

Abstract
Objective/context: We describe the second patient presenting the combination of two homoallelic homozygous nonsense mutations in two genes distant from 1.8 Mb in the chromosome 2p13-3, the methylmalonyl-CoA epimerase gene (MCEE) and the sepiapterin reductase gene (SPR).
Case report: The patient was born from consanguineous parents. He has presented a moderate but constant methylmalonic acid (MMA) excretion in urine associated with a mental retardation. The first homozygous mutation was identified in the MCEE gene (c.139C>T; p.Arg47*). Progressive dystonia and cataplexy narcolepsy led to diagnose the second homozygous mutation in the SPR gene: c.751A>T; p.Lys251*. Sepiapterin reductase deficiency (SRD) was characterized by a defect in tetrahydrobiopterin (BH4), the cofactor of several hydroxylases needed for the synthesis of neurotransmitters. A treatment with l-DOPA/carbidopa and 5-HTP dramatically improved the dystonic posture, the mood and the hypersomnia, proving that the pathogenesis was due to SRD. A supplementation with BH4 did not induce additional clinical benefit, although HVA and HIAA increased in CSF. The polyunsaturated fatty acids were measured in CSF as the markers of the neuronal stress. We have shown that DHA and its precursor EPA were high before and during the time course of the different treatments.
In conclusion: The patient has inherited two copies of the two mutations from his consanguineous parents in the MCEE and SPR genes in the chromosome 2p13-3. DHA and EPA increased in CSF as a response to the neuronal stress induced by the defect in neurotransmitters or the altered metabolism of the odd-chain fatty acids and cholesterol.
Keywords: BH4, CSF PUFA level, Methylmalonyl-CoA epimerase deficiency, Sepiapterin reductase deficiency
Introduction
Sepiapterin reductase (SPR MIM 612716) and methylmalonyl-CoA epimerase (MCEE MIM 608419) genes are mapped on chromosome 2p13-3, 1.8 Mb one of the other (Fig. 1). Catabolic pathways for the amino acids isoleucine, valine, methionine and threonine as well as for the odd-chain fatty acids and cholesterol proceeded via propionyl-CoA which is converted to (2S)-methylmalonyl-CoA by the propionyl-CoA carboxylase. Methylmalonyl-CoA is then isomerized into its (2R)-enantiomer by the action of the MCEE enzyme. In contrast to the (2R)-methylmalonyl-coenzyme A ([2R]-methylmalonyl-CoA) mutase (MIM 609058) deficiency which leads to a high excretion of methylmalonic acid, MCEE deficiency is responsible for a mild increase of methylmalonic aciduria, probably because of an in vivo shunt from the propionate-to-succinate pathway. Mutations in the MCEE gene were described in only five patients, identified by a moderate methylmalonic acid excretion in urine (Gradinger et al. 2007).
Fig. 1.
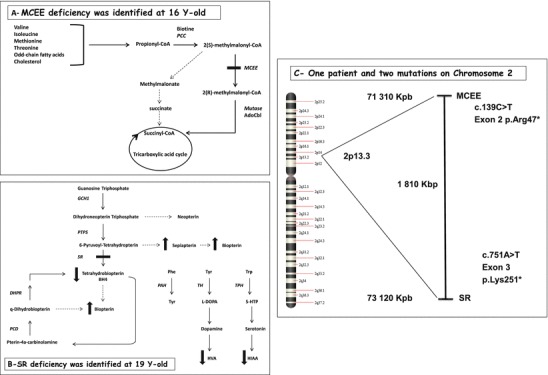
(a) The propionate-to-succinate pathway, propionyl-CoA carboxylase (PCC), 5′-deoxyadenosylcobalamin (AdoCbl); (b) metabolism of the BH4 cofactor, GTP cyclohydrolase I (GCH1), dihydropteridine reductase (DHPR), 6-pyruvoyl tetrahydropterin synthase (PTPS), pterin 4α-carbinolamine dehydratase (PCD), phenylalanine hydroxylase (PAH), tyrosine hydroxylase (TH), tryptophan hydroxylase (TPH); (c) localization of the SR and MCEE genes on chromosome 2. The hatched line represents a non-enzymatic step or an unknown shunt pathway
