initial investigation and further characterization of the biochemical phenotype of the patient. FV was involved in the patient care. GN created models to assess the impact of the SLC25A4 mutations on protein structure. HS was involved in writing the paper. AH performed the NMD experiments and was involved in the setup of the SLC25A4 sequencing. BB wrote the paper and is also a guarantor. All authors have critically revised the paper.
Name of One Author Who Serves as Guarantor
Irene Körver-Keularts
Conflict of Interest
Irene Körver-Keularts, Marianne de Visser, Henk Bakker, Ronald Wanders, Fleur Vansenne, Jasper Scholte, Bert Dorland, Gerry Nicolaes, Leo Spaapen, Bert Smeets, Alexandra Hendrickx, and Bianca van den Bosch have no conflict of interest.
There are no competing interests.
The author(s) confirm(s) independence from the sponsors.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the patient and his family for being included in the study.
Footnotes
Competing interests: None declared
Contributor Information
I. M. L. W. Körver-Keularts, Email: irene.keularts@mumc.nl
Collaborators: Johannes Zschocke
References
- Adrian GS, McCammon MT, Montgomery DL, Douglas MG. Sequences required for delivery and localization of the ADP/ATP translocator to the mitochondrial inner membrane. Mol Cell Biol. 1986;6:626–634. doi: 10.1128/mcb.6.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Durbin RM, Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker HD, Scholte HR, van den Bogert C, et al. Deficiency of the adenine nucleotide translocator in muscle of a patient with myopathy and lactic acidosis: a new mitochondrial defect. Pediatr Res. 1993;33:412–417. doi: 10.1203/00006450-199304000-00019. [DOI] [PubMed] [Google Scholar]
- Bakker HD, Scholte HR, van den Bogert C, et al. Adenine nucleotide translocator deficiency in muscle: potential therapeutic value of vitamin E. J Inherit Metab Dis. 1993;16:548–552. doi: 10.1007/BF00711678. [DOI] [PubMed] [Google Scholar]
- Cozens AL, Runswick MJ, Walker JE. DNA sequences of two expressed nuclear genes for human mitochondrial ADP/ATP translocase. J Mol Biol. 1989;206:261–280. doi: 10.1016/0022-2836(89)90477-4. [DOI] [PubMed] [Google Scholar]
- Deschauer M, Hudson G, Müller T, Taylor RW, Chinnery PF, Zierz S. A novel ANT1 gene mutation with probable germline mosaicism in autosomal dominant progressive external ophthalmoplegia. Neuromuscul Disord. 2005;15:311–315. doi: 10.1016/j.nmd.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005;579:633–637. doi: 10.1016/j.febslet.2004.12.034. [DOI] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Chassagne M, Ceresuela J, et al. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J Med Genet. 2012;49:146–150. doi: 10.1136/jmedgenet-2011-100504. [DOI] [PubMed] [Google Scholar]
- Fiore C, Trezeguet V, Le Saux A, et al. The mitochondrial ADP/ATP carrier: structural, physiological and pathological aspects. Biochimie. 1998;80:137–150. doi: 10.1016/S0300-9084(98)80020-5. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Heidkämper D, Müller V, Nelson DR, Klingenberg M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations on transport and the four ATP versus ADP exchange modes. Biochemistry. 1996;35:16144–16152. doi: 10.1021/bi960668j. [DOI] [PubMed] [Google Scholar]
- Houldsworth J, Attardi G. Two distinct genes for ADP/ATP translocase are expressed at the mRNA level in adult human liver. Proc Natl Acad Sci USA. 1988;85:377–381. doi: 10.1073/pnas.85.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. Membrane protein oligomeric structure and transport function. Nature. 1981;290:449–454. doi: 10.1038/290449a0. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. Molecular aspects of the adenine nucleotide carrier from mitochondria. Arch Biochem Biophys. 1989;270:1–14. doi: 10.1016/0003-9861(89)90001-5. [DOI] [PubMed] [Google Scholar]
- Komaki H, Fukazawa T, Houzen H, Yoshida K, Nonaka I, Goto Y. A novel D104G mutation in the adenine nucleotide translocator 1 gene in autosomal dominant progressive external ophthalmoplegia patients with mitochondrial DNA with multiple deletions. Ann Neurol. 2002;51:645–648. doi: 10.1002/ana.10172. [DOI] [PubMed] [Google Scholar]
- Ku DH, Kagan J, Chen ST, Chang CD, Baserga R, Wurzel J. The human fibroblast adenine nucleotide translocator gene. Molecular cloning and sequence. J Biol Chem. 1990;265:16060–16063. [PubMed] [Google Scholar]
- Li K, Warner CK, Hodge JA, et al. A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. J Biol Chem. 1989;264:13998–14004. [PubMed] [Google Scholar]
- Napoli L, Bordoni A, Zeviani M, et al. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology. 2001;57:2295–2298. doi: 10.1212/WNL.57.12.2295. [DOI] [PubMed] [Google Scholar]
- Narula N, Zaragoza MV, Sengupta PP, et al. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging. 2011;4:1–10. doi: 10.1016/j.jcmg.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckelmann N, Li K, Wade RP, Shuster R, Wallace DC. cDNA sequence of a human skeletal muscle ADP/ATP translocator: lack of a leader peptide, divergence from a fibroblast translocator cDNA, an coevolution with mitochondrial DNA genes. Proc Natl Acad Sci USA. 1987;84:7580–7584. doi: 10.1073/pnas.84.21.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Lawson JE, Klingenberg M, Douglas MG. Site-directed mutagenesis of the yeast mitochondrial ADP/ATP translocator. Six arginines and one lysine are essential. J Mol Biol. 1993;230:1159–1170. doi: 10.1006/jmbi.1993.1233. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Alberio S, Pisano I, et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]