

Abstract
Endoplasmic reticulum (ER) stress activates the unfolded protein response and its dysfunction is linked to multiple diseases. The stress transducer IRE1α is a transmembrane kinase endoribonuclease (RNase) that cleaves mRNA substrates to re-establish ER homeostasis. Aromatic ring systems containing hydroxy-aldehyde moieties, termed hydroxy aryl aldehydes (HAA), selectively inhibit IRE1α RNase and thus represent a novel chemical series for therapeutic development. We solved crystal structures of murine IRE1α in complex with three HAA inhibitors. HAA inhibitors engage a shallow pocket at the RNase active site through pi-stacking interactions with His910 and Phe889, an essential Schiff base with Lys907 and a H-bond with Tyr892. Structure activity studies and mutational analysis of contact residues define the optimal chemical space of inhibitors and validate the inhibitor binding site. These studies lay the foundation for understanding both the biochemical and cellular functions of IRE1α using small molecule inhibitors and suggest new avenues for inhibitor design.
Introduction
Endoplasmic reticulum (ER) stress and the resulting unfolded protein response (UPR) have gained significant interest as an area in which targeted molecules may have extensive therapeutic overlap in divergent disease types including neurodegeneration, inflammation, metabolic syndromes and cancer 1,2. The ER is a large intracellular compartment responsible for the production of secreted and cell surface proteins as well as cellular lipid biosynthesis and membrane generation. The ER is physically connected to both the nucleus and mitochondria and is fundamentally integrated with global cellular regulatory networks and metabolism. Protein folding, membrane and nutrient perturbations within the ER, arising from exogenous or internal stresses, induce the UPR 3. This cellular signaling response serves to alleviate the imposing stress and correct the imbalance to restore normal cellular homeostasis. However, accumulating evidences suggests the UPR, when chronically activated due to a myriad of stimuli, may be pathological resulting in inflammation, cell death or conversely may be exploited by cancer cells to ensure survival 1,3–5.
The three major arms of the fully integrated UPR include PERK (pancreatic eIF2alpha kinase), ATF6 (activating transcription factor 6) and IRE1 (inositol requiring enzyme 1), which function as a finely tuned triad. All three arms have overlapping yet distinct responsibilities and regulate a plethora of downstream genes 3. PERK, an ER resident transmembrane eIF-2α kinase, halts translation 6 to induce transcriptional programs via non-conventional translation of ATF4, which results in both pro-life and pro-death outcomes 7. During ER stress, ATF6 is translocated to the Golgi where it undergoes proteolysis at the membrane releasing it to the nucleus 8 where it transactivates genes required for the UPR 9
IRE1, the most conserved arm of the UPR, is a unique ER transmembrane dual fused kinase-endoribonuclease 10 that directly regulates HAC1 in yeast or its metazoan orthologue XBP1 3,4. Fungi possess a single isoform of IRE1 while higher eukaryotes generally possess two isoforms denoted IRE1α and β. The endoribonuclease (RNase) domain of IRE1, which appears unique to eukaryotes 11–15 is found in only one other paralogue, RNase L 16, an enzyme involved in innate immunity in vertebrates 17. UPR signaling initiates from the IRE1 luminal domain, which senses the accumulation of unfolded protein, proceeds by dimerization and oligomerization to activate the cytoplasmic auto-kinase activity 18,19 and in turn RNase activity 10,20. In metazoan organisms, selective cleavage of dual stem-loops within the XBP1 mRNA ensues and a 26-nucleotide intron is removed 21–23. The two exon ends are then ligated by an unknown process 24 allowing the spliced XBP1 (XBP1s) transcript to be translated into a functional transcription factor. XBP1s directs the transcription of chaperones, ERAD (Endoplasmic Reticulum-Associated protein Degradation) components and other targets involved in ER expansion, phospholipid synthesis and general homeostasis 25. Combined, the downstream activities of the XBP1s transcriptional network can enforce specialized cellular functions such as antibody secretion in plasma cells 26. In addition to XBP1 mRNA, IRE1 also cleaves a select set of ER targeted mRNAs 27. This process, termed RIDD (regulated IRE1 dependent decay) 28, attempts to rapidly decrease the secretory load during ER stress; however, little is known about its physiological significance 28,29. Recent evidence suggests IRE1α can be activated in response to specific signaling events independent of the UPR which have cell type or organ specific functions and can be dependent 30 or independent of XBP1 31,32. The kinetics of IRE1α signaling usually follows a rapid on-off response where activation is followed by rapid deactivation 33, which can have longer-term consequence 34. This is achieved by yeast IRE1 through dephosphorylation 35 or hyperphosphorylation 36 of the kinase.
