

Abstract
Background: Asparagine synthetase deficiency (ASD) is a newly identified neurometabolic disorder characterized by severe congenital microcephaly, severe global developmental delay, intractable seizure disorder, and spastic quadriplegia. Brain MRI showed brain atrophy, delayed myelination, and simplified gyriform pattern.
Methods: We report ASD deficiency in a 2- and 4-year-old sibling. On them, we described clinical, biochemical, and molecular findings, and we compared our results with previously reported cases.
Results: We identified a homozygous novel missense mutation in ASNS gene in both probands and we demonstrated low CSF and plasma asparagine in both patients.
Conclusions: Clinicians should suspect ASD deficiency in any newborn presented with severe congenital microcephaly followed by severe epileptic encephalopathy and global developmental delay. CSF asparagine level is low in this disorder while plasma may be low.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2014_405) contains supplementary material, which is available to authorized users.
Keywords: ASD, Asparagine, Asparagine synthetase deficiency, Inborn errors of metabolism, Neurometabolic
Introduction
Asparagine synthetase deficiency (ASD) is a new rare autosomal recessive neurometabolic disease which is caused by homozygous or compound heterozygous mutation in the ASNS gene on chromosome 7q21 (Ruzzo et al. 2013). It is described recently by Ruzzo et al. (2013) who reported nine cases from four unrelated families; they have a distinct form of severe encephalopathy associated with congenital microcephaly, progressive brain atrophy, intractable seizure, and profound developmental delay (Ruzzo et al. 2013). They had axial hypotonia with severe appendicular spasticity (Bourgeron et al. 1994; Ruzzo et al. 2013). As this is a newly described disease, there is not much data about clinical and biochemical phenotype. In this case report, we elaborate more on the clinical, biochemical, and molecular findings of 2 siblings with asparagine synthetase deficiency and we compared our results to the previously reported cases. To the best of our knowledge, this is the first time in the literature that confirmed low CSF asparagine in this disorder and the third report in the literature regarding this new inborn error of neurometabolic disorder.
Case Report
Patient 1 was a full-term baby boy born by cesarean section due to fetal distress and failure to progress to the first-cousin Saudi parents. His birth weight was 3.1 kg (10–25th percentile), length 45 cm (<5th percentile), head circumference 30 cm (<5th percentile), and Apgar score 8 and 9 at 5 and 10 min, respectively. Few hours after birth, he started to have intractable seizure. He stayed in the nursery for 6 days with myoclonic seizures until controlled with multiple antiepileptic medications. Then he was discharged on these medications and continued to have an on-and-off attack of myoclonic seizure. On examination, his growth parameters continued to be less than 5th percentile and growth arrested. Electroencephalogram (EEG) showed multifocal epileptiform discharges, diffuse slowing, and paroxysmal attenuation in sleep. Brain MRI showed severe microcephaly, brain atrophy, evidence of simplified gyral pattern, and delayed myelination (Fig. 1). Modified barium swallow showed minimal laryngeal penetration without aspiration. Upper GI study showed moderate to severe reflux with no malrotation.
Fig. 1.
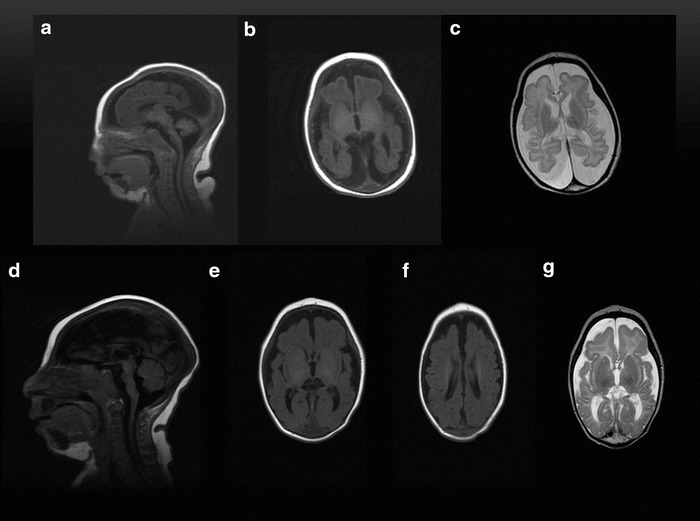
Brain MRI of a 14-day-old girl (a–c). (a) Sagittal T1 shows severe microcephaly and cerebral atrophy. (b) Axial T1 shows simplified gyriform pattern of the frontal lobes with delayed myelination. (c) Axial T2 shows normal basal ganglia. Brain MRI of a 4-month-old boy (d–g). (d) Sagittal T1 shows severe microcephaly and cerebral atrophy. (e, f) Axial T1 shows simplified gyriform pattern of the frontal lobes with delayed myelination. (g) Axial T2 shows normal basal ganglia, frontal lobes simplified gyriform pattern, and delayed myelination
Patient 2 was the sister born by normal spontaneous vaginal delivery. Her birth weight was 2.65 kg (below 10th percentile) and the head circumference was 26.5 cm (far below 5th percentile) and length was 42 cm (below 10th percentile). Apgar scores were 7 and at 1 min and 8 at 5 min. She was admitted to NICU after 5 min of age with severe microcephaly and seizure and she has the similar course and clinical phenotype as her brother.
