

Abstract
Background: Familial hypobetalipoproteinemia (FHBL) and abetalipoproteinemia (ABL) are rare inherited forms of hypolipidemia. Their differential diagnosis is important for predicting of the prognosis and selecting appropriate therapy.
Materials and Methods: Genetic analysis was performed in two patients with primary hypocholesterolemia born from consanguineous parents. The oral fat tolerance test (OFTT) was performed in one patient with FHBL (apoB-87.77) and one with ABL as well as in four normal control subjects. After overnight fasting, blood samples were drawn. Serum lipoprotein and remnant-like particle (RLP) fractions were determined by HPLC analysis.
Results: Both patients with homozygous FHBL were asymptomatic probably because of preserved levels of fat-soluble vitamins, especially vitamin E. The patients with FHBL were homozygous because of novel apoB-83.52 and apoB-87.77 mutations, and although one of them (apoB-87.77) had fatty liver disease, microscopic findings suggesting nonalcoholic steatohepatitis were absent. Fasting apoB-48 and RLP-triglyceride levels in the patient with homozygous FHBL, which were similar to those in normal control subjects, increased after OFTT both in normal control subjects and the patient with FHBL but not in the patient with ABL, suggesting that the fat load administered was absorbed only in the patient with FHBL.
Conclusion: Although lipid levels in the patients with homozygous FHBL and ABL were comparable, fasting, postoral fat loading of apoB-48, as well as RLP-triglyceride levels, may help in the differential diagnosis of FHBL and ABL and provide a prompt diagnosis using genetic analysis in the future.
Keywords: Abetalipoproteinemia, Familial hypobetalipoproteinemia, Oral fat tolerance test
Introduction
Hypobetalipoproteinemia is defined as low-density lipoprotein (LDL) cholesterol and apolipoprotein B (apoB) levels below the fifth percentile for age- and sex-matched general populations that have primary and secondary conditions, such as strict vegetarianism, in a hospital setting, cachexia, malnutrition, severe liver disease, and hyperthyroidism (Burnett et al. 2014a, b). Familial hypobetalipoproteinemia (FHBL) (OMIM 107730) is an autosomal dominant hereditary disorder characterized by low levels of LDL-cholesterol and apoB. FHBL heterozygotes are distributed at approximately one in 500–1,000 people in the general population; they are generally asymptomatic and are rarely accompanied by atherosclerosis, but instead by fatty liver (Burnett et al. 2014a; Hooper and Burnett 2014; Katsuda et al. 2009; Lee and Hegele 2014; Welty 2014). The best known molecular cause of FHBL is a mutation in APOB gene. A loss-of-function mutation in proprotein convertase subtilisin/kexin type 9 is also a cause of FHBL (Burnett et al. 2014b; Cohen et al 2005; Hooper and Burnett 2014; Welty 2014). On the other hand, FHBL homozygotes are very rare, and their lipid levels are similar to those of patients with abetalipoproteinemia (ABL) (OMIM 200100) (Burnett et al. 2014a; Hooper and Burnett 2014; Lee and Hegele 2014; Welty 2014).
ABL is an extremely rare recessive disorder characterized by undetectable or very low levels of LDL-cholesterol and apoB, failure to thrive, oral fat intolerance, diarrhea, steatorrhea, and acanthocytosis as well as fatty liver and lipid accumulation in enterocytes (Burnett et al. 2014a; Hooper and Burnett 2014; Lee and Hegele 2014; Welty 2014; Yang et al 1999). Neurological abnormalities such as spinocerebellar ataxia and retinitis pigmentosa are caused by deficiencies in fat-soluble vitamins. Although ABL is treatable by fat-soluble vitamins, especially vitamin E, their effectiveness is sometimes limited because of the lack of carrier lipoproteins (Burnett et al. 2014a). The molecular cause of ABL is genetic mutations of the microsomal triglyceride transfer protein (MTTP), which acts as a chaperone in the lipidation on nascent apoB (Burnett et al. 2014a; Hooper and Burnett 2014; Lee and Hegele 2014; Welty 2014; Wetterau et al 1992). Thus, deficiencies in MTP result in the lack of apoB lipidation and, consequently, a deficiency in all apoB-containing lipoproteins.
The frequencies of homozygous FHBL and ABL are estimated at less than 1 in a million in the general population. Although lipid levels in homozygous FHBL and ABL are similar, clinical manifestations of ABL are more severe than those of homozygous FHBL when the truncation of apoB is longer than apoB-48, probably because of lower levels of fat-soluble vitamins. On the other hand, homozygous FHBL shorter than apoB-48 represents a severe phenotype resembling ABL due to impaired formation of chylomicrons. Little is known about the difference in postprandial lipid levels, which could explain the difference between these two similar disorders. Thus, we performed genetic analysis in two patients with homozygous FHBL and then performed the oral fat tolerance test (OFTT) in one of them and in a patient with ABL in order to elucidate the differences between them with regard to postprandial lipid metabolism.