Small molecule modulators of IRE1 kinase and RNase functions have been reported with distinct mechanisms of action reflecting the engagement of three physically distinct binding sites. Kinase domain active site binders inhibit kinase output by virtue of ATP competition, and paradoxically can either potentiate 37,38 or inhibit 39 RNase output depending on the underlying chemotype. The structural basis for their differential effect on RNase function has not been resolved since X-ray crystal structures of RNase inhibitors bound to the IRE1 kinase domain specifically are lacking. Quercetin, representative of a second class of modulators, marginally influences the kinase output of yeast IRE1 while potently enhancing RNase output by promoting molecular dimerization through engagement of a composite binding pocket at the enzyme dimer interface 40. Salicylaldehyde derivatives represent a third class of modulators that potently, reversibly, and selectively inhibit IRE1 RNase activity 41, with weak effects on protein kinase function. Additional compounds using the adjacent hydroxy aldehyde motif and dual-ring biphenyl (WO 2008/154484 A1), naphthylene (WO 2008/154484 A1; WO 2011/056744 A1) and coumarin (WO 2011/127070 A2) aromatic systems, which we collectively refer to as hydroxy-aryl-aldehydes (HAA), were also found to be potent IRE1 RNase inhibitors 42–44. Notably, a combination of methods including chemical modification of the protein/HAA inhibitor complex demonstrated that the aldehyde forms a reversible Schiff base with lysine 907 in the active site region of the RNase domain 43. These findings were surprising and offered hope for the selective and potent targeting of IRE1 where RNase active sites in general have made poor drug targets as they possess hydrophobic pockets that are shallow or non-existent due to a requirement for extensive interactions with the RNA phosphodiester backbone.
Here we uncover the structural basis for HAA inhibitor mechanism of action through a detailed X-ray crystallographic study of three members, representative of two different chemical scaffolds, in complex with murine IRE1α (referred here generally as IRE1α). These structures and follow on analyses allow for a rationalization of structure-activity relationships and suggest avenues for future optimization.
Results
Co-crystallization and characterization of HAA inhibitors
To discern the mechanism of action of the HAA class of IRE1 specific endoribonuclease inhibitors, we pursued X-ray crystallographic analyses of HAA members in complex with the dual catalytic region of IRE1. In an extensive screening campaign sampling combinations of HAA inhibitors with nucleotides, protein kinase inhibitors, and IRE1α orthologues (yeast, zebrafish, human, and mouse) with different N-terminal domain boundaries and phosphorylation states, we obtained suitable co-crystals for three HAA inhibitors, namely MKC9989, OICR464 and OICR573 (see Fig. 1a for chemical structures and substituent numbering schemes). Each was obtained in complex with Mg2+/ADP and an auto-phosphorylated minimal dual catalytic domain fragment of murine IRE1α encompassing residues 549 to 977. The crystals diffracted to 2.9 Å, 3.0 Å, and 3.4 Å resolution, respectively and the co-structures were solved by molecular replacement using the human IRE1α crystal structure as a search model (see Methods for experimental details and Table 1 for data collection and refinement statistics).
Figure 1. Functional characterization of HAA inhibitors solved in complex with IRE1α.

(a) Chemical structure and substituent numbering scheme of HAA inhibitors.(b) Inhibition of IRE1 RNase activity by HAA inhibitors using a real time fluorescence readout assay. IC50s derived from fitted profiles represent the mean +/− s.d., n=6 (three experiments performed in duplicate) for murine IRE1α and yeast IRE1 and n=4 (two experiments performed in duplicate) for human IRE1α. Shown are representative profiles for one experiment performed in duplicate. (c) Inhibition of IRE1α auto-kinase activity by HAA inhibitors.
Table 1.