On examination of both siblings at 2 and 4 years of age, both have severe microcephaly and all growth parameters <3rd percentile. They have subtle dysmorphic features (microcephaly, brachycephaly, pear-like head shape, micrognathia). Neurological examination showed axial hypotonia and appendicular hypertonia with hyperreflexia (Fig. 2). They continued to have severe developmental delay and not acquiring any milestones. Other system examinations were unremarkable. Both continued to have intractable seizure refractory to antiepileptic medications. Milk scan showed recurrent gastroesophageal reflux.
Fig. 2.
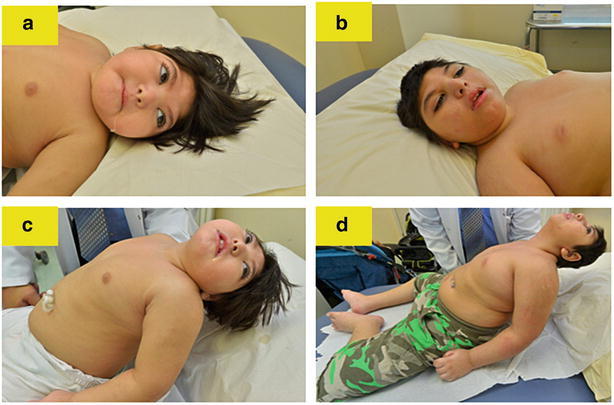
Subtle dysmorphic features: microcephaly, brachycephaly, pear-like head shape, micrognathia, and axial hypotonia (a–d)
All biochemical and molecular investigations including acylcarnitine profile and urine organic acids, creatine kinase (CK) level, total homocysteine, lactic acid, ammonia level, chromosomal analysis, and CGH microarray were unremarkable. The concentration of CSF neurotransmitters homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA) was moderately reduced. Molecular genetic testing of MC2R, PNKP, and ASPM was negative.
Further analysis by whole exome sequencing (WES) performed on both siblings and parents identified a previously unreported homozygous mutation in exon 10 of the asparagine synthetase (ASNS) gene (c.1160A>G[p.Tyr377Cys]) in both siblings. The parents confirmed to be carriers for this mutation and the discovered mutation validated by Sanger sequencing methodology (for detailed WES methodology, filtering strategy, and average coverage, please see supplementary materials).
Discussion
Our report showed the similar findings delineated by previous reports (Ruzzo et al. 2013; Ben-Salem et al. 2014). Table 1 compared the clinical findings in this report with the former ones. In 12 children described so far, all has the following cardinal features which are severe congenital microcephaly (100%), ranging between 26.5 and 31.5 cm; severe developmental delay and the patients just lying in the bed, not fixing or following and not acquiring any milestones; appendicular hypertonia; and hyperreflexia (100%). Majority of patients has additional clinical features including intractable seizure (9/12, 75%), axial hypotonia (8/12, 67%), and hyperekplexia (3/12, 25%). The type of seizure is not specific including myoclonic, tonic, spasm, and generalized tonic-clonic seizure which are refractory to antiepileptic medications. EEG pattern is reported differently but with main features such as multiple independent spike foci (8/12, 67%); other EEG findings were hypsarrhythmia, burst suppression, disorganized background. Radiologically, all patients have severe microcephaly, brain atrophy, and delayed myelination (100%). Other features include simplified gyriform pattern (9/12, 75%) and decreased size pons (7/12, 58%) (Fig. 2). The consanguinity found in 4/6 families (67%) suggests that this disorder is inherited as autosomal recessive pattern. Furthermore, the ethnicities are diverse including Iranian Jews, French Canadian, Bangladeshi, and Saudi Arabian in this report confirming that this disorder is pan-ethnic. In this report, we presented a novel missense mutation that was not described by previous reports in exon 10 of the ASNS gene. The pathogenicity and functional impact of this novel variant is supported by SIFT and MutationTaster software analyses. Furthermore, it is located in a moderately conserved nucleotide and highly conserved amino acid position, with large physiochemical differences between the amino acids tyrosine and cysteine. In addition, mutation-specific testing in both parents confirmed carrier status.