Methods
Genetic Analysis
Genetic analysis was performed after obtaining written informed consent from both patients in accordance with the guidelines of the Bioethical Committee on Human Genome and Genetic Analysis Study, School of Medicine, Kanazawa University. Genomic DNA was purified from peripheral leukocytes using a genomic DNA purification kit (Qiagen, Venlo, the Netherlands). We designed 63 primer pairs to cover all 29 exons and intronic junctions of the APOB gene using Primer 3 online software (http://simgene.com/Primer3). We screened the APOB gene by PCR-single-strand conformational polymorphism analysis of all exons. DNA sequencing was performed with fluorescently labeled dideoxy terminators using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Measurement of Lipids, Apolipoproteins, and Fat-Soluble Vitamins
Blood samples were drawn after overnight fasting. Serum total cholesterol, triglycerides, and high-density lipoprotein (HDL) cholesterol levels were measured by standard enzymatic methods; LDL-cholesterol levels were measured by a direct method (Sekisui Medical, Tokyo, Japan); and serum apolipoprotein levels were measured by a turbidimetric immunoassay, except for apoB-48, whose level was measured by ELISA using a monoclonal antibody against the C-terminal decapeptide of apoB-48 as described previously (Kinoshita et al 2005). Remnant-like particle (RLP) was estimated as the unbound fraction of serum after incubation with an immunoaffinity gel of apoB-100 monoclonal antibody and apoA-I monoclonal antibody (Japan Immunoresearch Laboratories, Takasaki, Japan) as described previously (Nakajima et al. 1993). Serum lipoprotein and RLP-cholesterol distributions were measured using an HPLC system using 0.05-M Tris-buffered acetate (pH 8.0) at a flow rate of 0.7 mL/min (Tosho, Tokyo, Japan) as described elsewhere (Usui et al 2002). Serum levels of vitamin A were measured by HPLC, and those of vitamin E were determined by a fluorimetric method and by RIA (Yang et al 1999), respectively.
Oral Fat Tolerance Test
OFTT was performed in the patients with FHBL and ABL, as well as normal control subjects according to an established method (Tada et al 2012; Inazu et al 2008). Briefly, 50 g per m2 of body surface of the OFTT cream (Jomo Shokuhin, Takasaki, Japan) was administered after overnight fasting (Tada et al 2012; Inazu et al 2008). The cream, which consists of 33% fat, 74 mg of cholesterol, and 341 kcal per 100 g, is rich in palmitic and oleic acids. Blood was drawn periodically before and 2, 4, and 6 h after fat loading. The normal control subjects were volunteers, and they also served as controls in our previous study (Tada et al 2012).
Case Report
Case 1 (FHBL): A 42-year-old Japanese male patient was referred to Showa University Hospital for further examination of his hypocholesterolemia in 2010. He was born from a first-cousin consanguineous marriage. He suffered from type 2 diabetes mellitus and hypertension since the age of 35 years and was treated with insulin injection therapy and pioglitazone, metformin, and carvedilol. His basal serum lipid and apolipoprotein levels, fat-soluble vitamin levels, and other biochemical parameters are shown in Table 1. His body measurements were as follows: height, 184 cm; body weight, 85.9 kg; and body mass index, 25.4 kg/m2. He was asymptomatic with no physical or neurological abnormalities, and no retinitis pigmentosa was found. The genetic analysis revealed that he was homozygous for a novel nucleotide duplication (c.11433dupT, p.E3812X) in exon 26 of the APOB gene (RefSeqNM_000384.2), resulting in the formation of a truncated apoB of 3,811 amino acids (apoB-83.52) (Fig. 1).
Table 1.