Data collection and refinement statistics for the IRE1α-MKC9989, IRE1α-OICR573, and IRE1α-OICR464 crystal complexes. Data was collected from a single crystal. Highest resolution shell is shown in parenthesis.
| IRE1 α MKC9989 | IRE1 α OICR573 | IRE1 α OICR464 | |
|---|---|---|---|
| Data collection | |||
| Space group | C 1 2 1 | C 1 2 1 | C 1 2 1 |
| Cell dimensions | |||
| a, b, c (Å) | 246.08, 90.5, 72.13 | 319.24, 63.03, 140.78 | 319.11, 62.31, 141.33 |
| α, β, γ (°) | 90.00, 91.88, 90.00 | 90.00, 99.49, 90.00 | 90.00, 99.57, 90.00 |
| Resolution (Å) | 72.09 – 2.90 (2.98 – 2.90) * | 48.27 – 3.40 (3.60 – 3.40) | 46.60 – 3.00 (3.18 – 3.00) |
| Rmeas (%) | 15.3 (99.8) | 17.2 (93.9) | 7.8 (91.2) |
| Rmerge (%) | 35.6 (172.7) | 30.9 (121.9) | 22.3 (158.0) |
| I/σI | 7.14 (1.17) | 8.69 (1.74) | 11.08 (1.33) |
| Completeness (%) | 96.2 (99.7) | 94.5 (97.3) | 96.2 (96.6) |
| Redundancy | 3.69 (3.34) | 3.71 (3.71) | 2.25 (2.24) |
| Refinement | |||
| Resolution (Å) | 72.09 – 2.90 | 48.27 – 3.40 | 46.60 – 3.00 |
| No. reflections | 21855 | 29201 | 40131 |
| Rwork/Rfree | 0.2015/0.2324 | 0.2219/0.2852 | 0.2100/0.2759 |
| No. atoms | |||
| Protein | 6220 | 12 448 | 12 316 |
| Ligand/ion | 102 | 165 | 152 |
| Water | 0 | 0 | 0 |
| B factors | |||
| Protein | 60.0 | 87.9 | 90.0 |
| Inhibitor | 64.6 | 115.9 | 107.7 |
| Other ligands | 48.5 | 71.5 | 69.3 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.018 | 0.006 | 0.012 |
| Bond angles (°) | 1.37 | 1.18 | 1.32 |
MKC9989, OICR464 and OICR573 inhibitors visualized in IRE1α co-structures potently inhibited the RNase activity of murine and human IRE1α, and to a lesser degree yeast IRE1 in the case of MKC9989 in vitro with IC50s ranging from 0.23 μM to 44 μM (Fig. 1b, upper panels). In contrast, the same inhibitors had no effect or weakly inhibited the auto-kinase activity of IRE1α at the highest concentrations tested (IC50s >> 10 μM) (Fig. 1c). In the case of OICR464 and OICR573 (and to a lesser extent MKC9989), inhibitor potency showed a significant dependency on pre-incubation time with IRE1α (Supplementary Fig. 1). This may reflect differences in the reactivity of the aldehyde group in each HAA scaffold required to support formation of a Schiff base interaction with Lys907. At 10 μM concentration, MKC9989 completely inhibited both basal and thapsigargin induced splicing of XBP1 mRNA (Fig. 2a). These effects were observed even in cells pre-treated with thapsigargin, indicating that MKC9989 can fully reverse the onset of XPB1 splicing after the UPR is initiated. In parallel analyses, MKC9989, markedly stabilized the RIDD target CD59 mRNA 45 when co-administered with thapsigargin relative to thapsigargin treatment alone and modestly increased levels of CD59 mRNA in non-stressed cells, the latter likely reflecting the inhibition of baseline RIDD activity. In contrast to effects on XBP1 splicing, MKC9989 moderately stabilized CD59 levels when administered 2 hour post treatment with thapsigargin. Lastly, the potency of MKC9989 against the splicing of XBP1 mRNA (EC50=0.33 μM) was comparable to its potency against RNA cleavage in vitro (Fig. 1b). These functional properties in total suggested that the MKC9989, OICR464 and OICR573 X-ray co-structures could help illuminate the mechanism of action of the HAA class of inhibitors in vitro and in cells.
Figure 2. Inhibition of IRE1α function by MKC9989 in human RPMI 8226 plasmacytoma cells.

(a) Time course analysis of MKC9989 inhibitory function on XBP1 splicing: RT-PCR PAGE analysis (top) and quantification (middle) of XBP1 spliced products from 3 independent experiments shown as +/− s.e.m. Quantification of CD59 transcript levels from the same RNA samples (bottom). (b) Dose response analysis of MKC9989 treatment on XBP1 splicing in human RPMI 8226 plasmacytoma cells. Shown is a representative profile with EC50 calculated as the average of 2 independent experiments.