Table 1.
Demographic and clinical data of the presented cases compared to previously reported cases
| Our report | Ruzzo et al. (2013) | Ben-Salem et al. (2014) | |
|---|---|---|---|
| Number of patients | 2 | 9 | 1 |
| Number of families | 1 | 4 | 1 |
| Age | (2 years, 4 years) | 9 months to 14 years | 5 years |
| Sex (M:F) | 1:1 | 8:1 | 1:0 |
| Ethnicity | Saudi Arabian | Iranian Jews, French Canadian, Bangladeshi | Emirati |
| Consanguinity | Yes | Yes in 2 families | Yes |
| Mutation in ASNS gene | c. 1160 A>G(p.Tyr377Cys) Saudi siblings | c.1084T>G(p. phe362Val) c.1648C>T (p.Arg550Cys c.1648C>T(p. Arg550Cys)/c.17C>A(p. Ala6Glu |
c.1193A>C(p. Tyr398Cys) |
| Types of mutation | Missense and homozygous | Missense, homozygous, and compound heterozygous | Missense and homozygous |
| Developmental delay | Severe | Severe | Severe |
| Progressive microcephaly | Both | Eight out of nine | Yes |
| Axial hypotonia | Both | Five out of nine | |
| Spastic quadriplegia | Both | All | Yes |
| Seizure | Both | Six out of nine | Yes |
| Type of seizure | Both have GTC and myoclonica | Three of them have spasm, tonic, myoclonic, and GTC Two of them have partial complex One of them has tonic and orobuccal |
Myoclonic |
| EEG pattern | MISFb | Two of them have bursts and MISF Three of them have hypsarrhythmia and MISF |
MISF |
| MRI | Both have severe microcephaly, brain atrophy and delayed myelination, and simplified gyriform pattern | All have the severe microcephaly, brain atrophy, and delayed myelination. 6/9 have decreased size pons and simplified gyriform pattern | Severe microcephaly, thin corpus callosum, ventriculomegaly, brain atrophy, decreased size pons, and simplified gyriform pattern |
M male, F female
aGeneralized tonic-clonic seizure
bMultiple independent spike foci
Biochemically, asparagine synthetase also known as aspartate-ammonia ligase, symbolic as ASNS, is an enzyme involved in the biosynthesis of asparagine from aspartate through an ATP-dependent transaminated reaction (Zhang et al. 1989). This conversion takes place in the presence of glutamine which acts as amino group donor in reaction (Fig. 3) (Zhang et al. 1989; Ruzzo et al. 2013). This suggested that asparagine would be low in body fluids if this enzyme is deficient and more specifically in the brain as ASNS is highly expressed in adult brain (Hongo et al. 1996; Ruzzo et al. 2013) Although this was not demonstrated by the Ruzzo et al. (2013) report because the CSF asparagine was not measured in their patients, our findings support this theory as CSF asparagine levels were low in both siblings. Plasma asparagine levels were low in the presented cases but were only low in 2/5 of previous patients (Table 2).
Fig. 3.
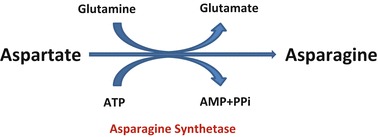
Asparagine synthetase enzyme converts aspartate to asparagine in the presence of glutamine which is the amino group donor in this reaction
Table 2.
Biochemical findings
| Amino acid levelsa | Patient 1 | Patient 2 | Ruzzo et al. (2013) and Ben-Salem et al. (2014) | % |
|---|---|---|---|---|
| Asparagine level (plasma) (33–68.4 μmol/L) | 10 μmol/L (low) | 6 μmol/L (low) | (11–57 μmol/l), 2/5 low, others: normal | 57 |
| Asparagine level (CSF) (1.1–6.9 μmol/L) | Not detected | 1 μmol/L | NA | 100 |
| Glutamine level (plasma) (254–823 μmol/L) | 339 μmol/L, normal | 328 μmol/L, normal | (439–1.250 μmol/l), 2/4 high, others: normal | 33 |
| Glutamine level (CSF) (356–680 μmol/L) | 922 μmol/L, high | 574 μmol/L, normal | NA | 50 |
NA not available
aReference ranges have been validated locally by our own population age related
ASD is one of aminoacidopathies that will be an another example which illustrate the principle of mechanism of occurring the inborn errors of metabolism which are either due to accumulation of substrate or deficiency of product or transport defect (Scriver 2001). Therefore, it will be added to other synthesis defects like creatine deficiency syndromes (Stockler et al 1994), glutamine synthetase deficiency (Haberle et al 2005), and the serine synthetic defects (van der Crabben et al. 2013). In all aforementioned disorders, the treatment is the administration of the deficient product to the affected individuals and the response was variable. It will be exciting to see the response of the patients with ASD to asparagine replacement; however, given that the phenotype is present since birth, it makes difficult to predict that such treatment will be curative (Ruzzo et al. 2013).
