

Abstract
We report about a patient with infantile-onset neurodegenerative disease associated with isolated mitochondrial respiratory chain complex III (cIII) deficiency. The boy, now 13 years old, presented with language regression and ataxia at 4 years of age and then showed a progressive course resulting in the loss of autonomous gait and speaking during the following 2 years. Brain MRI disclosed bilateral striatal necrosis. Sequencing of a panel containing nuclear genes associated with cIII deficiency revealed a previously undescribed homozygous rearrangement (c.782_786delinsGAAAAG) in TTC19 gene, which results in a frameshift with premature termination (p.Glu261Glyfs*8). TTC19 protein was absent in patient’s fibroblasts.
TTC19 encodes tetratricopeptide 19, a putative assembly factor for cIII. To date TTC19 mutations have been reported only in few cases, invariably associated with cIII deficiency, but presenting heterogeneous clinical phenotypes. We reviewed the genetic, biochemical, clinical and neuroradiological features of TTC19 mutant patients described to date.
Introduction
Isolated deficiency of mitochondrial respiratory chain (MRC) complex III (cIII) (MIM#124000) is a rare cause of mitochondrial disorder. Excluding mutations in MTCYB, the mitochondrial DNA (mtDNA) gene encoding cytochrome b, mutations in nuclear genes encoding other structural cIII subunits are extremely uncommon (Ghezzi and Zeviani 2012; Miyake et al. 2013). Contrariwise, several genetic defects have been described in cIII assembly factor genes, including BCS1L (De Lonlay et al. 2001), TTC19 (Ghezzi et al. 2011), and LYRM7 (Invernizzi et al. 2013). TTC19 mutations have been reported in few patients with heterogeneous phenotypes ranging from early onset neurodegenerative disorders (Ghezzi et al. 2011; Atwal 2013; Balasubramaniam et al. 2012) to adult forms with psychiatric manifestations and cerebellar ataxia (Nogueira et al. 2013; Morino et al. 2014). An overall evaluation of known TTC19 mutant cases including genotype/phenotype correlations has not been performed so far.
Bilateral striatal necrosis (BSN) includes a group of syndromes, usually with onset in infancy, characterized by bilateral and symmetrical degeneration of caudate and putamen nuclei (neostriatum). BSN results from either toxic exposure, infections and metabolic or neurodegenerative disorders (Zevit et al. 2007), including some mitochondrial diseases.
We report a novel deleterious mutation in TTC19, identified in a patient presenting with BSN and isolated mitochondrial cIII deficiency. In addition we report a review of previously published TTC19 patients.
Case Report
Our proband is a boy, second child of healthy related (first cousins) parents of Moroccan origin. Two younger sisters are in good health; another sister died 1 week after birth for unknown causes. He was born at term after uneventful pregnancy; psychomotor development was normal, but delay in language skills with possible deafness was referred. Since the age of 4 years, he presented with walking impairment and language regression that progressively worsened during the following 2 years leading to loss of autonomous gait and speaking. He was first examined at 9 years of age when he established in Italy. He showed low body weight and height (≤3rd percentile), diffuse muscle wasting, severe spastic tetraparesis with marked dystonic postures involving both upper and lower limbs and severe cognitive impairment. No epilepsy was reported. Blood routine exams, including copper and ceruloplasmin levels and urinary organic acids, were normal. Elevated levels of lactate in plasma (4,269 umol/L; n.v. 580–2,100) and CSF (3,806 umol/L; n.v. 800–2,100) and of pyruvate in plasma (208 umol/L; n.v. 55–145) and CSF (180 umol/L; n.v. 45–135) were detected.
Fundus oculi, motor and sensory nerve conduction velocities, and electroretinogram resulted normal. Visual and brainstem auditory evoked potentials showed bilateral increased latency and decreased amplitude. Electroencephalography showed normal background activity and few diffuse paroxysmal abnormalities during sleep. Brain MRI showed bilateral hyperintensities of putamen and caudate bodies (Fig. 1); no calcifications were present at CT.
Fig. 1.