Structure overview
In all three HAA co-structures, IRE1α crystalized in the same face-to-face dimer configuration adopted by human IRE1α 46, and not the back-to-back dimer or oligomer configuration adopted by yeast IRE1 11,37 (Fig. 3a). This conservation lends further support to the notion that the face-to-face dimer configuration is a functionally relevant conformation for IRE1α and raises the question why the human and mouse IRE1 orthologues did not adopt the back-to-back dimer configuration reflective of the ‘RNase active’ state. Each protomer of IRE1α consisted of a bilobal protein kinase domain rigidly fused through the kinase C-lobe to an α-helical RNase domain. All IRE1α protomers in each co-structure displayed unambiguous density for Mg2+/ADP in the catalytic cleft of the kinase domain (Supplementary Fig. 2a). Minimally one IRE1α protomer per co-structure displayed a well-ordered HAA inhibitor engaging the active site of the RNase domain (Supplementary Fig. 2a,b). The remaining RNase active sites were occluded by crystal packing or weak electron density precluded accurate modeling of the bound inhibitor. IRE1α protomers were well ordered with the exception of approximately 13 residues at both N and C-termini and also within the kinase domain activation segment (residues 719 to 730). IRE1α protomers were highly similar to each other (RMSD = 0.24 Å2 over 10 total molecules in the asymmetric unit of the three HAA co-structures), to human IRE1α protomers (PDB 3P23, RMSD= 1.28 Å2) and to yeast IRE1 protomers (PDB 3LJ0, RMSD = 1.48 Å2).
Figure 3. Crystal structure of IRE1α bound to MKC9989.

(a) Ribbons representation of a face-to-face dimer configuration of IRE1α in complex with Mg2+/ADP and MKC9989. ADP engaging the kinase active site and MKC9989 engaging the RNase active site are shown as sticks and highlighted by red boxes. For comparison, the dimer configurations adopted by human IRE1α (PDB 3P23) and yeast IRE1 (PDB 3LJ0) are shown at right. (b) Ribbons stereo representation of MKC9989 engaging the RNase active site of IRE1α. Invariant active site residues, Lys907, and other conserved residues in the binding pocket are coloured purple, yellow and pink respectively. Lys907 forms a Schiff base to the aldehyde moiety of MKC9989. (c) Zoom in surface view of the IRE1α-MKC9989 complex coloured as in (b). (d) Schematic of contact residues and notable interactions mediating MKC9989 binding to IRE1α.
Binding mode of HAA inhibitors to the RNase domain of IRE1
The structure of the IRE1α RNase domain consisted of 9 α-helices (denoted helices α1 to α8, and α3′). All three HAA inhibitors shared similar binding modes to a shallow pocket nestled at the nexus of helices α3′, α4, and α5, and lined by the side chains of Glu913, Leu914, Pro915, Phe889, Leu886, Lys907, Arg902, and the invariant or highly conserved catalytic residues Tyr892, Arg905, Asn906, His910 and Arg902 (Fig. 3b, Supplementary Figs. 3a, b). Using the human IRE1α structure (PDB = 3P23) as a reference for the apo state of the RNase domain, comparison to IRE1α in complex with HAA inhibitors revealed that the HAA binding pocket was largely preformed (Supplementary Fig. 3c,d). Small induced conformational changes consisted of: 1) enforcement of a single rotomer conformer for Asn906 and His910 that enlarged the binding pocket to accommodate the HAA ring structure, and 2) a clamping movement of the Phe889 side chain (5.1Å, Cα to Cα movement) onto the apical surface of the engaged HAA inhibitor.
Owing to overlap with the RNase active site (three of four invariant active site residues are exploited in the HAA binding interaction), the HAA binding pocket is highly conserved (Supplementary Fig. 3d) with only a conservative substitution of tyrosine for phenylalanine at position 889 differentiating the binding pocket between fungal and metazoan enzymes (Supplementary Fig. 4). This may explain the general cross reactivity of HAA inhibitors across IRE1α orthologues tested (yeast, mouse, and human) 41.