On the contrary, sepiapterin reductase deficiency (SRD) is a severe autosomal recessive disorder of tetrahydrobiopterin (BH4) metabolism (Friedman et al. 2012). BH4 is synthesized de novo from guanosine triphosphate (GTP) by three enzymatic steps: GTP cyclohydrolase I, 6-pyruvoyl-tetrahydrobiopterin synthase and the SPR enzyme to reduce 6-pyruvoyl-tetrahydropterin into BH4. BH4 is an essential cofactor required by phenylalanine, tyrosine and tryptophan hydroxylases which catalyse the rate-limiting steps in the biosynthesis of neurotransmitters, dopamine and serotonin, respectively. A defect in BH4 explains the neurological features. Most of the patients present symptoms in the first years of age, but the diagnosis is delayed when neurotransmitters in CSF are missed. Commonly, patients exhibit progressive psychomotor retardation, tremor, seizures, oculogyric crises and notably dystonia with diurnal fluctuations (Clot et al. 2009). Sleep disorders and marked hypersomnolence are also described (Friedman et al. 2006). To date, 13 different mutations for 44 subjects have been reported in the database (http://www.biopku.org) (Koht et al. 2014). SRD is a potentially treatable inborn error of pterin metabolism with a response to l-DOPA/carbidopa and 5-hydroxytryptophan (5-HTP).
Polyunsaturated long-chain fatty acids (PUFAs) were analysed in CSF as markers of neuronal deterioration (Engstrom et al. 2009) (Fig. 2). Here, we present the second patient described with combined SRD and MCEE deficiencies (Abeling et al. 2006; Bikker et al. 2006). Permanent but moderate methylmalonic aciduria led to firstly identify MCEE deficiency. A progressive neurological deterioration with a global hypotonia and hypersomnia led to secondarily diagnose SRD with neurotransmitters in CSF.
Fig. 2.
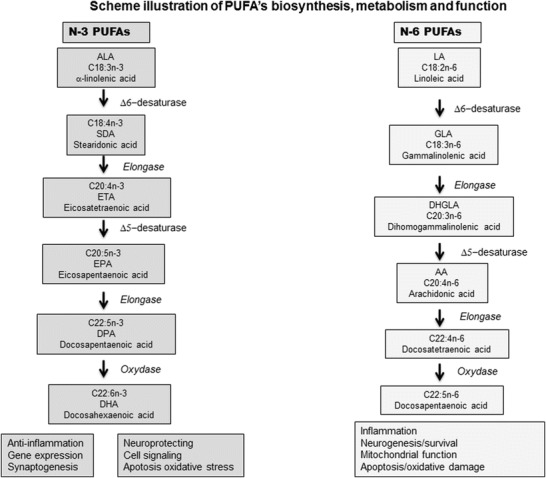
Scheme illustrating PUFA biosynthesis, metabolism and function. PUFA can be provided directly by the diet or can be synthesized from their respective essential dietary precursors, α-linolenic acid (ALA, 18:3n-3) and linoleic acid (LA, 18:2n-6). For example, DHA (22:6w3) is composed of 22 carbon atoms and six double bonds, and its upstream metabolic precursor is EPA (20:5w3). Because the first double bond, as counted from the methyl terminus, is at position 3, they belong to the omega-3 group. Two key enzymes, the Δ6 and Δ5 desaturases, catalyse the desaturation and elongation of ALA and LA in the endoplasmic reticulum and in one terminal cycle of β-oxidation in the peroxisome
Methods
Neurotransmitter Analysis in CSF
Biogenic amines, pterins, methyltetrahydrofolate and sepiapterin were analysed as previously described (Ormazabal et al. 2005; Zorzi et al. 2002).
Molecular Analysis
DNA was extracted from the white blood cells of the patient and his parents, after the informed consent. After the amplification, all the exons of the SPR and MCEE genes were sequenced and compared to the reference sequences (Big Dye TM Terminator v3.0 kit, cycle sequencing on the ABI 3130, Applied Biosystems Forster City, CA). Karyotype and sub-telomeric MLPA (kit SALSA P036) were used for genetic investigations.