Fasting serum levels of lipids, apolipoproteins, fat-soluble vitamins, and the other biochemical parameters of the study subjects
| Case | Case 1 | Case 2 | Case 3 | Control (n = 4) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV-1 | III-1 | III-2 | III-3 | III-4 | III-5 | III-6 | ||||||
| Sex | M | M | M | M | F | F | F | F | M | M | ||
| Gene mutation | ApoB-83.52/83.52 | ApoB-87.77/87.77 | ApoB-87.77/100 | ApoB-87.77/100 | ApoB-87.77/100 | ApoB-100/100 | ApoB-100/100 | ApoB-87.77/100 | c.1237-1G>A | Normal | ||
| Total cholesterol (mg/dL) | 55 | 63 | 181 | 150 | 180 | 208 | 280 | 161 | 39 | 173 | ± | 44 |
| LDL-cholesterol (mg/dL) | 5 | 1 | 96 | 80 | 96 | 110 | 197 | 85 | 2 | 88 | ± | 6 |
| HDL-cholesterol (mg/dL) | 43 | 51 | 74 | 55 | 64 | 77 | 39 | 67 | 24 | 66 | ± | 21 |
| Triglyceride (mg/dL) | 37 | 13 | 54 | 73 | 99 | 104 | 219 | 47 | 9 | 79 | ± | 45 |
| ApoA-I (mg/dL) | 119 | 117 | 160 | 150 | 156 | 164 | 114 | 153 | 54 | 156 | ± | 26 |
| ApoA-II (mg/dL) | 22 | 25.9 | 29.1 | 28.9 | 30.4 | 33.1 | 27.7 | 33.9 | 10.2 | 32.3 | ± | 4.2 |
| ApoB (mg/dL) | u.d. | u.d. | 56 | 52 | 63 | 79 | 142 | 54 | u.d. | 80 | ± | 13 |
| ApoB-48 (μg/mL) | 3.30 | 2.03 | n.d | n.d | n.d | n.d | n.d | n.d | 0.03 | 2.3 | ± | 1 |
| ApoC-II (mg/dL) | 1.5 | 0 | 3.1 | 1.7 | 3.1 | 3.5 | 8 | 2.6 | 0.1 | 4.7 | ± | 1.3 |
| ApoC-III (mg/dL) | 3.2 | 2.2 | 7.7 | 7.3 | 7.7 | 9.3 | 11.8 | 5.4 | 1.7 | 11.1 | ± | 3.1 |
| ApoE (mg/dL) | 1.4 | 2.3 | 4.1 | 3.1 | 3.9 | 4.2 | 6.1 | 3 | 5.6 | 4.3 | ± | 0.8 |
| AST (IU/L) | 44 | 72 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 96 | n.d. | ||
| ALT (IU/L) | 66 | 158 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 90 | n.d. | ||
| GGTP (IU/L) | 82 | 49 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 124 | n.d. | ||
| PG (mg/dL) | 116 | 101 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 84 | n.d. | ||
| HbA1c (%) | 9.2 | 5.4 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 4.4 | n.d. | ||
| Vit A (IU/dL) (97–316) | n.d. | 163 | 154 | 184 | 169 | 158 | 136 | 170 | 85 | n.d. | ||
| Vit E (mg/dL) (0.75–1.41) | n.d. | 0.41 | 1.05 | 0.84 | 1.00 | 1.13 | 1.79 | 0.89 | 0.37 | n.d. | ||
n.d. not determined, u.d. undetectable
Fig. 1.
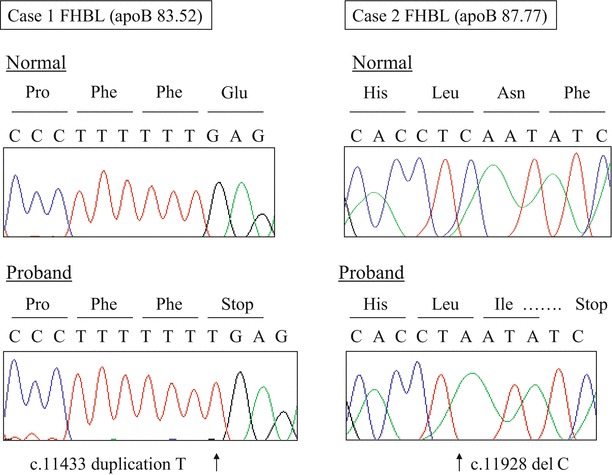
Genetic analysis of homozygous familial hypobetalipoproteinemia (FHBL) (a) apolipoprotein B (apoB)-83.52 and (b) apoB-87.77. Case 1 was homozygous for a novel nucleotide duplication (c.11433dupT, p.E3812X), resulting in the formation of a stop codon in exon 26 of the APOB gene and a truncated apoB of 3,811 amino acids (apoB-83.52). Case 2 was homozygous for a novel nucleotide deletion (c.11928delC, p.L3976fsX31) in exon 28 of the APOB gene, resulting in the formation of a stop codon at nucleotide 31 downstream and, thereafter, in a truncated apoB of 4,005 amino acids (apoB-87.77)
Case 2 (FHBL): A 23-year-old Japanese male patient was referred to Kanazawa University Hospital for further examination of his hypocholesterolemia in 2010. He was also born from a first-cousin consanguineous marriage (Fig. 2). His laboratory data and those of his family members are shown in Table 1. His body measurements were as follows: height, 172 cm; body weight, 58.5 kg; and body mass index, 19.8 kg/m2. He was also asymptomatic and had no physical or neurological abnormalities. These findings do not suggest that ABL was present. No acanthocytosis was found in the blood smear. No lipid accumulation was found in the upper and lower gastrointestinal tract by gastroscopy and colonoscopy, respectively, but a severe fatty liver was observed by ultrasonography. Severe steatosis with mild inflammatory changes and mild pericellular fibrosis was also found in the liver by histopathology, but no ballooning of hepatocytes or Mallory bodies were detected. These findings do not suggest that nonalcoholic steatohepatitis was present (Fig. 3). The genetic analysis revealed that case 2 was homozygous for a novel nucleotide deletion (c.11928delC, p.L3976fsX31) in exon 28 in the APOB gene, resulting in the formation of a downstream stop codon and, thereafter, of a truncated apoB of 4,005 amino acids (apoB-87.77) (Fig. 1).