Conserved interactions across the three co-structures that are predicted to contribute to affinity and specificity of the HAA inhibitor class include: a Schiff base interaction between the HAA aldehyde group with the amine group of Lys907; a hydrogen bond interaction between the HAA hydroxy group with the side chain of Tyr892; Pi stacking of the Phe889 side chain with the apical surface of the HAA aromatic ring directly harboring the hydroxy and aldehyde groups; and lastly, Pi stacking of the His910 side chain with the basal surface of flanking HAA aromatic rings. All three co-crystallized HAA inhibitors possessed methoxy moieties at a position immediately adjacent to the core hydroxy moiety, and these possess the capacity to hydrogen bond with the side chain of Asn906. Since the methoxy moiety is not a conserved feature of all HAA inhibitors 41–43, this interaction is not a general feature of the HAA inhibitor-binding mode.
Reflecting differences in chemical scaffold and substituent structure, each HAA inhibitor also made a limited number of distinct interactions with the binding pocket on IRE1α that may account for differences in potency. These unique interactions were accommodated in part by plasticity of the binding mode evident by a subtle (~1.5 Å) lateral shift of the single ring core of OICR464 and OICR573 relative to that of the planar fused ring core of MKC9989 (Supplementary Fig. 3d). Unique interactions exploited by MKC9989 include: hydrogen bonds between Lys907 and the oxygen heterocycle of the coumarin ring and between Tyr892 and the oxygen atom of the ether moiety at position 3; Van der Waals contacts involving the main chain Glu913 and the side chains of Pro915 and Leu914 with the coumarin C=O group; and lastly a hydrophobic interaction between the Phe889 side chain and the flexible poly ether moiety at position 7 (Fig. 3b,d). These inhibitor specific interactions may account for enhanced potency of MKC9989 relative to OICR464 and OICR573.
The aldehyde moiety of HAA inhibitors has been shown to selectively form a reversible Schiff base interaction with Lys907 in the RNase domain and to a lesser degree to Lys599 in the ATP binding site of the kinase domain, versus 20 other surface exposed lysine residues within the dual catalytic region of human IRE1α 43. This reactivity can be rationalized by the position of the Lys907 side chain at the back of a sheltered binding pocket. Due to the clamping action of the Phe889, His910, and Asn906 side chains on either side of the HAA planar ring structure, the Schiff base interaction once formed would be shielded from attack by water. Furthermore, reactivity of the aldehyde towards Lys907 may be enhanced by the electron polarizing H-bond between Tyr892 and the adjacent hydroxy moiety. These features are uniquely supported by the environment around Lys907. The fact that Lys599 displays some reactivity to HAAs may be explained by the sheltered environment of the kinase active site, which would also shield a Schiff base interaction from attack by water once formed. Using kinase inhibition as a proxy for kinase domain engagement, the interaction of MKC9989 and OICR464 with the kinase active site Lys599 is approximately 10 to 100 fold weaker than engagement with Lys907 whereas OICR573 does not appear to engage Lys599 to a detectable degree (Fig. 1c).
Basis for non-competitive mechanism of action
To discern how HAA inhibitors act non-competitively with respect to RNA substrate 41,43 while exploiting direct contacts to three of four invariant RNase active site residues, we modeled a di-nucleotide substrate into the active site region of IRE1α bound to MKC9989. As tRNA endonuclease, whose X-ray crystal structure was determined in complex with RNA 47, shares a highly similar active site architecture and inferred catalytic mechanism with IRE1 11, it provided an instructive template for modeling the IRE1α-RNA substrate-HAA inhibitor complex (see methods for modeling details). As modeled in Supplementary Fig. 3e, MKC9989 binds immediately opposite to the RNA along an axis linking Asn906, His910 and Arg902 side chains, without overt clashes. His910 has been proposed to act as a general acid that protonates the 5′-OH leaving group, where as Arg905 and Asn906, the latter supported by a H-bonding interaction with Tyr892, likely serve to position the scissile phosphate for nucleophilic attack. We reason that subtle changes to the position of three (His910, Asn906 and Tyr892) of these four conserved catalytic residues imposed by HAA inhibitor binding (Supplementary Fig. 3c) may be responsible in part for the resultant loss in RNase catalytic function. A more detailed understanding of HAA inhibitor mechanism of action awaits the determination of comparative structures of IRE1α productively bound to RNA and non-productively bound to both RNA and HAA inhibitor.