GC-MS PUFA Analysis
Total lipids were extracted from CSF (100 μl) by 10 ml of chloroform/methanol (1:2 v/v) (Bligh and Dyer 1959). The chloroform lipid-rich lower layer was evaporated to dryness. Dried lipids were derivatized to fatty acid methyl esters with 6 ml of methanol/sulphuric acid 2% (v/v) for 2 h at 70°C and then extracted by hexane. The extract (2 μl) was separated and quantified by GC-MS (Trace DSQ2, Thermo Electron, Les Ulis, France) using the positive chemical ionization mode. Fatty acid methyl esters (n = 17) were presently detected. The data were analysed using Qual Browser® software (Xcalibur® version 2.0.7, Thermo Electron, Les Ulis, France). The results were statistically analysed with a comparison of a single case to control normative samples by developing the Bayesian approach (Crawford and Garthwaite 2007).
Case Story
The patient was the second son born from French consanguineous parents. Two cousins presented methylmalonyl-CoA deficiency with neonatal coma at birth. At the age of 12 months, renal cysts led to the surgical ablation of the patient’s left kidney. From the first month of life, he was a floppy baby with many episodes of eyes rolling up. He walked at the age of 21 months. Cognitive impairment with an intelligence quotient of 55 and limited speech were noted at the age of 5. At the age of 7, axial hypotonia, postural instability, oculogyric crisis and fatigability with sleep disorders were persistent. A moderated but persisting excretion of urinary methyl malonic acid was identified as soon as the age of 9 (60 μmol/mmol creatinine/mmol creatinine; rv <2). C14 propionate incorporation into macromolecules was slightly decreased, suggesting a defect in the propionate-to-succinate pathways (data not shown). However, the methylmalonic-CoA mutase activity was normal.
From the age of 7 to 18, the patient presented hypersomnolence requiring daily long sleeps. The circadian rhythm was distorted with an ultradian sleep-wake activity. The abnormal movements induced dystonia with peripheral hypertonia worsening at the evening. The neurological deteriorations progressively induced limb blockades and abnormal eye movements. The boy became wheelchair-bound at the age of 16. Cystic thyroid nodules led to thyroidectomy. The brain MRI was normal. X-linked mental retardation, Smith-Magenis and Willi-Prader syndromes, chromosomal rearrangement and CGH array abnormalities were excluded. At the age of 16, a homozygous nonsense mutation in the MCEE gene, c139C>T inducing an early terminating signal (p.R47*) in the MCEE enzyme, led to identify MCEE deficiency, an unclassified form of methylmalonic aciduria. Both parents were heterozygous for the mutation and excreted normal amounts of methylmalonic acid. The supplementations with carnitine 4 g/day (Levocarnil®), oral hydroxocobalamin 1 mg/day and the antiepileptic sodium valproate up to 30 mg/kg/day did not improve the clinical evolution. The hypotonia, the psychomotor retardation and the fatigability were not considered to be suggestive of a neurotransmitter defect. At the age of 19, the sleep disturbances led to investigate long-term EEG and CSF neurotransmitters. A long-duration video-EEG sleep study (data not shown) clearly showed some asymptomatic spike wave discharges and an ultradian sleep-wake rhythm. The rapid eye movement sleeps represented more than 50% of the total sleep activity, and cataplexic narcolepsy was identified. Orexin A/hypocretin level was normal in CSF. A light elevation of prolactin level (31 ng/ml; normal range 2–20) was noted. We measured the low levels of HIAA and HVA associated with a high level of BP in CSF. We also quantified sepiapterin (15 nM) which is the specific marker for SRD, and thus, we have confirmed the diagnosis of SRD, explaining the aetiology of cataplexic narcolepsy (Table 1 and Fig. 1b). The homozygous nonsense mutation was identified in the SR gene c.751A>T and led to a truncated protein (p.Lys251*). The parents were also both heterozygous for this mutation. l-DOPA/carbidopa/benserazide 1.5 mg/kg/day (Modopar®, Sinemet®) and 5-HTP (Levotonine® 0.75 mg/kg/day) were gradually increased to 100 mg/day and were combined with sertraline (50–150 mg/day) and selegiline (5–10 mg/day). The treatment induced spectacular clinical improvements in respect to dystonia, fatigability and sleep. From the wheelchair condition, the boy was able to walk and practise sports. Circadian sleep-wake rhythm was restored. Despite a good response with an improvement in motor ability and in mood, he has continued to have learning difficulties with drug-induced mild dyskinesia. The treatment led to a gradual increase in the levels of HVA and HIAA which however did not reach the normal range after 18 months of treatment (Table 1). In SRD, the initial defect was the absence of BH4 with a low BH4/BH2 ratio. We have introduced BH4 (Kuvan® or sapropterin dihydrochloride) in addition to l-DOPA/5-HTP therapy. After the first dose of 5 mg/kg/day for 4 months followed by a lumbar puncture, the second dose reached 20 mg/kg/day for a further 4 months followed by the last lumbar puncture. HIAA and HVA increased by 60% under BH4 (Table 1). However, the treatment by BH4 was stopped since no additional clinical benefits were objectively noted. At the age of 20, the atonic episodes disappeared and he was self-sufficient, but he remained highly fatigable, not being able to work. However, the drug induced dyskinesia of the mouth and of the hands.