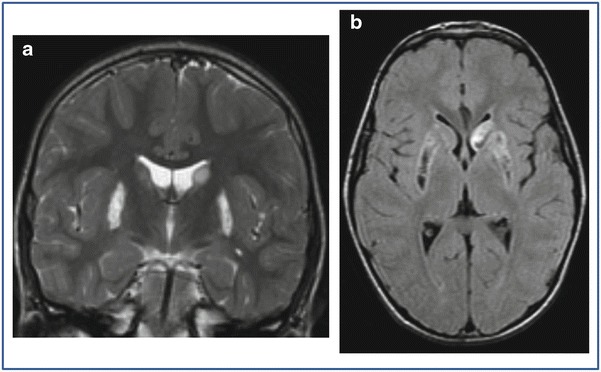
Bilateral hyperintensities of the putamen and caudate bodies (more evident in left side) in coronal T2-weighted image (a), with a cavitated appearance on transverse FLAIR sequence (b)
Histological analysis of patient’s muscle biopsy showed few hypotrophic fibers, with normal lipids and glycogen content. An isolated cIII deficiency (50%) in muscle homogenate was documented, whereas all MRC complex activities were normal in fibroblasts.
Treatment with coenzymeQ10, thiamine, and riboflavin was started, associated with symptomatic therapy with baclofen. The patient is now 13 years old, and his clinical condition appears stable.
Targeted resequencing of a panel containing nuclear genes associated with cIII deficiency (BCS1L, TTC19, LYRM7, UQCRB, UQCRQ) revealed the presence of a homozygous variant in TTC19, a novel rearrangement (c.782_786delinsGAAAAG) resulting in a frameshift with a predicted premature termination (p.Glu261Glyfs*8) (Fig. 2a, b). The mutation was found to be heterozygous in both parents and absent in two healthy siblings. Quantitative PCR revealed a marked reduction of the TTC19 transcript in patient’s fibroblasts (Fig. 2c), and Western blot analysis showed the absence of TTC19 protein (Fig. 2d).
Fig. 2.
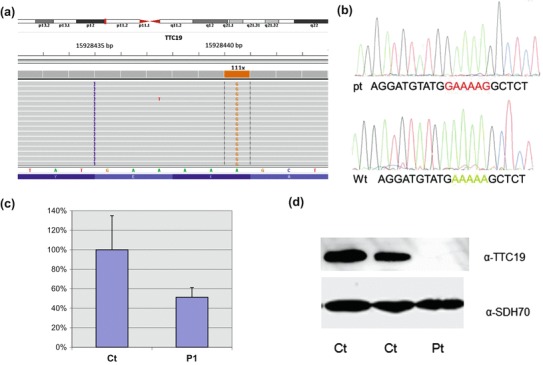
(a) Snapshot from IGV software of the mutation identified in the patient. A customized gene panel (TSCA, Illumina) was sequenced using a MiSeq system. (b) Electropherograms of TTC19 gene showing the homozygous rearrangement (c.782_786delinsGAAAAG) found in the patient (pt) resulting in a frameshift with premature termination (p.Glu261Glyfs*8); wild-type (wt) sequence in a control subject is also shown. (c) Quantitative real-time PCR of TTC19 mRNA in control (Ct) and patient’s (Pt) fibroblasts. (d) Immunoblot analysis of lysates from control and patient’s fibroblasts using α-TTC19 and α-SDH70 antibodies
Discussion and Conclusion
Mutations in TTC19 have been rarely described in patients with mitochondrial diseases. Eleven individuals, presenting with infantile or juvenile-adult onset, and eight different TTC19 mutations have been reported so far (Table 1).
Table 1.