In conclusion, we alert the clinicians to consider ASD deficiency in any child presented with congenital microcephaly, epileptic encephalopathy and severe developmental delay. For the first time in the literature, our report confirms low CSF asparagine level in patients with ASD deficiency.
Electronic Supplementary Material
Take-Home Message
Asparagine synthetase deficiency is characterized by severe congenital microcephaly followed by childhood global developmental delay, central hypotonia, spastic quadriplegia and intractable seizure disorder. CSF asparagine level is low in this disorder while plasma may be low. Brain MRI showed microcephaly, brain atrophy, and delayed myelination and simplified gyriform pattern.
Conflict of Interest
Majid Alfadhel, Muhammad Talal Alrifai, Daniel Trujillano, Hesham Alshaalan, Ali Al Othaim, Shatha Al Rasheed, Hussam Assiri, Abdulrhman A Alqahtani, Manal Alaamery, Arndt Rolfs, and Wafaa Eyaid declare that they have no conflict of interest.
Informed Consent
Informed consent was obtained from parents of the patients included in the study. Proof that informed consent was obtained is available upon request.
Authors’ Contributions
MAF performed the majority of work associated with preparing, writing, and submitting the manuscript and contributed to the clinical diagnosis and management of the patients. MTR edited the manuscript and contributed to the clinical diagnosis and management of the patients. DT performed the molecular testing and contributed to the diagnosis of the patients and editing the manuscript. HA assessed and described the radiological findings obtained from the patients. AA edited the manuscript and contributed to biochemical investigations of the patients. SR edited the manuscript and contributed to clinical management of the patients. HAS summarized the clinical findings and contributed to the writing of the first draft. AQ summarized the clinical findings and contributed to the writing of the first draft. AR edited the manuscript, performed the molecular testing, and contributed to the diagnosis of the patients. WE edited the manuscript and contributed to the clinical diagnosis and management of the patients.
Contributor Information
Majid Alfadhel, Email: dralfadhelm@gmail.com.
Collaborators: Johannes Zschocke
References
- Ben-Salem S, Gleeson JG, Al-Shamsi AM et al (2014) Asparagine synthetase deficiency detected by whole exome sequencing causes congenital microcephaly, epileptic encephalopathy and psychomotor delay. Metab Brain Dis. doi:10.1007/s1011-014-9618-0 [DOI] [PMC free article] [PubMed]
- Bourgeron T, Chretien D, Poggi-Bach J, et al. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J Clin Invest. 1994;93:2514. doi: 10.1172/JCI117261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Rutsch F, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. 2005;353:1926–1933. doi: 10.1056/NEJMoa050456. [DOI] [PubMed] [Google Scholar]
- Hongo S, Chiyo T, Takeda M. Cloning of cDNA for asparagine synthetase from rat Sertoli cell. Biochem Mol Biol Int. 1996;38:189–196. [PubMed] [Google Scholar]
- Ruzzo EK, Capo-Chichi J-M, Ben-Zeev B, et al. Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron. 2013;80:429–441. doi: 10.1016/j.neuron.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriver CR (2001) The metabolic & molecular bases of inherited disease. McGraw-Hill, New York/London
- Stockler S, Holzbach U, Hanefeld F, et al. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res. 1994;36:409–413. doi: 10.1203/00006450-199409000-00023. [DOI] [PubMed] [Google Scholar]
- van der Crabben SN, Verhoeven-Duif NM, Brilstra EH, et al. An update on serine deficiency disorders. J Inherit Metab Dis. 2013;36:613–619. doi: 10.1007/s10545-013-9592-4. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lambert MA, Cairney AE, Wills D, Ray PN, Andrulis IL. Molecular structure of the human asparagine synthetase gene. Genomics. 1989;4:259–265. doi: 10.1016/0888-7543(89)90329-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.