Table 1.
CSF neurotransmitter analysis before SRD diagnosis and during the time course of the different treatments. Biogenic amines, pterins and MTHF were analysed in CSF from 4 successive lumbar punctures: (1) before SRD diagnosis, (2) 1 year after l-DOPA/5-HTP therapy, (3) 4 months after the combined therapy of l-DOPA/5-HTP and BH4 (5 mg/kg/day) and (4) 4 months after the combined therapy of l-DOPA/5-HTP and BH4 (20 mg/kg/day). The MTHF level has decreased from the beginning of the treatment by l-DOPA because no Lederfolin was introduced. Supplementation by Lederfolin was then introduced
| Values (nM) | At the time of SRD diagnosis | 1 year after l-DOPA/5-HTP therapy | 4 months after l-dopa/5-HTP and BH4 5 mg/kg/day | 4 months after l-DOPA/5-HTP and BH4 20 mg/kg/day | Reference values |
|---|---|---|---|---|---|
| 5-HTP | 2 | 141 | 10 | 5 | 3–12 |
| HIAA | 7 | 16 | 26 | 29 | 63–185 |
| 3-OMD | 3 | 192 | 288 | 251 | 3–54 |
| HVA | 64 | 78 | 129 | 123 | 156–410 |
| MHPG | 2 | 14 | 11 | 16 | 11–46 |
| Neopterin | 25 | 19 | 37 | 28 | 10–24 |
| Biopterin | 51 | 37 | 73 | 54 | 14–36 |
| Sepiapterin | 15 | nd | nd | nd | 0 |
| MTHF | 75 | 62 | 56 | 39 | >44 |
PUFA Analysis
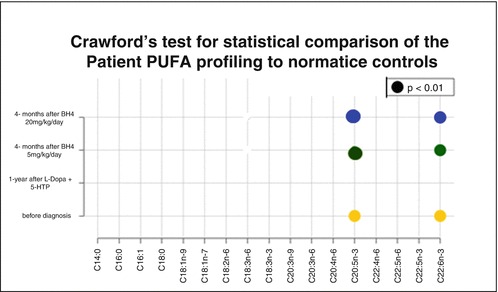
The plasma and erythrocyte PUFA levels were in the normal range (data not shown). The PUFA analysis has been set in CSF, and the reference values have been established in controls, excluding patients with neurotransmitter or BH4 defects. A statistical test comparing a single case to normative samples allowed us to find that DHA and EPA increased in 4/4 lumbar punctures of the patient, with a statistical significance in 3/4 (p < 0.01) and a trend for 1/4 lumbar punctures (Table 2). The other n-3 or n-6 fatty acids were never significantly different from controls. However, the levels of DHA and EPA did not normalize under different treatments.
Table 2.