Fig. 2.
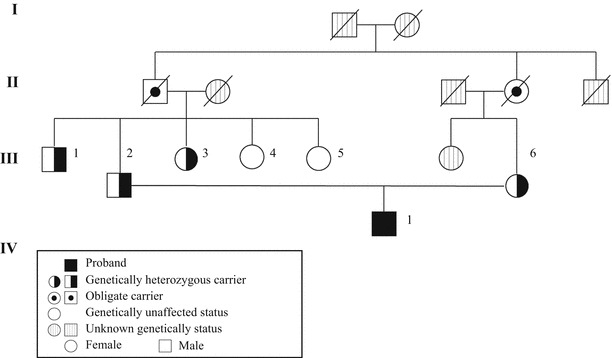
Pedigree of the patient with familial hypobetalipoproteinemia (FHBL) [apolipoprotein B (apoB-87.77)] (Case 2). Squares indicate males and circles indicate females. Closed and semi-closed symbols indicate homozygosity and heterozygosity for apoB-87.77, respectively. The paternal grandfather and maternal grandmother are siblings, indicating that the proband was born from a first-cousin consanguineous marriage
Fig. 3.
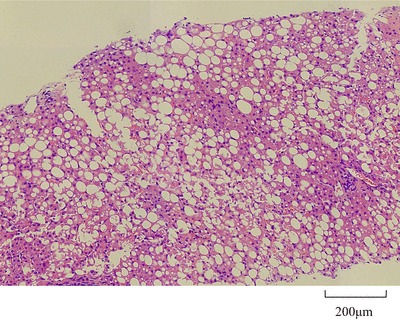
Liver biopsy of a patient with homozygous familial hypobetalipoproteinemia (FHBL) [apolipoprotein B (apoB-87.77)] (Case 2). Because severe fatty liver was observed by ultrasonography, a liver biopsy was performed. Severe steatosis with mild inflammatory changes and mild pericellular fibrosis was observed, but there was no ballooning of hepatocytes or Mallory bodies. These findings do not suggest the presence of nonalcoholic steatohepatitis
Case 3 (ABL): A 49-year-old Japanese male patient was admitted to Kanazawa University Hospital for regular evaluation of multiple organ complications due to ABL. He was born from a non-consanguineous marriage and was diagnosed with ABL caused by maternal uniparental disomy of intron 9 splice donor site mutation (c.1237-1G>A, p.Q413_K448del) of the MTTP gene (RefSeqNM_000253.1), which was previously reported (Yang et al 1999). His serum lipid and apolipoprotein levels, fat-soluble vitamin levels, and other biochemical parameters under vitamin E supplementation therapy are shown in Table 1. His body measurements were as follows: height, 152 cm; body weight, 51 kg; and body mass index, 22.0 kg/m2. He showed typical clinical manifestations of ABL, such as acanthocytosis, lipid accumulation in enterocytes, spinocerebellar ataxia, and retinitis pigmentosa (Yang et al 1999).
Although all three patients presented with an undetectable amount of apoB and extremely low levels of LDL-cholesterol, the patients with FHBL showed relatively high levels of HDL-cholesterol. Of note, the fasting serum apoB-48 level was almost undetectable in the patient with ABL, whereas in the patients with FHBL, the level was similar to that in normal control subjects. To elucidate the difference in lipoprotein metabolism after the postprandial state, we performed the OFTT in case 2 (FHBL; apoB-87.77), case 3 (ABL), and normal control subjects (n = 4).
As shown in Fig. 4, serum fasting triglyceride levels in the patients with FHBL (apoB-87.77) and ABL were lower than those in normal control subjects. After oral fat loading, although the peak time points were different, serum triglyceride levels increased in both normal control subjects and the patient with FHBL (apoB-87.77) but not in the patient with ABL. As a result, the incremental areas under the curve of serum triglyceride of the patients with FHBL and ABL were lower than those of control subjects. Similar tendencies after oral fat loading were observed for serum apoB-48, a specific carrier protein of chylomicrons. Similarly, RLP-triglyceride levels after oral fat loading were increased in the patient with FHBL (apoB-87.77) as well as in normal control subjects but not in the patient with ABL. Interestingly, the incremental areas under the curve of serum apoB-48 and RLP-triglyceride in the patients with FHBL and normal control subjects were similar, and those in patient with ABL were small. These observations suggest that no fat was absorbed in the patient with ABL, whereas fat was absorbed in the patient with FHBL (apoB-87.77). On the other hand, increases in the serum RLP-cholesterol level after oral fat loading were almost the same in normal control subjects and the patient with FHBL (apoB-87.77), despite the baseline level in the latter (apoB-87.77) being lower than that in the former. Interestingly, the RLP-cholesterol level in the patient with ABL was almost the same as that in normal control subjects before and after oral fat loading.