Mutational analysis of the HAA binding pocket
To explore HAA binding determinants on the RNase domain of IRE1α, we mutated the contact residues Phe889, Tyr892, Asn906, His910, and Lys907 in the context of a longer dual catalytic fragment of IRE1α encompassing residues 499 to 977 (Fig. 4a). Consistent with their immediate proximity to the RNase active site, all five mutations abolished RNase activity against a single hairpin substrate (Fig. 4b) while leaving protein kinase activity largely intact (Fig. 4c). This limited our ability to infer the effect of mutations on inhibitor binding by the analysis of enzymatic inhibition constants (Ki). However, using 600-fold higher concentrations of a double hairpin substrate produced in T7 RNA runoff reactions and 10,000 fold higher concentrations of IRE1α, the Lys907Ala and Phe889Ala but not His910Ala, Tyr892Ala and Asn906Leu mutants displayed detectable RNase activity. In contrast to the WT enzyme, the Lys907Ala but not the Phe889Ala mutant was insensitive to MKC9989 when assayed under these conditions (Fig. 4d).
Figure 4. Structure activity relationship of HAA inhibitors and their binding site on IRE1α.

(a) Schematic (left) and SDS PAGE analysis (right) of IRE1α RNase active site mutants. (b) Effect of RNase active site mutations on IRE1α RNase function as assessed using a real time fluorescence readout assay. (c) Effect of RNase active site mutations on IRE1α kinase function as assessed by auto-kinase (phosphor image top, quantification bottom). (d) Sensitivity of IRE1α WT and RNase active site mutants to inhibition of RNase activity by MKC9989 as assessed using a dual-hairpin RNA substrate.
Direct binding measurements using micro-scale thermophoresis revealed a close correlation between IRE1α binding activity and inhibitor potency, with MKC9989 binding more tightly than OICR464 and OICR573 (Supplementary Fig. 5a, Kd = 0.84 μM,15.9 μM and 35.2 μM, respectively). Notably, the IRE1α mutations Tyr892Ala (Kd = 10 μM), Asn906Leu (Kd = 8.9 μM), His910Ala (Kd = 5.3 μM) and Phe889Ala (Kd = 10.9 μM) perturbed binding to MKC9989 minimally by 10 fold while the IRE1α Lys907Ala mutation abolished detectable binding (Supplementary Fig. 5b). Together, these results demonstrated the importance of protein interactions visualized in the IRE1α-HAA inhibitor co-structures and highlighted the pre-eminent role for the Schiff base interaction with Lys907 in the HAA inhibitor mechanism of action.
Structure activity relationships
In order to unify our understanding of structure activity relationship data as well as validate the binding mode observed in the HAA co-structures, we assembled an HAA analogue series around a naphthalene scaffold 42 (WO 2008/154484 A1; WO 2011/056744 A1) (Fig. 5). This series offered the advantage of more sites around the dual fused ring core for addition of substituents while the chemical space largely overlaps with the biphenyl and coumarin HAA inhibitors. In keeping with chemical convention, we used a substituent numbering scheme with position 1 corresponding to the aldehyde moiety and position 2 proceeding along the direction of the adjacent hydroxy moiety (MKC3437). Consistent with previous findings for a single aryl ring scaffold 41, substitution of the aldehyde or hydroxy moieties at positions 1 and 2 were not tolerated (MKC3964, MKC3820). This likely reflected the importance of the Schiff base interaction with Lys907 and the hydrogen bond interaction with Tyr892, respectively. In accordance with position 3 and 7 facing towards solvent, addition of bulky substituents (MKC3426 and MKC3946) were well tolerated. Substitutions at position 8 were more restrictive owing to the partially buried nature of the interaction site with IRE1α. Addition of a bromine substituent at this position (MKC3987) was favoured relative to hydrogen (IC50 = 0.39 μM versus 2.2μM, respectively) while a bulkier methyl-piperizine-carbonyl-phenyl group was disfavoured (MKC4027) (IC50 = 5.7μM). Like the C=O group of the coumarin scaffold of MKC9989, bromine at position 8 would be accommodated in the narrow confines of the binding site. In contrast, the methyl-piperizine-carbonyl-phenyl group is predicted to sterically clash in the absence of a major shift in inhibitor binding mode. In summary, these structure activity relationships are consistent with the inhibitor-binding mode elucidated by X-ray co-structure and molecular modeling analyses and suggest further avenues for future optimizations.
Figure 5. Structure activity relationship analysis of a naphthol analogue series on IRE1α RNase activity.

Shown are analogue structures (left) and RNase inhibition profiles (right). IC50 values represent the average for n=4 (two experiments performed in duplicate) +/− s.d. Displayed profiles represent one experiment performed in duplicate. Substituent numbering scheme and RNase inhibitory activity against yeast IRE1 are shown on top for the minimal naphthol scaffold MKC3437.