PUFAs were analyzed in CSF obtained from four successive lumbar punctures: (1) before the SRD diagnosis, (2) 1 year after L-DOPA/5-HTP therapy, (3) 4 months after the combined therapy by L-DOPA/5-HTP and BH4 (5 mg/kg/day), and (4) 4 months after the combined therapy by L-DOPA/5-HTP and BH4 (20 mg/kg/day)
| PUFAs (%) | Patient | Controls n = 11 | |||||
|---|---|---|---|---|---|---|---|
| Before SRD diagnosis | 1 year after l-DOPA +5-HTP | 4 months after L-DOPA/5-HTTP and BH4 5 mg/kg/day | 4 months after L-DOPA/5-HT and BH4 20 mg/kg/day | Mean | SD | ||
| C14:0 | 1.05 | 1.35 | 1.39 | 1.41 | 2.36 | 1.11 |
|
| C16:0 | 29.89 | 28.93 | 28.35 | 32.32 | 30.98 | 5.99 | |
| C16:1 | 3.43 | 2.3 | 2.77 | 2.22 | 4.47 | 2.26 | |
| C18:0 | 22.12 | 25.39 | 23.41 | 24.97 | 19.95 | 4.08 | |
| C18:1 n-9 | 24.65 | 27.23 | 26.18 | 22.85 | 26.18 | 4.45 | |
| C18:1 n-7 | 5.1 | 2.94 | 3.38 | 3.37 | 4.32 | 1.42 | |
| C18:2 n-6 LA | 6.45 | 7.69 | 7.83 | 5.98 | 8.11 | 2.95 | |
| C18:3 n-6 | 0.003 | 0.01 | 0.04 | 0.04 | 0.04 | 0.03 | |
| C18:3 n-3 ALA | 0.18 | 1.23 | 0.24 | 0.15 | 0.51 | 0.46 | |
| C20:3 n-9 | 0.06 | 0.05 | 0.05 | 0.05 | 0.06 | 0.02 | |
| C20:3 n-6 | 0.28 | 0.1 | 0.29 | 0.27 | 0.17 | 0.09 | |
| C20:4n-6 ARA | 2.71 | 1.15 | 2.62 | 2.75 | 1.51 | 0.66 | |
| C20:5 n+3 EPA | 0.08 * | 0.03 | 0.07 * | 0.06 * | 0.02 | 0.01 | |
| C22:4 n-6 | 0.32 | 0.11 | 0.31 | 0.3 | 0.22 | 0.18 | |
| C22:5 n-6 | 0.34 | 0.56 | 0.23 | 0.61 | 0.51 | 0.29 | |
| C22:5 n-3 | 0.12 | 0.05 | 0.12 | 0.11 | 0.08 | 0.07 | |
| C22:6 n-3 DHA | 3.19 * | 0.92 | 2.73 * | 2.53 * | 0.53 | 0.27 | |
Significance was analysed by Crawford’s test: EPA and DHA p < 0.01 was indicated by circles
Fatty acids of the main interest are in bold. DHA (C22 6n-3) and EPA (C20:5n-3) were significantly higher in the patient than in controls. Crawford’s test was used for statistical comparison of the patient PUFA profiling to normative controls: EPA and DHA p < 0.01 was indicated by*
Discussion
Our patient has turned out to be homozygous for two different autosomal recessive disorders, i.e. MCEE deficiency and SRD. Both diseases are extremely rare. The consanguineous parents were heterozygous for both mutations. One coincidence was the fact that the two genes are mapped in the same region of the chromosome 2p13-3 and distant for only 1.8 Mb. More surprisingly, our patient was the second patient identified with combined MCEE and SPR mutations: the MCEE gene mutations were similar, whereas the mutations in the SR gene were different (Abeling et al. 2006; Bikker et al. 2006). The possibility of a contiguous gene syndrome was ruled out by CGH array. Uniparental disomy was excluded since both parents were heterozygous for the two mutations. However, their union was consanguineous, and the closer is the parental relationship, the greater is the risk of the child inheriting 2 copies of a deleterious gene mutation from his parents (Kearney et al. 2011). The mild methylmalonic aciduria and the decrease in [14C] propionate incorporation were attributed to the MCEE gene mutation. Five other cases were reported without clinical feature (Gradinger et al. 2007). A functional role for MCEE or epimerase has been debated in humans (Montgomery et al. 1983). For our patient, the MCEE defect has no clinical effect and the treatment by vitamins was inefficient. So MCEE deficiency can be considered as a genetic variant. In contrast, the establishment of the SRD fully explained the neurological picture. All the clinical symptoms resulting from the low production of dopamine and serotonin in SRD dramatically improved under treatment by l-DOPA and 5-HTP. The dystonia has regressed; the excessive sleepiness and the narcolepsy were suppressed. The addition of BH4 led to an increase in HVA and HIAA by 60%. The amount of BH4 being able to cross the uninjured brain blood barrier is still under debate. SRD is characterized by a high level of BH2 and a low level of BH4. However, we have only measured BP which is the sum of BH4 and BH2. In these conditions, we could not