

Abstract
Polo-like kinases (Plks) are a family of serine-threonine kinases that regulate multiple intracellular processes including DNA replication, mitosis, and stress response. Plk1, the most well understood family member, regulates numerous stages of mitosis and is overexpressed in many cancers. Plk inhibitors are currently under clinical investigation, including phase III trials of volasertib, a Plk inhibitor, in acute myeloid leukemia and rigosertib, a dual inhibitor of Plk1/phosphoinositide 3-kinase signaling pathways, in myelodysplastic syndrome. Other Plk inhibitors, including the Plk1 inhibitors GSK461364A, TKM-080301, GW843682, purpurogallin, and poloxin and the Plk4 inhibitor CFI-400945 fumarate, are in earlier clinical development. This review discusses the biologic roles of Plks in cell cycle progression and cancer, and the mechanisms of action of Plk inhibitors currently in development as cancer therapies.
Introduction
During the cell cycle, diverse molecular signals are integrated to initiate and maintain checkpoints that halt the progression of cell growth and allow time for DNA repair [1]. These checkpoints, which occur at the G1/S phase transition, the G2/M phase transition, and during mitosis before cell division, are tightly regulated by nuclear serine-threonine kinases, including cyclin-dependent kinases (Cdks), Polo-like kinases (Plks), and Aurora kinases [2]. In cancer, these kinases are often dysregulated, promoting uncontrolled cell proliferation and aberrant cell division [1]. Dysregulated expression of different Plk family members has been documented in many cancer types and has been associated with poor prognosis, leading to an enhanced interest in these kinases as promising targets for anticancer drug development [3]. This review will discuss the functions of the Plk family members in cell cycle progression and their dysregulation in cancer. The mechanisms of action and preclinical findings of promising Plk inhibitors, some of which are now in clinical trials, will also be discussed.
Structure of Plks
The Polo gene was first cloned from Drosophila melanogaster in 1988, and it was observed that mutations in Polo induced abnormal spindle poles during mitoses [4]. Five mammalian homologues for Polo, named Plk1 to Plk5, were soon identified and have been reviewed by Strebhardt [3]. The Plk1 to Plk4 proteins have similar structures, with a conserved serine-threonine kinase domain located at the amino-terminal and a regulatory domain consisting of one (as in Plk4) or two (as in Plk1 to Plk3) polo-box domains (PBDs) at the carboxyl-terminal (Figure 1) [5]. Crystal structures of the kinase and PBDs of human Plk1 are displayed in Figure 2. PBD-dependent binding is important for subcellular localization and targeting of Plk activity toward specific subcellular domains [6]. Distinct from the other Plk family members, Plk5 is a PBD-containing protein that lacks the kinase domain [7].
Figure 1.
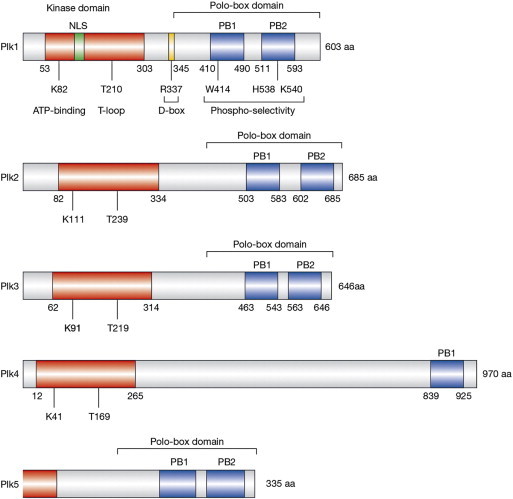
Domain structures of the human Plk family of proteins. Open reading frame amino acid (aa) lengths are shown on the right, and positions of the kinase domains (red) and polo-boxes 1 and 2 (blue) are indicated. Nuclear localization (NLS) sequences are indicated in green, and the D-box domain is indicated in yellow. Residues that are critical for ATP-binding and enzymatic activation (T-loop) within the kinase domains and phospho-selectivity within the polo-boxes are indicated. Adapted by permission from Macmillan Publishers Ltd, copyright (2006) [5].
Figure 2.
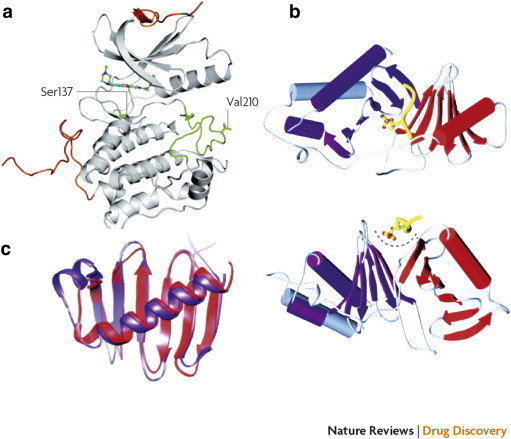
Crystal structure of the N-terminal kinase domain (a) and C-terminal PBD (b) and (c) of human Plk1. (a) The crystal structure of the kinase domain was determined in complex with the pyrrolo-pyrazole inhibitor PHA 680626 at 2.4-Å resolution. The N-terminal and C-terminal extensions are shown in orange, and the activation loop is shown in green. The regulatory phosphorylation site Thr210 was mutated to Val210 to reduce conformational heterogeneity. The position of Ser137, which has been proposed as an additional phosphorylation site for the activation of the kinase activity of Plk1, is also indicated. (b) The crystal structure of the PBD is shown as a ribbon diagram from two different angles in complex with a phosphothreonine-containing peptide (shown in yellow). Polo-box 1 and polo-box 2 are shown in red and purple, respectively. The polo-cap at the N-terminal end of polo-box 1 (gray) folds around polo-box 2, tethering it to polo-box 1 and forming a pocket to accommodate the phosphopeptide. (c) A superposition of the polo-box 1 and polo-box 2 structures is shown (colors indicated in b). Each polo-box consists of a six-stranded β-sheet and an α-helix, which associate to form a 12-stranded β-sandwich domain. This structure documents an interaction along a positively charged cleft formed between the two polo-boxes. Reprinted with permission from Macmillan Publishers Ltd, copyright (2010) [3].
Functions of Plks in the Cell Cycle
Plk1 is expressed in normal dividing cells and has a pivotal role in regulating many key stages of the cell cycle [3]. In mouse models, homozygous loss of Plk1 resulted in early embryonic lethality, with embryos displaying developmental delays and a lack of mitotic spindle assembly at embryonic day 3.5, ultimately failing to survive past the eight-cell stage [8]. In contrast, mice heterozygous for Plk1 (Plk1+/−) were healthy at birth and displayed no obvious defects from the loss of one Plk1 allele [8]. Preclinical studies in human cell lines have demonstrated that Plk1 activity is essential in numerous stages of mitosis, including functional maturation of centrosomes and bipolar spindle assembly, M phase entry, nuclear envelope breakdown (NEBD), sister chromatid cohesion and formation of kinetochore-microtubule attachments, and finally mitotic exit and cytokinesis (Figure 3) [5].
Figure 3.