Fig. 4.
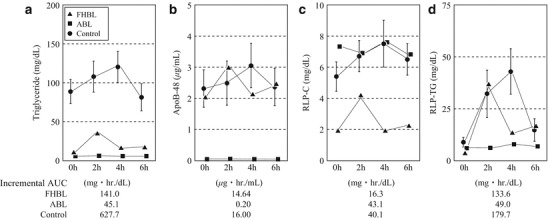
Chronological changes in serum lipid and apolipoprotein levels after oral fat loading. Serum levels of (a) triglyceride, (b) apolipoprotein B-48 (apoB-48), (c) remnant-like particle (RLP)-cholesterol, and (d) RLP-triglyceride were measured periodically after oral fat loading. Triangles, squares, circles, and bars indicate familial hypobetalipoproteinemia (FHBL), abetalipoproteinemia (ABL), control, and 1SD, respectively. The incremental area under the curve (AUC) is indicated below the figures. Fasting serum levels of triglyceride in the patients with FHBL (apoB-87.77) and ABL were lower than those in normal control subjects. The incremental AUC of apoB-48 in the patient with FHBL (apoB-87.77) and normal control subjects was similar. On the other hand, in the patient with ABL, it was extremely low. This indicated that the patient with FHBL (apoB-87.77) absorbed fat to the same degree as normal control subjects and the patient with ABL absorbed no fat
To elucidate the more detailed changes in the lipoprotein subfraction before and after oral fat loading, HPLC analyses of cholesterol and RLP fractions were performed (Fig. 5). The HPLC method can separate lipoproteins according to their particle size, with the larger-sized lipoproteins being eluted earlier. In normal control subjects, a small but significant RLP-cholesterol peak eluted at around 15 min 2–6 h after oral fat loading, indicating the presence of chylomicrons, and two large peaks eluted at around 21 and 25 min, indicating the presence of the LDL and HDL fractions, respectively (Fig. 5c). As expected from the lack of increase in apoB-48 after oral fat loading in the patient with ABL (Fig. 4b), no RLP-cholesterol peak was eluted at around 15 min, suggesting the absence of chylomicron formation (Fig. 5b). Moreover, there was an RLP-cholesterol peak between the original LDL and HDL peaks, suggesting the presence of apoE-rich large HDL particles (Fig. 5b). In contrast, a relatively wide slope of RLP-cholesterol was observed in the patient with FHBL (apoB-87.77), suggesting the presence of unusually small chylomicron particle (Fig. 5a).
Fig. 5.
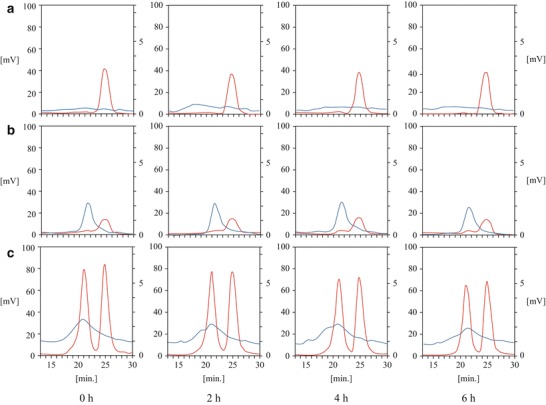
HPLC analysis of serum from homozygous familial hypobetalipoproteinemia (FHBL) [apolipoprotein B (apoB-87.77)], abetalipoproteinemia (ABL), and representative normal control subject. The HPLC method can separate lipoproteins according to their particle size, with the larger-sized lipoproteins being eluted earlier. In the normal control subjects, a small but significant remnant-like particle (RLP)-cholesterol peak eluted at around 15 min 2–6 h after oral fat loading, indicating the presence of chylomicrons, and two large peaks eluted at around 21 and 25 min, indicating the low-density lipoprotein (LDL) and high-density lipoprotein (HDL) fractions, respectively. The HPLC analyses of serum cholesterol levels and RLP fractions are shown as red and blue lines, respectively. (a) FHBL, (b) ABL, and (c) normal control
Serum levels of vitamins A and E were measured in some patients and their family members. Although the patient with ABL was supplemented with weekly 1,200-mg vitamin E drip intravenous injections, his serum vitamin A and vitamin E levels were lower than the normal range. The serum vitamin A level of the patient with FHBL (apoB-87.77) was within the normal limit and was almost the same as that of his family members, which included a patient with heterozygous FHBL and normal subjects. On the other hand, his serum vitamin E level was lower than the normal range and that of all his family members, but it was higher than that of the patient with ABL who was supplemented with vitamin E.