interpret the increment due to BH4. Finally, the clinical benefits did not objectively improve and BH4 supplementation was stopped. To further investigate the neuronal deterioration of our patient, we have pointed out that the PUFA status in CSF represented a marker of its neuronal stress. We have shown that the levels of DHA and its precursor EPA, but not the other n-3 or n-6 PUFA, increased in CSF. The only study of fatty acid levels in CSF was recently reported in Alzheimer’s patients (Fonteh et al. 2014). An increase in the PLA2 activity has liberated DHA from the breakdown of the neuronal lipid membrane and was related to the abnormal oxidative metabolism (Fonteh et al. 2013). The Alzheimer disorder is a neurodegenerative disorder thus radically different in terms of pathogenesis and genetic background. Thus, our results represented the first study of CSF PUFA status in an inherited metabolic disease. Some further investigations will be necessary to elucidate whether the increase in DHA was a common response to a neuronal stress, itself coming from either the odd-chain fatty acids and the cholesterol metabolism or the neurotransmitter disorders.
The diet has been reported to modulate the brain PUFA level and consequently biogenic amine metabolism (Lavialle et al. 2008; Jiang et al. 2009). The high level of EPA and DHA in red blood cells is associated with a slowed hippocampal and overall brain atrophy in humans (Pottala et al. 2014). A supplementation with DHA and EPA has been reported to enhance memory and cognitive function though DHA treatment remains somewhat unproven (Bauer et al. 2014). However, the peripheral status of PUFA in blood was normal in our patient. Thus, the high level of DHA in CSF could better result from a remodelling of PUFA metabolism in the brain. DHA mainly esterifies the brain and the retina phospholipids and thus plays a structural role for the membranes (Kim 2007). DHA is also the precursor for bioactive signalling molecules such as neuroprotectin D1, identified as a neuroprotective mediator to counteract neuronal apoptosis (Bazan 2006; Hong et al. 2014). DHA is thus considered to be essential for proper neuronal development and function.
Conclusion
We have reported the second case with the double MCEE/SPR homozygous mutations: the MCEE mutation was similar, but the SPR mutations were different. Our observation underlined that the pathogenesis was not due to the MCEE variant, but to the SRD which induced a defect in BH4 and consequently a defect in neurotransmitter metabolism. Indeed, the l-DOPA/carbidopa and 5-HTP treatment dramatically improved the clinical outcome. The supplementation with BH4 did not induce additional clinical benefits, although the level of HVA and HIAA increased in CSF. The high level of DHA and EPA in CSF probably constituted a marker in response to its neuronal stress.
Acknowledgements
We thank Clotilde Robin for correcting the English language. Claude Wolf for helpful discussions about PUFA and Rafel Laboissiere for statistical advising.
Abbreviations
- 5-HIAA
5-Hydroxyindoleacetic acid
- 5-HT
5-Hydroxytryptamine/serotonin
- 5-HTP
5-Hydroxytryptophan
- BH2
Dihydrobiopterin
- BH4
Tetrahydrobiopterin
- BP
Biopterin
- CSF
Cerebrospinal fluid
- DA
Dopamine
- DHA
Docosahexaenoic acid (22:6n-3)
- EPA
Eicosapentaenoic acid (20:5n-3)
- GC-MS
Gas chromatography-mass spectrometry
- HGH
Human growth hormone
- HVA
Homovanillic acid
- l-DOPA
3,4-Dihydroxyphenylalanine
- MCEE
Methylmalonyl-CoA epimerase gene
- MMA
Methylmalonic acid
- MTHF
Methyltetrahydrofolate
- NOS
Nitric oxide synthase
- OMD
3-Orthomethyl-Dopa or 3-methoxytyrosine
- PUFA
Polyunsaturated fatty acid
- RV
Reference value
- SPR
Sepiapterin reductase gene
- SRD
Sepiapterin reductase deficiency
Take-Home Message
We present a complete description of the medical and the genetic history for the patient with a combination of the two rare MCEE/SPR homozygous mutations, and we present the first investigation of the level of DHA and EPA in CSF for metabolic diseases.