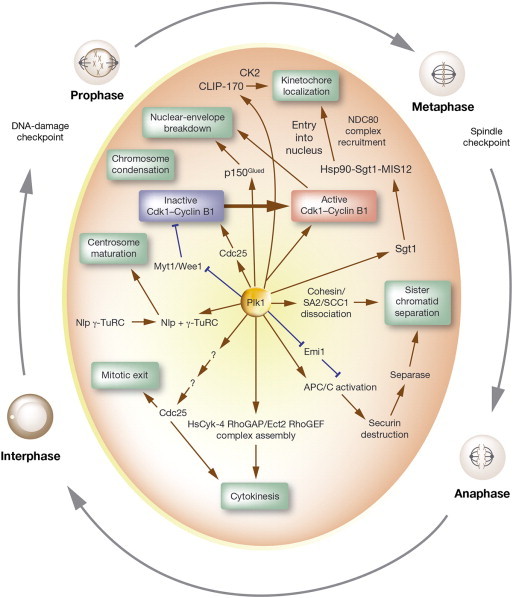
Functional roles of Plk1 in cell cycle progression. The figure is a schematic diagram depicting Plk1 as a regulator of several stages during mitotic progression. This view is not intended to be complete but represents frequently studied aspects of Plk1 activities along with several newly ascribed functional interactions. Stimulatory interactions are shown in brown and inhibitory interactions are shown in blue.
Early studies in human immortalized HeLa cells demonstrated the requirement of Plk1 activity in centrosome maturation and establishment of a bipolar spindle before mitotic entry [9]. Microinjection of anti-Plk1 antibodies into cells resulted in reduced centrosome size and decreased accumulation of γ-tubulin, now known to form the γ-tubulin ring complex required for microtubule nucleation. Later studies further demonstrated that Plk1 phosphorylation of the centrosome-associated ninein-like protein, involved in microtubule assembly, first dissociates ninein-like protein from the centrosome, allowing recruitment of the γ-tubulin ring complex (Figure 3) [10]. Using RNA interference (RNAi) in human osteosarcoma cell lines, Plk1 was also shown to regulate the centrosomal localization of Aurora A kinase, another key regulator of centrosome maturation and function [11].
Plk1 is part of a regulatory network controlling Cdk1/cyclin B complex activity and entry into mitosis at the G2/M transition [2]. Before active complex formation with cyclins, Cdk activation requires the removal of inhibitory phosphates by Cdc25 phosphatases. Preclinical studies in human Jurkat cells have shown that Plk1 directly phosphorylates and activates Cdc25, resulting in subsequent dephosphorylation of the inactive Cdk1/cyclin B complex [12]. Further, Plk1 was shown to phosphorylate and stimulate degradation of Wee1, a negative regulator of Cdk1 (Figure 3) [13]. Plk1-mediated degradation of Wee1 was also shown to be an important step in the regulation of mitotic entry following DNA damage response, suggesting an important role for Plk1 in DNA damage recovery following G2 phase arrest [13].
Another essential step for mitotic entry that occurs during G2/M transition is NEBD, a process regulated by both microtubules and microtubule-associated motor proteins, such as the dynein/dynactin complex [14]. In recent studies in mammalian cells, Plk1 was shown to interact with and phosphorylate p150Glued, a major component of the dynein/dynactin complex, during NEBD at prophase [14]. This Plk1-mediated phosphorylation of p150Glued positively regulated the accumulation of p150Glued at the nuclear envelope (Figure 3). Further, expression of a Plk1-unphosphorylatable mutant of p150Glued (p150Glued-S179A) resulted in reduced NEBD and G2 phase arrest, suggesting that Plk1 phosphorylation of p150Glued may be an important regulatory step of NEBD and mitotic entry [14].
Once cells have entered mitosis, the correct segregation of sister chromatids at anaphase requires the dissociation of cohesin complexes from the replicated chromosomes and the formation of kinetochore-microtubule attachments. In a Xenopus model, Plk1 was shown to phosphorylate the SA2 subunit of cohesin, resulting in dissociation of the cohesin complex from chromosomes during prophase [15]. In addition, recruitment of Plk1 to kinetochore/centromere regions of chromosomes has been observed, suggesting a role in kinetochore-microtubule attachment [16]. In this context, Plk1 was shown to phosphorylate CLIP-170, a protein that directly interacts with the dynein/dynactin complex to promote kinetochore-microtubule attachments [17]. Plk1 phosphorylation of CLIP-170 at S195 is thought to create a docking site for additional phosphorylation by casein kinase 2, which promotes dynactin-mediated kinetochore localization of CLIP-170 during prometaphase (Figure 3). Inhibition of CLIP-170 phosphorylation by Plk1 and casein kinase 2 through mutation of the specific phosphorylation residues resulted in defects in kinetochore formation [17]. Plk1 was also shown to phosphorylate and regulate the localization of Sgt1, a co-chaperone of heat shock protein 90, to kinetochores during prometaphase [18]. Plk1 phosphorylation of Sgt1 enhanced the association of heat shock protein 90–Sgt1 with the MIS12 complex. This interaction stabilized the MIS12 complex at kinetochores, facilitating the recruitment of the NDC80 complex and subsequent formation of microtubule-binding sites at the kinetochores (Figure 3) [18]. Inhibition of Plk1 phosphorylation of Sgt1 decreased recruitment of the MIS12 and NDC80 complexes at kinetochores and impaired microtubule attachment, ultimately resulting in chromosome misalignment and delayed onset of anaphase.
Complete chromosome segregation and exit from mitosis is regulated by the anaphase-promoting complex/cyclosome (APC/C) [19]. Along with the Cdk1/cyclin B complex, Plk1 has been shown to phosphorylate subunits of the APC/C, jointly activating the ubiquitin ligase activity of the APC/C that is required for degradation of mitotic cyclins, facilitation of sister chromatid segregation, and initiation of mitotic exit [19]. Plk1 was also shown to regulate the localization and destruction of a negative regulator of APC/C, early mitotic inhibitor 1 [20]. In coordination with chromosome segregation, Plk1 has been shown to localize to the central spindle [21] and regulate the assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex, facilitating cleavage furrow formation in late-stage mitosis, which is necessary for the final stage of the cell cycle, cytokinesis (Figure 3) [22].
Multiple levels of control are in place to ensure that Plk1-dependent phosphorylation of its various substrates is properly coordinated in time and space [23]. Plk1 transcription is regulated by several transcription factors, including forkhead box protein M1, p53, and the E2F family, during various stages of the cell cycle [3,23]. Regulation of Plk1 protein activity by phosphorylation on a conserved Thr residue (Thr210) in the kinase activation loop was shown to be mediated by Bora/Aurora A during normal mitotic entry and following DNA damage checkpoint recovery [24]; other kinases and phosphatases have also been implicated in the regulation of Plk1 at this residue [23]. In addition to regulation of Plk1 activity by kinases/phosphatases, spatial regulation of Plk1 through protein interactions with the PBD domain has been demonstrated with numerous proteins [23]; details regarding these interactions are beyond the scope of this review. Like many other proteins, Plk1 is also a target of ubiquitination and proteosome-dependent degradation, with the APC/C as the requisite ubiquitin ligase for Plk1 at mitotic exit [25].