ADP as a model for dimer breaking IRE1α RNase inhibitors
Back-to-back dimerization and subsequent polymerization of the fused kinase-endoribonuclease catalytic module, is required for the RNase activity of yeast IRE1 11 and presumably all orthologues. The binding of nucleotide and small molecule protein kinase inhibitors to the kinase domain can influence dimer/oligomer formation and as a result RNase activity; ADP, JAK Inhibitor I, CDK inhibitor I, and APJ promoted the dimerization/oligomerization of yeast IRE1 and concomitantly its RNase activity 37,38,40, whereas Compound 3 (1-(4-(8-amino-3-isopropylimidazo[1,5-a]pyrazin-1-yl)naphthalen-1-yl)-3-(3-(trifluoromethyl)phenyl)urea) inhibited the dimerization/oligomerization of human IRE1α and concomitantly its RNase function 39. We posit that the structure of murine IRE1α reported here and human IRE1α reported previously, which do not adopt the back-to-back dimer configuration reflective of the ‘RNase active’ state, provides insight into how in general dimer-breaking agents might act.
Contrary to expectation based on its effect on yeast IRE1, we discovered that ADP is an inhibitor of both the RNase and dimer/oligomerization activities of murine IRE1α in vitro. As assessed by velocity analytical ultra centrifugation (AUC), in the absence of ADP Ire1 undergoes concentration dependent oligomerization (Fig. 6a). Addition of ADP (Fig. 6a) and JAK inhibitor I (Fig. 6a), antagonized and enhanced this behavior respectively (see Fig. 6b for chemical structures). Consistent with these effects on oligomerization, ADP inhibited (IC50= 117μM) and JAK inhibitor I activated (EC50= 0.19μM) murine IRE1α RNase function in vitro (Fig. 6c,d). JAK inhibitor I exerted its influence on yeast IRE1 by engaging the kinase active site 40, and consistent with a common site of action on murine IRE1α, JAK inhibitor I potently inhibited auto-kinase activity with an IC50 of 1.1 μM (Fig. 6e). Supporting the notion that murine IRE1α samples the same back-to-back dimer and higher order polymer configurations demonstrated for yeast IRE1, murine IRE1α oligomerization was similarly dependent on 1) the N-terminal region preceding the kinase domain (Fig. 6a), 2) the integrity of the back-to-back dimer interface as assessed by a Asp620Ala mutation (Fig. 6a) and 3), enzyme phosphorylation status (Fig. 6a). Confirming the predicted importance of the yeast IRE1 back-to-back dimer configuration for murine IRE1α RNase function, dimer interface mutations in murine as shown for yeast IRE1 11, also perturbed the RNase but not the kinase functions of murine IRE1α (Fig. 6f,g). These findings demonstrated that many of the regulatory features discerned for yeast IRE1 were general to murine IRE1α with the exception of the specific response to ADP. The findings further suggested that the crystal structure of murine IRE1α bound to ADP might provide a useful platform to discern the basis by which dimer breaking inhibitors of IRE1 RNase function act.
Figure 6. Effects of kinase domain active site binders on murine IRE1α function.

(a) Analysis of oligomerization status. Velocity analytical ultracentrifugation analyses were performed on murine IRE1α preparations (labeled on left) at 18μM protein concentration. See Supplementary Figure 6 for full protein concentration series. ADP and JAK inhibitor I concentrations were 500 μM and 30 μM respectively. SDS-PAGE analysis of murine IREα pre and post treatment with lambda phosphatase is shown in the right panel. (b) Chemical structures of ADP and JAK inhibitor I. (c). Effect of ADP on the RNase activity of murine IRE1α using a model single hairpin substrate labeled with fluorescein. Reaction products were separated via PAGE, detected using a GE Healthcare Typhoon Variable Mode Imager (top panel), and quantified using ImageQuant 5 software (bottom panel). Shown is a representative profile. IC50 +/− s.d., was calculated for n=3. (d) Dose response analysis of JAK inhibitor I on the RNase activity of murine IRE1α assayed as in (c). Shown is a representative profile. EC50 +/− s.d., was calculated for n=3. (e) Dose response analysis of JAK inhibitor I on the auto-kinase activity of murine IRE1α as assessed using P32-γ-ATP. Quantification plot presented in lower panel represents the 10 minute time point from auto-radiographs in the top panel. Shown is a representative profile. IC50 was calculated for n=2 (two experiments performed in singlicate). (f) Effect of the indicated back-to-back dimer interface mutations on murine IRE1α RNase function, as assayed in Fig. 1b. (g) Effect of the indicated back-to-back dimer interface mutations on murine IRE1α auto-kinase function (phosphor image top, quantification bottom), as assayed in Fig. 1c.