Disclosure
The authors have nothing to disclose.
Compliance with Ethics Guidelines
Conflict of Interest
Michel Mazzuca, Marie-Anne Maubert, Léna Damaj, Fabienne Clot, Marylène Cadoudal, Christele Dubourg, Sylvie Odent, Jean François Benoit, Nadia Bahi-Buisson, Laurence Christa and Pascale de Lonlay declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
For medical care of the patient: Michel Mazzuca, Léna Damaj, Nadia Bahi-Buisson and Pascale de Lonlay were the referring physicians.
For biochemical analysis: Marie-Anne Maubert (PUFA analysis) and Marylène Cadoudal and Laurence Christa (neurotransmitter analysis) designed and performed the experiments.
For molecular genetic analysis: Fabienne Clot, Christele Dubourg, Sylvie Odent and Jean François Benoit designed and performed the experiments.
Conduct and reporting of the work described in the article: Michel Mazzuca, Sylvie Odent, Laurence Christa and Pascale de Lonlay analysed the data and wrote the manuscript.
All these authors equally contributed to this work.
Footnotes
Competing interests: None declared
Contributor Information
Laurence Christa, Email: Laurence.christa@parisdescartes.fr.
Collaborators: Johannes Zschocke
References
- Abeling NG, Duran M, Bakker HD, Stroomer L, Thony B, Blau N, Booij J, Poll-The BT. Sepiapterin reductase deficiency an autosomal recessive DOPA-responsive dystonia. Mol Genet Metab. 2006;89:116–120. doi: 10.1016/j.ymgme.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Bauer I, Crewther S, Pipingas A, Sellick L, Crewther D. Does omega-3 fatty acid supplementation enhance neural efficiency? A review of the literature. Hum Psychopharmacol. 2014;29:8–18. doi: 10.1002/hup.2370. [DOI] [PubMed] [Google Scholar]
- Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–271. doi: 10.1016/j.tins.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Bikker H, Bakker HD, Abeling NG, Poll-The BT, Kleijer WJ, Rosenblatt DS, Waterham HR, Wanders RJ, Duran M. A homozygous nonsense mutation in the methylmalonyl-CoA epimerase gene (MCEE) results in mild methylmalonic aciduria. Hum Mutat. 2006;27:640–643. doi: 10.1002/humu.20373. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabrol B, Landrieu P, Nguyen K, Ponsot G, Abada M, Doummar D, Damier P, Gil R, Thobois S, Ward AJ, Hutchinson M, Toutain A, Picard F, Camuzat A, Fedirko E, San C, Bouteiller D, LeGuern E, Durr A, Vidailhet M, Brice A. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain. 2009;132:1753–1763. doi: 10.1093/brain/awp084. [DOI] [PubMed] [Google Scholar]
- Crawford JR, Garthwaite PH. Comparison of a single case to a control or normative sample in neuropsychology: development of a Bayesian approach. Cogn Neuropsychol. 2007;24:343–372. doi: 10.1080/02643290701290146. [DOI] [PubMed] [Google Scholar]
- Engstrom K, Saldeen AS, Yang B, Mehta JL, Saldeen T. Effect of fish oils containing different amounts of EPA, DHA, and antioxidants on plasma and brain fatty acids and brain nitric oxide synthase activity in rats. Ups J Med Sci. 2009;114:206–213. doi: 10.3109/03009730903268958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonteh AN, Chiang J, Cipolla M, Hale J, Diallo F, Chirino A, Arakaki X, Harrington MG. Alterations in cerebrospinal fluid glycerophospholipids and phospholipase A2 activity in Alzheimer’s disease. J Lipid Res. 2013;54:2884–2897. doi: 10.1194/jlr.M037622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS One. 2014;9:e100519. doi: 10.1371/journal.pone.0100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hyland K, Blau N, MacCollin M. Dopa-responsive hypersomnia and mixed movement disorder due to sepiapterin reductase deficiency. Neurology. 2006;67:2032–2035. doi: 10.1212/01.wnl.0000247274.21261.b4. [DOI] [PubMed] [Google Scholar]