The functional roles of the other Plk members in normal cell biology are not yet fully understood. Plk2, a centrosomal kinase expressed primarily in G1 phase, has been shown to regulate centriole duplication in cooperation with Plk4 [26] and may play a role in the G2/M checkpoint following activation by p53 in response to genotoxic damage [27]. Unlike Plk1 null mice (Plk1−/−) mice, Plk2−/− mice are viable, albeit with observable retardation of skeletal development and growth [28]. Despite these developmental delays, no differences in 12-month survival rates compared with wild-type mice were observed.
Among the Plk family members, Plk3 appears to be most similar to Plk2, with murine homozygous loss of Plk3 having no effect on viability [29]. Interestingly, Plk3−/− mice displayed no obvious growth impairment and were larger than age- and sex-matched, wild-type littermates by 20 months of age. In human cell lines, Plk3 expression was shown to peak in G1 phase [30]. RNAi-mediated depletion of Plk3 in these cells resulted in attenuation of cyclin E expression and inhibition of S phase entry, suggesting a role for Plk3 in the G1/S phase transition. Plk3 was also shown to localize to centrosomes during interphase and spindle poles during mitosis and was detected at the midbody during cytokinesis [31]. Overexpression of Plk3 in human cells resulted in mitotic arrest and defects in cytokinesis. As such, Plk3 has been implicated in the regulation of various stages of mitosis and response to genotoxic stress [31].
Similar to findings with Plk1, homozygous loss of Plk4 resulted in embryonic lethality in mouse models [32]. Plk4−/− embryos ceased to grow following gastrulation at embryonic day 7.5, displaying cells that appeared to be arrested in late-stage mitosis. In mammalian cell lines, Plk4 expression was shown to increase at the G1/S phase transition, persisting until late M phase and declining in early G1 phase [33]. The Plk4 protein has been shown to localize to the nucleolus during G2 phase, to centrosomes during early M phase, and to the cleavage furrow during cytokinesis [32]. In addition to the established role of Plk4 in centriole duplication [26], Plk4 was implicated in the regulation of APC/C-mediated destruction of cyclin B1 during mitotic exit in Plk4−/− mouse models [32].
Plk5 was cloned in 2010 [34] and little is known about its functions. The Plk5 protein lacks the kinase domain found in other Plk family members and appears to have no catalytic activity (Figure 1). To date, the effects of genetic deletion of Plk5 in mouse models have yet to be reported. In mouse and human cell lines, ectopic expression of Plk5 resulted in a G1 phase cell cycle arrest and subsequent apoptosis [34]. Plk5 levels were shown to be downregulated in proliferating human cells compared with quiescent cells [7]. Interestingly, Plk5 was shown to function in neuron biology and was expressed primarily in brain of both mice and humans [7].
Plk Expression in Human Tumors
Cell cycle dysregulation is a common feature of human cancer, with cancer cells frequently displaying unscheduled proliferation, as well as genomic and chromosomal instability [1]. As Plk1 plays an important role in cell cycle progression in normal proliferating tissues, it is not surprising that Plk1 is overexpressed in many cancer types, including melanoma, breast, non–small cell lung, colorectal, prostate, pancreatic, ovarian, and head and neck cancers, as well as non-Hodgkin’s lymphomas and acute myeloid leukemia (AML) as reviewed by Strebhardt [3] and Cholewa et al. [35]. In contrast to the low levels of Plk1 expression observed during interphase in normal cells, localization of Plk1 to the nucleus in cancer cells occurs before G2/M phase and persists through G1/S phase [35]. Plk1 overexpression is associated with a worse prognosis, including lower overall survival in several cancer types, such as non–small cell lung, head and neck, melanoma, and prostate cancers [3,35]. Interestingly, Plk1 expression is low in surrounding normal, non-dividing tissue. No chromosomal translocations or mutations have been found in the Plk1 gene [35].
In contrast to Plk1 overexpression observed in some human cancers, Plk2 expression was found to be decreased in B-cell neoplasms due to CpG methylation–dependent silencing [36]. Methylation in the Plk2 CpG island has also been observed in other hematologic malignancies and was correlated with chemotherapeutic sensitivity in cancer cell lines and outcome in patients with ovarian cancer treated with platinum- and taxane-based therapy [37]. In core-binding factor AML, overexpression of the microRNA miR-126/126*, a negative regulator of Plk2, was observed, potentially suggesting a role for Plk2 as a tumor suppressor in these malignancies [38]. Plk3 has also been suggested as a possible tumor suppressor since it is often downregulated in certain tumors, such as lung and head and neck cancers [39,40]. Expression levels of Plk4 in human cancers are less consistent, with reported overexpression in colon and breast cancers [41,42] and down-regulation in hepatocellular carcinoma [18]. In breast cancer cells overexpressing Plk4, RNAi depletion of Plk4 was shown to inhibit cell growth in vitro and in vivo [42]. Finally, Plk5 was shown to be significantly downregulated in human brain tumors, with frequent silencing of Plk5 by hypermethylation observed in astrocytomas and glioblastomas [7]. Re-expression of Plk5 prevented cell proliferation of these tumor cells in vitro.
Interaction of Plks with Other Cancer-Associated Pathways
Crosstalk between Plk1 to Plk4 and the p53 tumor suppressor has been reported in cancer cells under conditions of genotoxic stress [3]. Plk1 was also shown to be required for the viability of cells harboring activated Ras [43] or inactivated p53, particularly in the presence of genotoxic damage [44]. Accumulating evidence suggests an important role for Plk1 in the negative regulation of p53 activity through two novel Plk1 targets: G2 and S phase–expressed protein 1 (GTSE1) and DNA topoisomerase 1 binding protein (Topors) [45,46]. GTSE1 is a negative regulator of p53 that directly binds and shuttles p53 out of the nucleus, inducing its degradation and allowing for G2 checkpoint recovery [46]. Plk1 was shown to phosphorylate GTSE1 and promote its nuclear localization, facilitating the interaction of GTSE1 with p53 and subsequent checkpoint recovery. Topors is a dual ubiquitin and sumoylation E3 ligase for p53, wherein ubiquitination of p53 results in its degradation but sumoylation of p53 results in increased p53 stability [45]. Plk1-mediated phosphorylation of Topors was shown to modulate the activity of this ligase, resulting in decreased p53 sumoylation and increased p53 ubiquitination by Topors. Taken together, these data suggest that Plk1 is an important negative regulator of both p53 protein stability and nuclear localization [45,46].