In summary, although lipid levels in the patients with homozygous FHBL and ABL were comparable, the former patients (whose apoB was longer than apoB-48) were asymptomatic. The fasting, postoral fat loading of apoB-48, as well as RLP-triglyceride levels of homozygous FHBL, was higher than those of the patient with ABL.
Discussion
There were three main findings of this study. First, we identified novel APOB gene mutations (apoB-83.52 and apoB-87.77) in two Japanese patients with homozygous FHBL. Second, these two patients were almost asymptomatic, although the former suffered from type 2 diabetes mellitus and developed complications of fatty liver. Third, for the first time, OFTTs were performed in a patient with ABL and with homozygous FHBL (apoB-87.77), revealing clear, clinically noteworthy differences in lipoprotein metabolism after oral fat loading.
Since the cloning of the APOB gene, more than 60 different gene mutations have been identified as the cause of FHBL (Tarugi et al 2007). Most of them result in truncated proteins of various lengths ranging from apoB-2 to apoB-89 due to nonsense or frameshift mutations. Previously, we detected two novel APOB gene mutations in 14 Japanese patients with FHBL (apoB-13.7 and apoB-82) and demonstrated that such a gene mutation is not rare in patients with Japanese primary hypocholesterolemia (Katsuda et al 2009). In the present study, we screened and found APOB gene mutations in two patients who resembled ABL in terms of extremely low LDL-cholesterol levels and birth from consanguineous parents. As a result, we identified two novel mutations in the APOB gene: apoB-83.52 and apoB-87.77.
Although some cases of homozygous or heterozygous FHBL with type 2 diabetes mellitus (Groenewegen et al 1994; Ohashi et al 1998; Pulai et al 1998; Turk et al 2012) have been reported, no predominant causality has been proposed. Ohashi et al. (1998) reported an unusual Japanese patient with homozygous FHBL (apoB-38.7) presenting with retinitis pigmentosa and some atypical neurological abnormalities despite a normal plasma level of vitamin E, such as paresthesia in both hands, glove-/stocking-type hypesthesia, the absence of deep tendon reflexes in the lower extremities, and a positive Romberg’s sign. Moreover, unlike in ABL, this specific patient presented with normal pyramidal, cerebellar, and posterior column functions. In general, most complications associated with ABL or FHBL can be explained by deficiencies in fat-soluble vitamins, especially vitamin E (Lee and Hegele 2014; Tarugi et al 2007). Thus, the absence of physical abnormalities in the two patients with homozygous FHBL in the present study, in contrast to the patient with ABL, may be explained by the fact that truncation of apoB is longer than apoB-48 and by the resultant existence of lower-than-normal plasma fat-soluble vitamin levels.
The metabolism of apoB in heterozygous FHBL due to several truncated apoBs has been studied using a stable isotope technique (Parhofer and Barrett 2006). These results indicated that apoB-containing lipoproteins showed both decreased production rates and increased clearance rates. Truncations that retain the LDL receptor-binding domain, such as apoB-89, apoB-87, and apoB-75, have increased fractional catabolic rates, whereas every 1% of apoB truncation results in an approximately 1.4% reduction of VLDL-apoB secretion (Parhofer et al 1996).
OFTT is easy to perform and enables the detection of postprandial lipoprotein metabolism. Averna et al. (1993) performed OFTT in a patient with heterozygous FHBL caused by an apoB truncation. They compared fat absorption between patients with heterozygous FHBL with apoB <48 and apoB >48 and found no difference between them suggesting that one allele of intestinal apoB-48 is sufficient for normal fat absorption. Hooper et al. (2007) performed OFTT and detected postprandial apoB-48 kinetics in patients with heterozygous FHBL (apoB-6.9, apoB-25.8, and apoB-40.3). As a result, these patients with heterozygous FHBL, caused by truncations shorter than apoB-48, showed a lower production and normal clearance of triglyceride-rich lipoproteins.