- Friedman J, Roze E, Abdenur JE, Chang R, Gasperini S, Saletti V, Wali GM, Eiroa H, Neville B, Felice A, Parascandalo R, Zafeiriou DI, Arrabal-Fernandez L, Dill P, Eichler FS, Echenne B, Gutierrez-Solana LG, Hoffmann GF, Hyland K, Kusmierska K, Tijssen MA, Lutz T, Mazzuca M, Penzien J, Poll-The BT, Sykut-Cegielska J, Szymanska K, Thony B, Blau N. Sepiapterin reductase deficiency: a treatable mimic of cerebral palsy. Ann Neurol. 2012;71:520–530. doi: 10.1002/ana.22685. [DOI] [PubMed] [Google Scholar]
- Gradinger AB, Belair C, Worgan LC, Li CD, Lavallee J, Roquis D, Watkins D, Rosenblatt DS. Atypical methylmalonic aciduria: frequency of mutations in the methylmalonyl CoA epimerase gene (MCEE) Hum Mutat. 2007;28:1045. doi: 10.1002/humu.9507. [DOI] [PubMed] [Google Scholar]
- Hong SH, Belayev L, Khoutorova L, Obenaus A, Bazan NG. Docosahexaenoic acid confers enduring neuroprotection in experimental stroke. J Neurol Sci. 2014;338:135–141. doi: 10.1016/j.jns.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Shi Y, Wang LS, Yang ZR. The influence of orally administered docosahexaenoic acid on cognitive ability in aged mice. J Nutr Biochem. 2009;20:735–741. doi: 10.1016/j.jnutbio.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Kearney H.M., Kearney J.B., Conlin L.K. (2011) Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 31:595-613, ix. DOI: 10.1016/j.cll.2011.08.003. [DOI] [PubMed]
- Kim HY. Novel metabolism of docosahexaenoic acid in neural cells. J Biol Chem. 2007;282:18661–18665. doi: 10.1074/jbc.R700015200. [DOI] [PubMed] [Google Scholar]
- Koht J, Rengmark A, Opladen T, Bjornara KA, Selberg T, Tallaksen CM, Blau N, Toft M (2014) Clinical and genetic studies in a family with a novel mutation in the sepiapterin reductase gene. Acta Neurol Scand Suppl 7–12. doi:10.1111/ane.12230 [DOI] [PubMed]
- Lavialle M, Champeil-Potokar G, Alessandri JM, Balasse L, Guesnet P, Papillon C, Pevet P, Vancassel S, Vivien-Roels B, Denis I. An (n-3) polyunsaturated fatty acid-deficient diet disturbs daily locomotor activity, melatonin rhythm, and striatal dopamine in Syrian hamsters. J Nutr. 2008;138:1719–1724. doi: 10.1093/jn/138.9.1719. [DOI] [PubMed] [Google Scholar]
- Montgomery JA, Mamer OA, Scriver CR. Metabolism of methylmalonic acid in rats. Is methylmalonyl-coenzyme a racemase deficiency symptomatic in man? J Clin Invest. 1983;72:1937–1947. doi: 10.1172/JCI111158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormazabal A, Garcia-Cazorla A, Fernandez Y, Fernandez-Alvarez E, Campistol J, Artuch R. HPLC with electrochemical and fluorescence detection procedures for the diagnosis of inborn errors of biogenic amines and pterins. J Neurosci Methods. 2005;142:153–158. doi: 10.1016/j.jneumeth.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Pottala JV, Yaffe K, Robinson JG, Espeland MA, Wallace R, Harris WS. Higher RBC EPA+DHA corresponds with larger total brain and hippocampal volumes: WHIMS-MRI study. Neurology. 2014;82:435–442. doi: 10.1212/WNL.0000000000000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorzi G, Redweik U, Trippe H, Penzien JM, Thony B, Blau N. Detection of sepiapterin in CSF of patients with sepiapterin reductase deficiency. Mol Genet Metab. 2002;75:174–177. doi: 10.1006/mgme.2001.3273. [DOI] [PubMed] [Google Scholar]