Since the p53 tumor suppressor is a critical regulator of DNA damage checkpoints and cell cycle control, the negative regulation of p53 by Plk1 may provide a potential mechanism for the tumorigenic potential of Plk1. In this context, depletion of Plk1 using small interfering RNA in human cancer cell lines resulted in a G2/M phase arrest and subsequent apoptosis, which was characterized by activation of the p53 pathway [47–49]. Interestingly, the cytotoxicity observed with Plk1 depletion was enhanced in cancer cells with either no p53 or inactivated p53 compared with those harboring wild-type p53 protein. DNA damage was detected in Plk1-depleted cells regardless of p53 status, suggesting that the combined loss of Plk1 and p53 checkpoint activity may contribute to enhanced cytotoxicity in the presence of accumulating genotoxic damage [47,48]. Importantly, normal cells were shown to be largely unaffected by Plk1 depletion in these studies, supporting the feasibility of Plk1 as a potential target for cancer therapy [47–49].
As discussed, Plks are susceptible to aberrant DNA methylation in many tumor types. In a recent study by Ward et al., oxidative stress induced by hypoxic conditions or reactive oxygen species (ROS) in mouse embryonic fibroblasts resulted in significant hypermethylation of Plk1 and Plk4 promoters and a reduction in the corresponding protein levels [50]. In human hepatocellular carcinoma cell lines, hypermethylation of Plk1 and Plk4 appeared to be dependent on the presence of functional p53, and Plk4 up-regulation was observed in p53-deficient cells in the presence of hypoxia and ROS. Plk4 expression and protein levels were also elevated in response to ROS treatment in p53-deficient osteosarcoma cells, wherein the Plk4 promoter was observed to be hypomethylated [50]. These findings highlight the complex and cell-specific activities of Plks and suggest a model whereby oxidative stress may lead to epigenetic silencing of Plks in normal and cancer cells in a p53-dependent manner. However, in the absence of p53, enhanced expression of Plk1 and Plk4 could promote aberrant cell cycle progression, genomic instability, and tumorigenesis [50].
Plk2 and Plk3 were also shown to play important roles in mediating response to genotoxic stress [3]. In preclinical studies, both Plks have demonstrated a functional interaction with the p53 pathway; however, unlike Plk1, the expression of Plk2 and Plk3 was induced in response to cellular stresses [27,51]. A p53-dependent induction of Plk2 was shown in response to genotoxic damage in irradiated cells, and depletion of Plk2 enhanced cell death in response to mitotic catastrophe [27]. Plk3 was shown to phosphorylate and activate p53 in an ataxia telangiectasia mutated–dependent manner in response to DNA damage [51]. These studies suggest tumor suppressive roles for Plk2 and Plk3 in response to genotoxic stress.
Preclinical Data Demonstrating Validity of Plk1 as a Target in Cancer
The critical roles of Plk1 in cell cycle progression and response to cellular stresses implicate this Plk family member in the development of genomic instability and aberrant cell proliferation and survival associated with tumorigenesis [35]. Although homozygous loss of Plk was lethal in mouse models, Plk1+/− mice aged 50 to 70 weeks were shown to harbor tumors in various organs, predominantly consisting of lymphomas that had invaded the lung and liver, and lung carcinomas [8]. As described above, preclinical studies have shown that normal human cells are largely unaffected by loss of Plk1, whereas Plk1 depletion promotes enhanced apoptosis in cancer cells [49]. Depletion of Plk1 was also shown to decrease xenograft tumor growth of human cancer cells [52]. These preclinical studies suggest that Plk1 is both a feasible and relevant target for suppressing tumor cell growth.
Tumors with both p53 deficiency and/or RAS mutations and high Plk1 expression may be particularly sensitive to Plk1 inhibitors [43,44]. In this context, cancer cells with wild-type p53 were shown to be less sensitive to loss of Plk1 activity than p53-deficient cells [48]. In addition to its interactions with the p53 tumor suppressor pathway, Plk1 was shown to facilitate survival in phosphatase and tensin homologue (PTEN)–depleted prostate cancer cells [53]. Loss of PTEN in prostate cancer cells lines was associated with increased aneuploidy and mitotic stress, as well as an increase in Plk1 expression. This overexpression of Plk1 was shown to be required for PTEN-depleted cells to adapt to mitotic stress for survival, and reintroduction of PTEN activity reduced the survival dependence on Plk1 [54]. Further, inhibition of Plk1 suppressed xenograft tumor growth of PTEN-depleted prostate cancer cells. These preclinical findings suggest that Plk1 could be a particularly relevant target for cancers characterized by other cancer-associated mutations.
In addition to Plk1, heterozygous loss of Plk4 and homozygous loss of Plk3 have been associated with spontaneous tumor development in mice. In Plk4+/− mice aged > 18 months, the incidence of spontaneous liver and lung tumor development was approximately 15 times greater compared with wild-type littermates [55]. In Plk3−/− mice aged > 18 months, the incidence of spontaneous tumor development was more than six times higher compared with wild-type littermates, with tumors reported in the lung, kidney, liver, and uterus [29]. Plk3+/− mice were not reported to have elevated tumor formation compared with wild-type mice in this study.
Plk Inhibitors as Anticancer Therapies
Inhibition of cell proliferation and induction of apoptosis are basic principles of anticancer therapy. Antimitotic therapy is standard for many cancer types, but lack of selectivity is associated with adverse effects, such as neurotoxicity and myelosuppression [3]. The key role of Plk1 in oncogenic events gave impetus to the development of potent and specific small molecule Plk1 inhibitors (Table 1). In this context, there are two potential sites of Plk1 to target: the adenosine-5′-triphosphate (ATP)–binding site in the kinase domain (a classic target for the design of inhibitors of the human kinome) and the PBD [63]. Targeting Plk1 RNA and the interaction of Plk1 with a key binding partner are other possible approaches. Preclinical studies with several different Plk inhibitors have been published, and some are under clinical investigation. The majority of these inhibitors target Plk1 but also Plk2 and Plk3 to a lesser extent. In addition, there is one Plk4 inhibitor currently in clinical development.
Table 1.
Small Molecule Plk1 Inhibitor Structures, Mechanisms of Action, and Preclinical Activities
| Agent/Structure | Mechanism of Action | IC50 |
|---|---|---|
Rigosertib (ON 01910.Na) (benzylstyryl sulphone)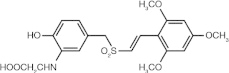
|
Affects microtubule dynamics | Plk1 = 9-10 nM 10- to 20-fold higher concentrations were required for inhibition of Plk2 [56] |
Volasertib (BI 6727) (dihydropteridinone derivative)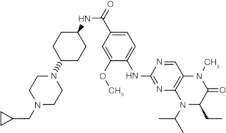
|
ATP-competitive inhibitor | Plk1 = 0.87 nM Plk2 = 5 nM Plk3 = 56 nM [57] |
GSK 461364 (thiophene derivative)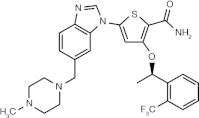
|
ATP-competitive inhibitor | Plk1 ≤ 0.5 nM⁎ Plk2 = 860 nM⁎ Plk3 = 1000 nM⁎[58] |
GW843682 (benzimidazole thiophene)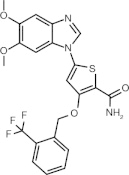
|
ATP-competitive inhibitor | Plk1 = 2.2 nM Plk2 = N/A Plk3 = 9.1 nM [59] |
PLHS-Pmab ((2S,3R)-2-amino-3-methyl-4-phosphonobutyric acid)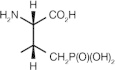
|
Interferes with Plk1 PBD functions in vitro and in vivo | Not reported [60] |
PPG (benzotropolone-containing compound)
|
Inhibits PBD-dependent binding in vitro and in vivo | Plk1 = ~ 0.3 μM (in glutathione S-transferase pull-down assay) Plk2 = N/A Plk3 = N/A [61] |
Poloxin (thymoquinone derivative)
|
Interferes with Plk1 PBD functions in vitro and in vivo | Plk1 = 4.8 μM Plk2 = 18.7 μM Plk3 = 53.9 μM [62] |
N/A, not applicable.