Clinical, instrumental, and laboratory findings in patients carrying TTC19 mutations
| Patients | P1a | P2a | P3a | P4a | P5b | P6b | P7b | P8b | P9c | P10d | P11e | P12f |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age of onset/gender | 5 year/f | 10 year/m | 5 year/f | 42 year/m | 27 year/m | Adolescence/f | Adolescence/m | 34 year/f | 13 month/ m | 31 year/f | ?/f | 4 year/m |
| Clinical presentation at onset | Cognitive impairment ataxia | Cognitive impairment ataxia | Cognitive impairment (language regression, lack of interest), ataxia | Subacute muscle weakness | Psychiatric symptoms, ataxia | Psychiatric symptoms | Psychiatric symptoms | Psychiatric symptoms | Development delay, language regression | Dysarthria | ? | Cognitive impairment (language regression) ataxia |
| Evolution | Slowly progressive | Rapidly progressive | Progressive and rapid worsened by cardiorespiratory arrest | Rapidly progressive | Rapidly progressive | Rapidly progressive | Rapidly progressive | Rapidly progressive | Slowly progressive | Rapidly progressive | Progressive | Rapidly progressive |
| Clinical presentation at diagnosis time | Cerebellar signs, pyramidal signs (right side) | Cerebellar signs, focal dystonia, bilateral hearing loss, severe cognitive deterioration | Cerebellar signs, focal dystonia, severe cognitive deterioration | Gait apraxia, dysarthria bradykinesia, dystonia, paraparesis, psychiatric symptoms | Ataxia, pyramidal signs, psychiatric symptoms | Ataxia, pyramidal signs, muscle atrophy, psychiatric symptoms | Mild ataxia, mild pyramidal and extrapyramidal signs, psychiatric symptoms | Mild ataxia, mild pyramidal and extrapyramidal signs, psychiatric symptoms | Development delay | Ataxia, cognitive impairment, pes cavus | Encephalomyopathy, failure to thrive | Spastic tetraparesis dystonic posture, severe cognitive impairment |
| Outcome (last observation) | Wheelchair, upper limb ataxia (37 years) | Bedridden, fluctuating comatose status, PEG-tracheostomy (26 years) | Bedridden, fluctuating comatose status, PEG-assisted ventilation | Deceased (45 years) | Deceased for respiratory insufficiency (49 years) | Deceased for respiratory insufficiency (33 years) | Deceased for cardiac arrest (30 years) | Stable (38 years) | Stable (4 years) | Wheelchair (34 years) | Encephalomyopathy, failure to thrive (8 years) | Stable |
| MRI features | CA, L, IO, SN, PG, both C, left P | n.a. | IO, PG, T, CA | CoA, right C, both P | OPCA, BSN, MO, D CoA | OPCA, BSN, MO, D CoA | OPCA, BSN, MO, D | OPCA, BSN, MO, D | BSN, involvement of brainstem | CA, IO | Bilateral P, Pa | BSN |
| TTC19 mutations | c.656T>G/c.656 T>G | c.656T>G/c.656T>G | c.656T>G/c.656T>G | c.517C>T/c.517C>T | c.963_966delTGGC/c.963_966delTGGC | c.963_966delTGGC/c.963_966delTGGC | c.963_966delTGGC/ c.963_966delTGGC | c.963_966delTGGC/c.963_966delTGGC | c.577G>A/c.964_967delGGCT | c.829C>T/c.829C>T | c.937C>T/c.829C>T | c.782_786delinsGAAAAG/c.782_786delinsGAAAAG |
| ENG | Axonal motor neuropathy | n.a. | Axonal motor neuropathy | Axonal motor neuropathy | Axonal motor neuropathy | n.a. | Axonal motor neuropathy | n.a | n.a. | n.a. | n.a. | Normal |
| Lactate level | Normal (plasma) | n.a. | High | n.a. | High | n.a. | n.a | n.a | n.a. | Normal/high 6 y after onset | Normal | High |
| Complex III deficit in muscle | 19% | 14% | 17% | 8% | 30% | 31% | 33% | 39% | I + III 79%, II 52%, II + III 36%, IV 46% | n.a. | n.a. | 50% |
CA cerebellar atrophy, L leukodystrophy, OPCA olivopontocerebellar atrophy, CoA cerebral cortical atrophy, Profound gray matter involvement/necrosis in IO inferior olives, SN substantia nigra, PG periaqueductal gray matter, C caudate nuclei, P putamina, Pa pallidus nuclei, T thalami, P pons, D dentate nuclei, MO medullary olives, BSN bilateral striatal necrosis
aGhezzi et al. (2011)
bNogueira et al. (2013)
cAtwal (2013)
dMorino et al. (2014)
eBalasubramaniam et al. (2012)