In the present study, for the first time, we directly compared lipid and apolipoprotein levels after OFTT in a patient with homozygous FHBL (apoB-87.77) and ABL (Figs. 4 and 5). The serum triglyceride level in the former patient was similar to that in the latter patient at fasting, and it increased 2 h after OFTT only in the former patient. This result suggests that the loaded fat was not absorbed in the patient with ABL. This concept was confirmed by measuring apoB-48 levels, a specific carrier protein of chylomicrons (Fig. 4) and performing an HPLC analysis (Fig. 5) over time. The HPLC analysis revealed small but significant peaks of RLP-cholesterol that eluted at around 15 min, indicating the presence of chylomicrons in the patient with homozygous FHBL (apoB-88.29) at 2, 4, and 6 h after oral fat loading, but not in the patient with ABL. Because the truncated length of apoB was longer than apoB-48 in the patient with homozygous FHBL, it was speculated that the loaded fat was absorbed to the same degree as that in normal control subjects. Although lipid levels in the patients with homozygous FHBL and ABL are similar, fasting or postoral fat loading apoB-48 levels as well as RLP-triglyceride levels may help in the differential diagnosis of these two rare disorders and provide an opportunity to promptly diagnose them using genetic analysis in the future.
The present study has several limitations. First, a relatively small number of subjects were included in the current study because of the extreme rarity of the disorder. The results from a single patient are not suitable for statistical analysis. However, the differences in OFTT-derived FHBL and ABL data are quite distinct. Second, the truncation length of apoB in both the patients with FHBL exceeded apoB-48, making it impossible to compare the OFTT data between apoB >48 and apoB <48.
Acknowledgment
We express sincere gratitude to Kazuko Honda (staff of Kanazawa University) for her outstanding technical assistance. The authors would like to thank Enago (www.enago.jp) for the English language review.
Take Home Message
Measuring apolipoprotein B-48 and remnant-like particle triglyceride levels before and after the oral fat tolerance test may help in the differential diagnosis of homozygous familial hypobetalipoproteinemia and abetalipoproteinemia.
Compliance with Ethics Guidelines
Conflict of Interest
We hereby certify that this paper, which consists of unpublished original observations, is not under consideration for publication elsewhere. All coauthors have been involved in drafting and revising the article, and finally, this manuscript has been read and approved by all coauthors. Atsushi Nohara and Hiroshi Mabuchi have received research grants from MSD K.K., Sanofi K.K., Shionogi & Co., Ltd., Kowa Co., Ltd., Astellas Pharma Inc., AstraZeneca K.K., Keiaikai Medical Corp., and Biopharm of Japan Co. Masakazu Yamagishi has received research grants from Astellas Pharma Inc., Daiichi Sankyo Co., Ltd., and Ono Pharmaceutical Co., Ltd. Masa-aki Kawashiri, Hayato Tada, Marowa Hashimoto, Matsuo Taniyama, Takamitsu Nakano, Katsuyuki Nakajima, Takeshi Inoue, Mika Mori, Chiaki Nakanishi, Tetsuo Konno, Kenshi Hayashi, Akihiro Inazu, Junji Koizumi, Hirotaka Ishihara, Junji Kobayashi, and Tsutomu Hirano have no financial or other relations that could lead to a conflict of interest. All coauthors agreed to submit this article to the Journal of Inherited Metabolic Disease.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all participants for being included in the study. Additional informed consent was obtained from all patients for whom identifying information is included in this article.
Author Contributions
Masa-aki Kawashiri designed and performed all functions throughout the research and wrote the paper. Hayato Tada collected clinical data and performed the genetic analysis and fat-loading test. Marowa Hashimoto performed the genetic analysis. Takamitsu Nakano and Katsuyuki Nakajima performed the biochemical assay. Takeshi Inoue, Mika Mori, Chiaki Nakanishi, Tetsuo Konno, Kenshi Hayashi, and Atsushi Nohara collected the clinical data and performed the genetic analysis. Akihiro Inazu performed and supervised the genetic and biochemical analyses. Junji Koizumi collected the clinical data and supervised the genetic analysis. Hirotaka Ishihara collected the clinical data. Junji Kobayashi collected and supervised the clinical data and wrote the paper. Tsutomu Hirano collected and supervised the clinical data. Hiroshi Mabuchi and Masakazu Yamagishi supervised all aspects through the research period and wrote the paper.
Footnotes
Competing interests: None declared
Contributor Information
Masa-aki Kawashiri, Email: mk@med.kanazawa-u.ac.jp.