IC50 values for GSK461364A were determined on the basis of the intrinsic binding constant (Ki⁎app), which was calculated by applying the Cheng-Prusoff relationship for a competitive inhibitor (ATP Kmapp = 16 μM) to the IC50 value obtained following a 60-minute preincubation in the presence of GSK461364A [58].
Plk Inhibitors in Clinical Development
Rigosertib (ON 01910.Na)
Rigosertib (ON 01910.Na) is a non–ATP-competitive, small-molecule, dual inhibitor of the phosphoinositide 3-kinase (PI3K) and Plk signaling pathways [56]. In recent preclinical studies by Reddy et al., rigosertib was shown to bind the Ras-binding domain of Raf, resulting in the inactivation of Raf and inhibition of the Raf-Plk1 interaction that activates Plk1 activity [64]. Rigosertib also binds to a similar Ras-binding domain in PI3K, a signaling protein involved in the regulation of various biologic processes, including cell cycle progression, metabolism, and survival [65]. In human cancer cells in vitro and in xenograft tumor models, rigosertib at a half maximal inhibitory concentration [IC50] of 50 to 200 nM resulted in mitotic arrest, characterized by spindle abnormalities and subsequent apoptosis [56]. In vivo studies in rats and dogs to examine the safety profile of rigosertib showed that this agent was not associated with hematotoxicity, liver damage, or neurotoxicity [56]. Phase I studies have been completed in patients with various tumor types, and rigosertib is now in phase III trials for second-line treatment of high-risk myelodysplastic syndrome (NCT01928537 and NCT00906334) and phase II trials for first-line treatment of low-risk myelodysplastic syndrome (NCT01584531 and NCT01904682). A phase III study of rigosertib in combination with gemcitabine is also underway in patients with metastatic pancreatic cancer (NCT01360853).
Volasertib
Volasertib (BI 6727) is an ATP-competitive kinase inhibitor from the dihydropteridinone class of compounds [57]. Unlike the dual PI3K/Plk pathway inhibitor rigosertib, volasertib is a potent and selective Plk inhibitor that did not target > 50 additional kinases tested [57]. Volasertib targets the ATP-binding pocket of Plk1, binding to the hinge region between the amino-terminal and the carboxyl-terminal lobes of the kinase domain through hydrogen bonds from the dihydropteridinone core to the backbone amino and carbonyl groups of Cys133 [57]. As residues in the ATP-binding pocket of Plks are highly conserved, volasertib also targets Plk2 and Plk3, although to a lesser extent than Plk1 (IC50 values of 0.87, 5, and 56 nM for Plk1, Plk2, and Plk3, respectively).
Volasertib was shown to induce the characteristic prometaphase arrest (“Polo arrest”) associated with the inhibition of Plk1 activity and subsequent apoptosis in a panel of human cancer cell lines in vitro and in xenograft tumor models [57]. Importantly, the effects of volasertib on xenograft tumors were not observed in normal intestinal cells of mice, suggesting that volasertib may have less of an effect on normal proliferating tissues. Volasertib was also efficacious in a taxane-resistant xenograft model [57] and in a human AML xenograft model [66]. In animal models, volasertib demonstrated a high volume of distribution, which indicated good tissue penetration, a long half-life, and good oral bioavailability [57]. These preclinical observations are supported by early clinical trial data in numerous studies of patients with solid tumors and AML [67–69].
In addition to the single-agent activity of volasertib shown in preclinical studies, volasertib demonstrated synergy with Aurora kinase inhibition in cell lines generated from patients with relapsed pediatric leukemia [70]. Volasertib is currently in a phase III trial in combination with low-dose cytarabine in patients aged ≥ 65 years with previously untreated AML who are ineligible for intensive remission induction therapy (NCT01721876). Phase II trials of volasertib in patients with various tumor types, including AML [71] and urothelial [72] and ovarian cancers, have been completed [73], and a phase II trial in advanced non–small cell lung cancer (NCT00824408) is ongoing.
GSK461364A
GSK461364A is a selective and reversible ATP-competitive inhibitor of Plk1 that induced growth inhibition in a panel of human solid and peripheral blood tumor cell lines and xenograft tumor models [58]. The cell cycle effects of GSK461364A were dose-dependent, primarily resulting in the characteristic prometaphase Polo arrest and subsequent apoptosis at doses of 10 to 250 nM. At higher concentrations (> 250 nM), GSK461364A induced a G2 phase arrest followed by a gradual progression into terminal mitoses, which correlated with a decrease in apoptosis in some cases [58]. Loss of p53 activity in human cancer cells was associated with increased sensitivity to GSK461364A compared with cells harboring wild-type p53 [74]. Furthermore, treatment with GSK461364A was shown to increase the radiosensitivity of glioblastoma multiforme cells, with no effect on normal cells [75]. Clinical activity of GSK461364A was reported in some patients with advanced solid tumors in a phase I study [76].
CFI-400945 fumarate
CFI-400945 fumarate is a Plk4 inhibitor discovered using a virtual screening program of ligands designed based on a novel (E)-3-((1H-indazol-6-yl)methylene)indolin-2 structure [77]. CFI-400945 demonstrated an IC50 of 0.6 nM for Plk4, at least two orders of magnitude higher than that observed for any other kinases, including other Plk family members (> 10 μM). In human cancer cell lines and xenograft tumor models, CFI-400945 inhibited cell proliferation and tumor growth [42,77]. CFI-400945 was shown to suppress phosphorylation of Plk4 and histone H3, leading to failure of centrosome clustering and formation of multipolar spindles and resulting in cell death [42]. Interestingly, response to Plk4 inhibition appeared to differ between breast cancer subtypes and may be influenced by hormone receptor status (e.g., estrogen receptor) and the presence of specific mutations (e.g., PTEN) [42]. A phase I trial in patients with advanced cancer is currently recruiting (NCT01954316).
TKM-080301 (TKM-PLK1)
TKM-080301 (TKM-PLK1) is a lipid nanoparticle formulation of a small interfering RNA directed against Plk1 [78,79]. In preclinical studies, TKM-080301 induced Plk1 mRNA cleavage and silencing of Plk1 expression in human cancer cell lines and potent antiproliferative activity in various cancer cell lines. Antitumor activity in xenograft tumor models was also observed [79]. Further, in vivo silencing of Plk1 expression by TKM-080301 was detected for up to 7 to 10 days following single administration. TKM-080301 is being evaluated in a first-in-human, dose-escalation, phase I study in patients with advanced solid tumors or lymphoma, which will include study of the pharmacodynamic effects of Plk1 inhibition in patient biopsy samples [78].