fThis report
Ghezzi et al. first reported in 2011 the presence of mutations in TTC19 in four Italian patients. Three of these cases (P1–P3) carried a homozygous nucleotide change (c.656T>C; p.Leu219*) predicting the synthesis of a truncated protein and presented with infantile onset of cognitive impairment and ataxia. Two patients (P2, P3) showed a rapidly progressive course characterized by severe cognitive regression, cerebellar and extrapyramidal signs, hearing loss and severe axonal neuropathy. They became bedridden, presented a fluctuating comatose state requiring assisted ventilation and percutaneous endoscopic gastrostomy. Brain MRI of P3 showed progressive necrotic lesions in the brainstem and thalami and cerebellar atrophy. P1, sister of P2 and carrying the same mutation, presented a slowly progression of cerebellar and pyramidal signs and developed axonal motor neuropathy, becoming wheelchair bound since the age of 24 years. Brain MRI showed multifocal involvement of the deep gray matter, severe cerebellar atrophy and leukodystrophy. The fourth patient (P4) carried another homozygous nonsense mutation (c.517C>T; p.Gln173*). He presented with an adulthood subacute onset and rapidly progressive multisystemic neurological disease resulting in death 3 years later. Brain MRI showed diffuse cortical atrophy and right caudate and bilateral putamina involvement. Blood lactate was normal in P1, increased in P3 and not investigated in the others. A marked reduction of cIII activity in muscle homogenate was revealed in all patients. Decreased TTC19 mRNA and virtual absence of the protein in muscle tissue and/or fibroblasts were also detected.
Nogueira et al. 2013 reported on a consanguineous family with four affected siblings (P5–P8) carrying a novel mutation (c.600_604delTGGC) predicting a frameshift and the synthesis of a truncated TTC19 protein (p.Ala200Alafs*8). The clinical phenotype was characterized by late onset (ranging from adolescence to adulthood) with psychiatric symptoms; subsequently patients displayed cerebellar signs and mild pyramidal and extrapyramidal signs. In two siblings (P5, P7) an axonal motor neuropathy was also present. Three patients (P5–P7) showed a progressive course resulting in death about 20 years after onset; the only alive patient (P8) resulted stable 4 years after the disease onset. MRI showed a severe olivopontocerebellar atrophy associated with necrosis in the caudate, putamina and medullary olives. High lactate levels are reported only in one patient (P5), while a marked isolated cIII deficiency was documented in all muscle samples.
An additional case (P9), compound heterozygous for the c.600_603delTGGC and a novel nonsense mutation (c.195G>A; p.Trp65*), was reported by Atwal. He presented with psychomotor delay, followed by language regression at 13 months, and subsequent stable clinical course until the last observation (4 years of age). Brain MRI disclosed necrosis in the putamina, caudate and brainstem. Muscle biopsy showed decreased activity of MRC complexes I + III, II, II + III and IV.
More recently, Morino at al. described a woman (P10) with a spinocerebellar phenotype. She had an adulthood onset with dysarthria at 31 years of age and rapidly developed cognitive impairment, ataxia, and peripheral signs (pes cavus) leading to loss of autonomous gait 3 years later. MRI showed involvement of inferior olives and cerebellar atrophy. Lactate level was normal at onset, but resulted elevated in subsequent measurements. Muscle biopsy was not performed. Exome sequencing revealed a novel homozygous nonsense mutation (c.829C>T; p.Gln277*) in TTC19.
Finally, Balasubramaniam et al. reported the case of a girl (P11) affected with a slowly progressive encephalomyopathy and MRI abnormalities in lentiform nuclei associated with a novel homozygous nonsense mutation (c.574C>T; p.Gln192*). Plasma and CSF lactate levels were reported normal.