Collaborators: Johannes Zschocke
References
- Averna M, Seip RL, Mankowitz K, Schonfeld G. Postprandial lipemia in subjects with hypobetalipoproteinemia and a single intestinal allele for apoB-48. J Lipid Res. 1993;34:1957–1967. [PubMed] [Google Scholar]
- Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: abetalipoproteinaemia - Update 2014. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: familial hypobetalipoproteinaemia (APOB) - Update 2014. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- Groenewegen WA, Averna MR, Pulai J, Krul ES, Schonfeld G. Apolipoprotein B-38.9 does not associate with apo[a] and forms two distinct HDL density particle populations that are larger than HDL. J Lipid Res. 1994;35:1012–1025. [PubMed] [Google Scholar]
- Hooper AJ, Burnett JR. Update on primary hypobetalipoproteinemia. Curr Atheroscler Rep. 2014;16:423. doi: 10.1007/s11883-014-0423-3. [DOI] [PubMed] [Google Scholar]
- Hooper AJ, Robertson K, Barrett PH, Parhofer KG, van Bockxmeer FM, Burnett JR. Postprandial lipoprotein metabolism in familial hypobetalipoproteinemia. J Clin Endocrinol Metab. 2007;92:1474–1478. doi: 10.1210/jc.2006-1998. [DOI] [PubMed] [Google Scholar]
- Inazu A, Nakajima K, Nakano T, et al. Decreased post-prandial triglyceride response and diminished remnant lipoprotein formation in cholesteryl ester transfer protein (CETP) deficiency. Atherosclerosis. 2008;196:953–957. doi: 10.1016/j.atherosclerosis.2007.02.028. [DOI] [PubMed] [Google Scholar]
- Katsuda S, Kawashiri MA, Inazu A, et al. Apolipoprotein B gene mutations and fatty liver in Japanese hypobetalipoproteinemia. Clin Chim Acta. 2009;399:64–68. doi: 10.1016/j.cca.2008.09.021. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Kojima M, Matsushima T, Teramoto T. Determination of apolipoprotein B-48 in serum by a sandwich ELISA. Clin Chim Acta. 2005;351:115–120. doi: 10.1016/j.cccn.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis. 2014;37:333–339. doi: 10.1007/s10545-013-9665-4. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Saito T, Tamura A, et al. Cholesterol in remnant-like lipoproteins in human serum using monoclonal anti apo B-100 and anti apo A-I immunoaffinity mixed gels. Clin Chim Acta. 1993;223:53–71. doi: 10.1016/0009-8981(93)90062-9. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Ishibashi S, Yamamoto M, Osuga J, Yazaki Y, Yukawa S, Yamada N. A truncated species of apolipoprotein B (B-38.7) in a patient with homozygous hypobetalipoproteinemia associated with diabetes mellitus. Arterioscler Thromb Vasc Biol. 1998;18:1330–1334. doi: 10.1161/01.ATV.18.8.1330. [DOI] [PubMed] [Google Scholar]
- Parhofer KG, Barrett PH. Thematic review series: patient-oriented research. What we have learned about VLDL and LDL metabolism from human kinetics studies. J Lipid Res. 2006;47:1620–1630. doi: 10.1194/jlr.R600013-JLR200. [DOI] [PubMed] [Google Scholar]
- Parhofer KG, Barrett PH, Aguilar-Salinas CA, Schonfeld G. Positive linear correlation between the length of truncated apolipoprotein B and its secretion rate: in vivo studies in human apoB-89, apoB-75, apoB-54.8, and apoB-31 heterozygotes. J Lipid Res. 1996;37:844–852. [PubMed] [Google Scholar]
- Pulai JI, Latour MA, Kwok PY, Schonfeld G. Diabetes mellitus in a new kindred with familial hypobetalipoproteinemia and an apolipoprotein B truncation (apoB-55) Atherosclerosis. 1998;136:289–295. doi: 10.1016/S0021-9150(97)00222-0. [DOI] [PubMed] [Google Scholar]
- Tada H, Kawashiri MA, Tanaka A, et al. Post-prandial remnant lipoprotein metabolism in autosomal recessive hypercholesterolaemia. Eur J Clin Invest. 2012;42:1094–1099. doi: 10.1111/j.1365-2362.2012.02700.x. [DOI] [PubMed] [Google Scholar]
- Tarugi P, Averna M, Di Leo E, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195:e19–e27. doi: 10.1016/j.atherosclerosis.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Turk U, Basol G, Barutcuoglu B, Sahin F, Habif S, Tarugi P, Bayindir O. A 54-year-old diabetic man with low serum cholesterol. Clin Chem. 2012;58:826–829. doi: 10.1373/clinchem.2011.163543. [DOI] [PubMed] [Google Scholar]
- Usui S, Hara Y, Hosaki S, Okazaki M. A new on-line dual enzymatic method for simultaneous quantification of cholesterol and triglycerides in lipoproteins by HPLC. J Lipid Res. 2002;43:805–814. [PubMed] [Google Scholar]
- Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol. 2014;25:161–168. doi: 10.1097/MOL.0000000000000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- Yang XP, Inazu A, Yagi K, Kajinami K, Koizumi J, Mabuchi H. Abetalipoproteinemia caused by maternal isodisomy of chromosome 4q containing an intron 9 splice acceptor mutation in the microsomal triglyceride transfer protein gene. Arteioscler Thromb Vasc Biol. 1999;19:1950–1955. doi: 10.1161/01.ATV.19.8.1950. [DOI] [PubMed] [Google Scholar]