Plk Inhibitors in Preclinical Development
GW843682
GW843682, a selective thiophene benzimidazole ATP-competitive inhibitor of Plk1 (IC50 of 2.2 nM) and Plk3 (IC50 of 9.1 nM), demonstrated > 100-fold greater selectivity for Plk1 compared with other non-Plk kinases tested [59]. GW843682 inhibited the proliferation of a variety of human solid tumor cell lines, demonstrating a transient G2/M arrest and mitotic spindle defects and resulting in apoptosis. While GW843682 also induced a G2/M arrest in normal human diploid fibroblasts, little apoptosis was observed [59].
Plk1 PBD inhibitors (PLHS-Pmab, purpurogallin, and poloxin)
Structural studies of Plk1 suggest that the two polo-box motifs of the PBD contain identical β6α folds (consisting of a six-stranded antiparallel β-sheet and an α-helix), forming a heterodimeric phosphopeptide-binding module [80]. The Lee laboratory showed that a phosphopeptide, PLHSpT, derived from the T78 motif of the PBD-binding centromere protein PBIP1 [16], binds to the PBD in a cleft formed between the two polo-box motifs through direct hydrogen bonding and inhibits the interaction between the PBD and its binding targets [60]. On the basis of these findings, a nonhydrolyzable p-Thr78 mimetic peptide (PLHS-Pmab) was synthesized and its activity was assessed in HeLa cells. In glutathione S-transferase pull-down assays, PLHS-Pmab interacted with Plk1 but not Plk2 or Plk3 [60]. Similar to the phenotype observed with loss of PBD function, cells treated with PLHS-Pmab demonstrated a chromosome congression defect followed by mitotic catastrophe. Further, reduced Plk1 localization at the centrosomes and kinetochores was observed [60].
In addition to PBD-binding phosphopeptides, the compounds purpurogallin (PPG) and poloxin have been described as small, drug-like Plk1 PBD inhibitors [81]. However, these compounds demonstrate different PBD-binding properties and are associated with distinct cellular phenotypes. PPG, a natural benzotropolone compound extracted from nutgalls, was found in a high-throughput screening of natural products to identify inhibitors of PBD-dependent binding [61]. Structurally, PPG was proposed to fill the SpT pocket of the PBD through π-π stacking, electrostatic, and hydrogen bonding interactions [81]. PPG was shown to inhibit PBD binding of Plk1 and Plk2 but not Plk3 [61]. PPG inhibited PBD-dependent binding of Plk1 in vitro and in HeLa cells, decreasing centrosomal and kinetochore localization of Plk1 [61]. Although PPG did not appear to impair the formation of bipolar spindles, chromosome alignment at metaphase plates was perturbed, which activated the spindle assembly checkpoint pathway and prolonged mitotic progression.
Poloxin is a synthetic derivative of thymoquinone, a constituent of black seed (Nigella sativa) known for its antineoplastic activity [62]. Unlike PPG, poloxin does not appear to target the SpT pocket of the PBD but may fill a different site within the PBD through covalent bonding [81]. Poloxin inhibits the PBD of Plk1 in vitro (IC50 of 4.8 μM) and Plk2 and Plk3 to a slightly lesser extent (IC50 of 18.7 and 53.9 μM, respectively). Inhibition of Plk1 by poloxin results in Plk1 mislocalization, chromosomal defects, mitotic arrest, and apoptosis in cancer cell lines and xenograft tumor models [62,82]. There has been some concern that poloxin causes unacceptable levels of cytotoxicity, and a critical assessment of the efficacy and safety of these PBD inhibitors should be conducted [3].
Conclusion
Plks appear to be relevant and feasible targets for cancer treatment, and Plk inhibitors are currently under investigation to determine their efficacy and safety profiles in diverse tumor types. Preclinical studies have demonstrated a particular sensitivity to Plk1 inhibition in human cancer cells harboring specific genetic abnormalities, including mutations in p53, Ras, and PTEN [43,44,54]. These studies provide a rationale for potential patient selection in the clinic, as patients with tumors showing these mutations may particularly benefit from Plk1 inhibitor therapy. In addition to exploring genetic biomarkers for patient selection, there is a critical need for further evaluation of pharmacodynamic endpoints and functional biomarkers of response to Plk inhibition. Despite the existence of many downstream targets of Plk1 activity, there are currently no defined biomarkers of response developed for clinical use [83]. The identification and development of biomarkers associated with sensitivity and clinical response to Plk inhibitors could not only improve patient selection and provide a means to monitor treatment efficacy but also open the door to new therapeutic combination strategies.
In addition to the current focus on Plk1, a greater understanding of the roles of Plk2 and Plk3 in cancer development is needed. If they are truly tumor suppressors, partial inhibition by Plk1 inhibitors could support long-term malignant progression [3]. For those Plks affected by methylation events, such as Plk2 [36,37], it has been postulated that drugs used to reverse hypermethylation may represent an effective strategy to restore Plk tumor suppressive activities, particularly in combination treatments [50].
Acknowledgements
The author was fully responsible for all content and editorial decisions, was involved at all stages of manuscript development, and has approved the final version of this review that reflects the author’s interpretation and conclusions.
Footnotes
Medical writing assistance, supported financially by Boehringer Ingelheim Pharmaceuticals, Inc, was provided by Katie McClendon of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this review. Boehringer Ingelheim was given the opportunity to review for factual accuracy only. This work was supported by the National Institutes of Health (R01CA157429), the American Cancer Society (RSG-13-073-01-CNE), and the National Science Foundation (1049693-MCB). The author declares no conflicts of interest.