Our patient (P12 in Table 1) is the fifth infantile case associated with TTC19 mutation. The clinical onset and the rapidly progressive course leading to spastic tetraparesis, dystonic postures, and marked cognitive impairment were similar to other infantile cases. Both our patient and previous cases had no seizures. Axonal motor neuropathy, described in some TTC19 mutant individuals, was not present in P12.
The biochemical profile with marked cIII deficiency was consistent in all reported cases, although in P9 reduction of other MRC complex activities were found.
P12 brain MRI showed BSN. In 10/11 cases reported to date, brain MRI was available; the most common alterations were basal ganglia necrosis and subtentorial involvement, including deep gray matter and cerebellar atrophy. MRI alterations, always severe, were similar in infantile and adult cases, in spite of a different clinical phenotype. Our patient and P11 were the only cases without subtentorial involvement.
BSN has been already reported as a typical MRI feature in mitochondrial diseases caused by mutations in either mitochondrial genes (ATP6, ND1, and ND6 encoding subunits of respiratory chain complex V and I, respectively) (Lal et al. 2013; Campos et al. 2013; Solano et al. 2003) or in nuclear genes (NDUFV1 and NDUFS4 encoding complex I subunits) (Lal et al. 2013; Budde et al. 2003); AIFM1 encoding apoptosis-inducing factor, mitochondrial 1 (Ghezzi et al. 2010); and SLC25A19 encoding a mitochondrial thiamine pyrophosphate transporter (Spiegel et al. 2009). Thus, TTC19 must be added to the list of genes associated with infantile mitochondrial disorders and bilateral striatal necrosis.
Our report and the review of previous cases carrying TTC19 mutations demonstrate that ataxia and impairment of cortical functions leading to language or cognitive regression are the clinical hallmarks of infantile-onset forms, whereas psychiatric symptoms are typical of juvenile-adult forms. Axonal motor neuropathy is frequent but not always present. MRI pattern, characterized by both supra- or subtentorial gray matter involvement and cerebellar atrophy, shows some common features in spite of different age of onset.
Decreased cIII activity was present in all patients reported to date. Nevertheless, lactate levels may be normal and lactic acidosis is not a reliable biomarker.
The review of published TTC19 mutant cases does not suggest a strict and univocal genotype/phenotype correlation, but underlines that TTC19-related diseases can be severely disabling. Since almost all known TTC19 mutations were associated with or are predicted to result in the absence of the protein, the differences in disease severity could not be easily ascribed to diverse deleterious effects of different mutations. A larger set of TTC19 mutant patients should be examined, and the use of unbiased approaches for genetic analysis, such as targeted resequencing of broad gene panel or exome sequencing, would allow to better define the clinical spectrum of diseases caused by TTC19 mutations.
Acknowledgments
This work was supported by Fondazione Pierfranco e Luisa Mariani (CM23), Fondazione Telethon (grant GGP11011), Cell line and DNA Bank of Genetic Movement Disorders and Mitochondrial Disesases of Telethon Network of Genetics Biobanks (grant GTB12001), Italian Ministry of Health (GR 2010–2316392) and the Italian Association of Mitochondrial Disease Patients and Families (Mitocon ONLUS)
Synopsis
We describe a novel deleterious mutation in TTC19 associated with early onset severe and progressive neurological deterioration. The review of previous cases does not suggest a strict and univocal genotype/phenotype correlation, but demonstrates that some clinical hallmarks and common MRI pattern are available in spite of different age of onset. Biochemical (decreased cIII activity) and molecular data (all known TTC19 mutations were associated with or are predicted to result in the absence of the protein) are common in spite of different phenotypes.
Compliance with Ethics Guidelines
Conflict of Interest
Dr. Ardissone reports no conflict of interest.
Dr. Granata reports no conflict of interest.
Dr. Legati reports no conflict of interest.
Dr. Diodato reports no conflict of interest.
Dr. Melchionda reports no conflict of interest.
Dr. Lamantea reports no conflict of interest.