References
- 1.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 2.Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol. 2013;14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- 3.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 4.Sunkel CE, Glover DM. Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89:25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 5.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 6.Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, Lee KS. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277:32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 7.de Cárcer G, Escobar B, Higuero AM, García L, Ansón A, Pérez G, Mollejo M, Manning G, Melendez B, Abad-Rodríguez J. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol Cell Biol. 2011;31:1225–1239. doi: 10.1128/MCB.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu LY, Wood JL, Minter-Dykhouse K, Ye L, Saunders TL, Yu X, Chen J. Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol Cell Biol. 2008;28:6870–6876. doi: 10.1128/MCB.00392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–1713. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casenghi M, Barr FA, Nigg EA. Phosphorylation of Nlp by Plk1 negatively regulates its dynein-dynactin-dependent targeting to the centrosome. J Cell Sci. 2005;118:5101–5108. doi: 10.1242/jcs.02622. [DOI] [PubMed] [Google Scholar]
- 11.De Luca M, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle. 2006;5:296–303. doi: 10.4161/cc.5.3.2392. [DOI] [PubMed] [Google Scholar]
- 12.Roshak AK, Capper EA, Imburgia C, Fornwald J, Scott G, Marshall LA. The human polo-like kinase, PLK, regulates cdc2/cyclin B through phosphorylation and activation of the cdc25C phosphatase. Cell Signal. 2000;12:405–411. doi: 10.1016/s0898-6568(00)00080-2. [DOI] [PubMed] [Google Scholar]
- 13.van Vugt MA, Brás A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Liu XS, Yang X, Song B, Wang Y, Liu X. Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proc Natl Acad Sci U S A. 2010;107:14633–14638. doi: 10.1073/pnas.1006615107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol Cell. 2002;9:515–525. doi: 10.1016/s1097-2765(02)00473-2. [DOI] [PubMed] [Google Scholar]
- 16.Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, Seong YS, Yu H, Garfield S, Veenstra TD. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell. 2006;24:409–422. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Liu XS, Yang X, Wang Y, Wang Y, Turner JR, Liu X. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments. EMBO J. 2010;29:2953–2965. doi: 10.1038/emboj.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XS, Song B, Tang J, Liu W, Kuang S, Liu X. Plk1 phosphorylates Sgt1 at the kinetochores to promote timely kinetochore-microtubule attachment. Mol Cell Biol. 2012;32:4053–4067. doi: 10.1128/MCB.00516-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golan A, Yudkovsky Y, Hershko A. The cyclin-ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1-cyclin B and Plk. J Biol Chem. 2002;277:15552–15557. doi: 10.1074/jbc.M111476200. [DOI] [PubMed] [Google Scholar]
- 20.Hansen DV, Loktev AV, Ban KH, Jackson PK. Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP-dependent destruction of the APC inhibitor Emi1. Mol Biol Cell. 2004;15:5623–5634. doi: 10.1091/mbc.E04-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neef R, Gruneberg U, Kopajtich R, Li X, Nigg EA, Sillje H, Barr FA. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat Cell Biol. 2007;9:436–444. doi: 10.1038/ncb1557. [DOI] [PubMed] [Google Scholar]
- 22.Wolfe BA, Takaki T, Petronczki M, Glotzer M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 2009;7:e1000110. doi: 10.1371/journal.pbio.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 24.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 25.Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol. 2004;164:233–241. doi: 10.1083/jcb.200309035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cizmecioglu O, Warnke S, Arnold M, Duensing S, Hoffmann I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell Cycle. 2008;7:3548–3555. doi: 10.4161/cc.7.22.7071. [DOI] [PubMed] [Google Scholar]
- 27.Burns TF, Fei P, Scata KA, Dicker DT, El-Deiry WS. Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol. 2003;23:5556–5571. doi: 10.1128/MCB.23.16.5556-5571.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma S, Charron J, Erikson RL. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol Cell Biol. 2003;23:6936–6943. doi: 10.1128/MCB.23.19.6936-6943.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, Jiang N, Alberts GF, Costa M, Lu L. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008;68:4077–4085. doi: 10.1158/0008-5472.CAN-07-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmerman WC, Erikson RL. Polo-like kinase 3 is required for entry into S phase. Proc Natl Acad Sci U S A. 2007;104:1847–1852. doi: 10.1073/pnas.0610856104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang N, Wang X, Jhanwar-Uniyal M, Darzynkiewicz Z, Dai W. Polo box domain of Plk3 functions as a centrosome localization signal, overexpression of which causes mitotic arrest, cytokinesis defects, and apoptosis. J Biol Chem. 2006;281:10577–10582. doi: 10.1074/jbc.M513156200. [DOI] [PubMed] [Google Scholar]
- 32.Hudson JW, Kozarova A, Cheung P, Macmillan JC, Swallow CJ, Cross JC, Dennis JW. Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr Biol. 2001;11:441–446. doi: 10.1016/s0960-9822(01)00117-8. [DOI] [PubMed] [Google Scholar]
- 33.Fode C, Binkert C, Dennis JW. Constitutive expression of murine Sak-a suppresses cell growth and induces multinucleation. Mol Cell Biol. 1996;16:4665–4672. doi: 10.1128/mcb.16.9.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrysik Z, Bernstein WZ, Deng L, Myer DL, Li YQ, Tischfield JA, Stambrook PJ, Bahassi EM. The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res. 2010;38:2931–2943. doi: 10.1093/nar/gkq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cholewa BD, Liu X, Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res. 2013;73:6848–6855. doi: 10.1158/0008-5472.CAN-13-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Syed N, Smith P, Sullivan A, Spender LC, Dyer M, Karran L, O'Nions J, Allday M, Hoffmann I, Crawford D. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood. 2006;107:250–256. doi: 10.1182/blood-2005-03-1194. [DOI] [PubMed] [Google Scholar]
- 37.Coley HM, Hatzimichael E, Blagden S, McNeish I, Thompson A, Crook T, Syed N. Polo like kinase 2 tumour suppressor and cancer biomarker: new perspectives on drug sensitivity/resistance in ovarian cancer. Oncotarget. 2012;3:78–83. doi: 10.18632/oncotarget.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B, Ouyang B, Pan H, Reissmann PT, Slamon DJ, Arceci R, Lu L, Dai W. Prk, a cytokine-inducible human protein serine/threonine kinase whose expression appears to be down-regulated in lung carcinomas. J Biol Chem. 1996;271:19402–19408. doi: 10.1074/jbc.271.32.19402. [DOI] [PubMed] [Google Scholar]
- 40.Dai W, Li Y, Ouyang B, Pan H, Reissmann P, Li J, Wiest J, Stambrook P, Gluckman JL, Noffsinger A. PRK, a cell cycle gene localized to 8p21, is downregulated in head and neck cancer. Genes Chromosomes Cancer. 2000;27:332–336. doi: 10.1002/(sici)1098-2264(200003)27:3<332::aid-gcc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 41.Macmillan JC, Hudson JW, Bull S, Dennis JW, Swallow CJ. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann Surg Oncol. 2001;8:729–740. doi: 10.1007/s10434-001-0729-6. [DOI] [PubMed] [Google Scholar]
- 42.Mason J, Wei S, Luo X, Nadeem V, Kiarash R, Huang P, Awrey D, Leung G, Beletskaya I, Feher M. Inhibition of Polo-like kinase 4 as an anti-cancer strategy. Cancer Res. 2011;71 [Abstract LB-215] [Google Scholar]
- 43.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr., Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Li H, Zhou Z, Wang WH, Deng A, Andrisani O, Liu X. Plk1-mediated phosphorylation of Topors regulates p53 stability. J Biol Chem. 2009;284:18588–18592. doi: 10.1074/jbc.C109.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu XS, Li H, Song B, Liu X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010;11:626–632. doi: 10.1038/embor.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A. 2003;100:5789–5794. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 2005;65:2698–2704. doi: 10.1158/0008-5472.CAN-04-2131. [DOI] [PubMed] [Google Scholar]
- 50.Ward A, Hudson JW. p53-Dependent and cell specific epigenetic regulation of the polo-like kinases under oxidative stress. PLoS One. 2014;9:e87918. doi: 10.1371/journal.pone.0087918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie S, Wu H, Wang Q, Cogswell JP, Husain I, Conn C, Stambrook P, Jhanwar-Uniyal M, Dai W. Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J Biol Chem. 2001;276:43305–43312. doi: 10.1074/jbc.M106050200. [DOI] [PubMed] [Google Scholar]
- 52.Spankuch B, Matthess Y, Knecht R, Zimmer B, Kaufmann M, Strebhardt K. Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J Natl Cancer Inst. 2004;96:862–872. doi: 10.1093/jnci/djh146. [DOI] [PubMed] [Google Scholar]
- 53.Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, Ahmad N, Liu X. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286:35795–35800. doi: 10.1074/jbc.C111.269050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Choy E, Harmon D, Yang S, Yang C, Mankin H, Hornicek FJ, Duan Z. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anticancer Drugs. 2011;22:444–453. doi: 10.1097/CAD.0b013e32834513f4. [DOI] [PubMed] [Google Scholar]
- 55.Ko MA, Rosario CO, Hudson JW, Kulkarni S, Pollett A, Dennis JW, Swallow CJ. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat Genet. 2005;37:883–888. doi: 10.1038/ng1605. [DOI] [PubMed] [Google Scholar]
- 56.Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 57.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 58.Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG, Madden L, Hassler DF, Smith GK, Gontarek RR, Courtney MP. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009;69:6969–6977. doi: 10.1158/0008-5472.CAN-09-0945. [DOI] [PubMed] [Google Scholar]
- 59.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 60.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem. 2009;284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 62.Reindl W, Yuan J, Kramer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol. 2008;15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 63.Garuti L, Roberti M, Bottegoni G. Polo-like kinases inhibitors. Curr Med Chem. 2012;19:3937–3948. doi: 10.2174/092986712802002455. [DOI] [PubMed] [Google Scholar]
- 64.Reddy EP. Deciphering the novel mechanism of rigosertib. 2014. http://globenewswire.com/news-release/2014/01/17/603196/10064629/en/Onconova-Therapeutics-Inc-to-Host-Webcast-Regarding-Insights-Into-Rigosertib-Mechanism-of-Action-With-Scientific-Founder-Dr-E-Premkumar-Reddy.html Webcast.
- 65.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gjertsen BT, Schöffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11–19. doi: 10.1038/leu.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin C-C, Su W, Yen C-J, Hsu C-H, Su W-P, Yeh K-H, Lu Y-S, Cheng AL, Huang DC, Fritsch H. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous Polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer. 2014;110:2434–2440. doi: 10.1038/bjc.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schöffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, Taton M, Fritsch H, Glomb P, Munzert G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer. 2012;48:179–186. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 69.Döhner H, Bug G, Müller-Tidow C, Krämer A, Lübbert M, Krug U, Schlenk RF, Voss F, Taube T, Liu D. Phase I/II study of volasertib, an intravenous Polo-like kinase inhibitor (Plk), in patients with relapsed/refractory acute myeloid leukemia (AML): updated phase I results for volasertib monotherapy. Haematologica. 2014;99 [Abstract S649] [Google Scholar]
- 70.Jayanthan A, Cooper T, Dunn SE, Lewis VA, Narendran A. Cooperative lethality of Polo-like kinases (PLK) and Aurora kinases (AK) in refractory pediatric leukemia. Blood. 2012;120 [Abstract 3573] [Google Scholar]
- 71.Döhner H, Lübbert M, Fiedler W, Fouillard L, Haaland A, Brandwein JM, Lepretre S, Reman O, Turlure P, Ottmann OG. Randomized, phase 2 trial comparing low-dose cytarabine with or without volasertib in AML patients not suitable for intensive induction therapy. Blood. 2014;124:1426–1433. doi: 10.1182/blood-2014-03-560557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stadler WM, Vaughn DJ, Sonpavde G, Vogelzang NJ, Tagawa ST, Petrylak DP, Rosen P, Lin CC, Mahoney J, Modi S. An open-label, single-arm, phase 2 trial of the polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer. 2014;120:976–982. doi: 10.1002/cncr.28519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pujade-Lauraine E, Weber BE, Ray-Coquard I, Vergote I, Selle F, Del Campo JM, Sufliarsky J, Tschope I, Garin Chesa P, Nazabadioko S. Phase II trial of volasertib (BI 6727) versus chemotherapy (CT) in platinum-resistant/refractory ovarian cancer (OC) J Clin Oncol. 2013;31 doi: 10.1200/JCO.2015.62.1474. [Abstract 5504] [DOI] [PubMed] [Google Scholar]
- 74.Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther. 2010;9:2079–2089. doi: 10.1158/1535-7163.MCT-10-0095. [DOI] [PubMed] [Google Scholar]
- 75.Tandle AT, Kramp T, Kil WJ, Halthore A, Gehlhaus K, Shankavaram U, Tofilon PJ, Caplen NJ, Camphausen K. Inhibition of polo-like kinase 1 in glioblastoma multiforme induces mitotic catastrophe and enhances radiosensitisation. Eur J Cancer. 2013;49:3020–3028. doi: 10.1016/j.ejca.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res. 2011;17:3420–3430. doi: 10.1158/1078-0432.CCR-10-2946. [DOI] [PubMed] [Google Scholar]
- 77.Laufer R, Forrest B, Li SW, Liu Y, Sampson P, Edwards L, Lang Y, Awrey DE, Mao G, Plotnikova O. The discovery of PLK4 inhibitors: (E)-3-((1H-Indazol-6-yl)methylene)indolin-2-ones as novel antiproliferative agents. J Med Chem. 2013;56:6069–6087. doi: 10.1021/jm400380m. [DOI] [PubMed] [Google Scholar]
- 78.Northfelt DW, Hamburg SI, Borad MJ, Seetharam M, Curtis KK, Lee P, Crowell B, Vocila L, Fredlund P, Gilbert MJ. A phase I dose-escalation study of TKM-080301, a RNAi therapeutic directed against polo-like kinase 1 (PLK1), in patients with advanced solid tumors: Expansion cohort evaluation of biopsy samples for evidence of pharmacodynamic effects of PLK1 inhibition. J Clin Oncol. 2013;31 [Abstract TPS2621] [Google Scholar]
- 79.Semple SC, Judge AD, Robbins M, Klimuk S, Eisenhardt M, Crosley E, Leung A, Kwok R, Ambegia E, McClintock K. Preclinical characterization of TKM-080301, a lipid nanoparticle formulation of a small interfering RNA directed against polo-like kinase 1. Cancer Res. 2011;71:2829. [Google Scholar]
- 80.Cheng KY, Lowe ED, Sinclair J, Nigg EA, Johnson LN. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J. 2003;22:5757–5768. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao C, Park JE, Bang JK, Nicklaus MC, Lee KS. Exploring potential binding modes of small drug-like molecules to the polo-box domain of human polo-like kinase 1. ACS Med Chem Lett. 2010;1:110–114. doi: 10.1021/ml100020e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan J, Sanhaji M, Kramer A, Reindl W, Hofmann M, Kreis NN, Zimmer B, Berg T, Strebhardt K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol. 2011;179:2091–2099. doi: 10.1016/j.ajpath.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yim H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs. 2013;24:999–1006. doi: 10.1097/CAD.0000000000000007. [DOI] [PubMed] [Google Scholar]