Dr. Garavaglia received funding and grants for research from Fondazione Mariani (CM23), Telethon Network of Genetics Biobank (GTB12001).
Dr. Ghezzi received funding and grants for research from the Italian Ministry of Health, Telethon Italy (GGP11011), and CARIPLO foundation.
Dr. Moroni received funding and grants for research from Telethon Italy (GUP11001; GUP13006).
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from patient’s parents for being included in the study.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Details of the Contributions of Individual Authors
AA, TG, and IM evaluated the patient and wrote the case report. LM and DD performed genetic analyses; AL analyzed targeted resequencing data. EL performed biochemical analyses under the supervision of BG. DG monitored genetic/protein analyses. AA and DG wrote the manuscript; IM critically revised the manuscript for important intellectual content. All authors read and approved the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Anna Ardissone, Email: anna.ardissone@istituto-besta.it.
Collaborators: Johannes Zschocke
References
- Atwal PS. Mutations in the complex III assembly factor tetratricopeptide 19 gene TTC19 are a rare cause of Leigh syndrome. JIMD Rep. 2013;25:43–45. doi: 10.1007/8904_2013_282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam et al. (2012) Human Genetics Society of Australasia Meeting 2012
- Budde SM, van den Heuvel LP, Smeets RJ, et al. Clinical heterogeneity in patients with mutations in the NDUFS4 gene of mitochondrial complex I. J Inherit Metab Dis. 2003;200326(8):813–815. doi: 10.1023/B:BOLI.0000010003.14113.af. [DOI] [PubMed] [Google Scholar]
- Campos Y, Martín MA, Rubio JC, Gutiérrez del Olmo MC, Cabello A, Arenas J. Bilateral striatal necrosis and MELAS associated with a new T3308C mutation in the mitochondrial ND1 gene. Neurogenetics. 2013;14(1):85–87. doi: 10.1007/s10048-013-0355-z. [DOI] [PubMed] [Google Scholar]
- De Lonlay P, Valnot I, Barrientos A, et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29(1):57–60. doi: 10.1038/ng706. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Arzuffi P, Zordan M, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43(3):259–263. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86(4):639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- Invernizzi F, Tigano M, Dallabona C, et al. A homozygous mutation in LYRM7/MZM1L associated with early onset encephalopathy, lactic acidosis, and severe reduction of mitochondrial complex III activity. Hum Mutat. 2013;34(12):1619–1622. doi: 10.1002/humu.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D, Becker K, Motameny S, et al. Homozygous missense mutation of NDUFV1 as the cause of infantile bilateral striatal necrosis. Neurogenetics. 2013;14(1):85–87. doi: 10.1007/s10048-013-0355-z. [DOI] [PubMed] [Google Scholar]
- Miyake N, Yano S, Sakai C, et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum Mutat. 2013;34:446–452. doi: 10.1002/humu.22257. [DOI] [PubMed] [Google Scholar]
- Morino H, Miyamoto R, Ohnishi S, Maruyama H, Kawakami H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014;14:5. doi: 10.1186/1471-2377-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira C, Barros J, Sá MJ, et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics. 2013;14(2):153–160. doi: 10.1007/s10048-013-0361-1. [DOI] [PubMed] [Google Scholar]
- Solano A, Roig M, Vives-Bauza C, Hernandez-Peña J, et al. Bilateral striatal necrosis associated with a novel mutation in the mitochondrial ND6 gene. Ann Neurol. 2003;54(4):527–530. doi: 10.1002/ana.10682. [DOI] [PubMed] [Google Scholar]
- Spiegel R, Shaag A, Edvardson S, et al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66(3):419–424. doi: 10.1002/ana.21752. [DOI] [PubMed] [Google Scholar]
- Zevit N, Steinmetz A, Kornreich L, Straussberg R. Acute infantile bilateral striatal necrosis: single-photon emission computed tomography (SPECT) imaging and review. J Child Neurol. 2007;22(10):1222–1226. doi: 10.1177/0883073807304194. [DOI] [PubMed] [Google Scholar]