The kinase domain of murine IRE1α possessed a prototypical bilobal architecture consisting of an N-lobe rich in β-strands and a single regulatory helix αC and a C-lobe composed predominantly of α helices. In their active states, protein kinase domains can sample a closed conformation that aligns parallel regulatory and catalytic spines to productively bind and coordinate ATP within the inter-lobe cleft and to facilitate the phospho-transfer reaction 48. The murine IRE1α kinase domain is notable in adopting a ‘kinase-inactive’ conformation in which the catalytic spine is intact but the regulatory spine is disrupted (Fig. 7a, left panel). The catalytic spine, consisting of N-lobe residues Val586, Ala597, the adenine ring of ADP, and C-lobe residues Leu649, Ile694, Leu695, Leu696, Val763 and Val767, forms a continuous hydrophobic column that spans the vertical length of the kinase domain. The regulatory spine in contrast, consisting of the N-lobe residues Leu616 and Tyr628 and the C-lobe residues His686 and Phe712, forms a broken hydrophobic column partitioned midway by a mispositioned Leu616 side chain, which projects from a non-productive, laterally displaced helix αC. A non-productive orientation of helix αC also manifests in a loss of the ATP coordinating salt bridge between the kinase domain invariant residues Lys599 (subdomain II) and Glu612 (subdomain III). Specifically, a 9Å lateral shift of Glu612, prohibited the close (≅ 3.5 Å) interaction with Lys599 observed for the corresponding residues in yeast ‘kinase-active’ IRE1 structures (Fig. 7b). These two non-productive features of the murine IRE1α kinase domain are shared with the human IRE1α kinase domain structure also bound to ADP 46 and differ from all yeast IRE1 co-structures bound to dimer promoting small molecule effectors (Fig. 7a, right panel).
Figure 7. Structure analysis of the kinase domain of murine IRE1α.

(a) Ribbons representation of IRE1α kinase domains highlighting regulatory (yellow) and catalytic (red) spine alignments. All yeast structure solved to date display productive alignment of spines (right) while HAA bound murine IRE1α structures and the dephospho-human IRE1α structure adopt non-productive alignments of the regulatory spine. PDB code and the respective ligand engaging the kinase active site are indicated. (b) Zoom in view of the kinase active site of murine IRE1α superimposed on yeast IRE1 (PDB 2RIO) highlighting the displaced position of helix αC. (c) Zoom in view of the projected back-to-back dimer interface of murine IRE1α (only one protomer shown) superimposed on the yeast IRE1 back-to-back dimer (PDB 2RIO) highlighting the physical link between helix αC and the back-to-back dimer interface. Side chains composing an essential intra-dimer salt bridge and the subdomain III glutamate side chain are shown as sticks.
The non-productive orientation of helix αC is notable as helix αC forms an integral component of the back-to-back dimer interface of ‘RNase-active’ yeast IRE1 structures 11,37 and may provide a link between ligand binding to the kinase active site and back-to-back dimerization. IRE1 invariant residues Arg697, Asp723, and Arg730 in yeast IRE1 (corresponding to Arg594, Asp620, and Arg627 in murine IRE1α) form an essential three center salt bridge across the back-to-back dimer interface (Fig. 7c) that is required for the RNase function of both yeast 11 and murine (Fig. 6f) enzymes. In the murine IRE1α structure, the lateral shift in helix αC displaces Asp620 10.8 Å from the required position to interact with Arg594 and Arg627 (Fig. 7c). Since the three center intermolecular salt bridge is essential for the back-to-back dimerization and RNase function of murine IRE1α (Fig. 6a,f), we reason that the non-productive conformation of helix αC is responsible for the decreased propensity of murine IRE1α to adopt the back-to-back dimer configuration when bound to ADP.
Discussion
Our studies reveal how HAA inhibitors engage the RNase active site of IRE1α with high affinity and specificity by exploiting a shallow complementary pocket and an essential Schiff base interaction between the aldehyde moiety of the inhibitor and the side chain amino group of Lys907. The presented HAA co-structures allow for a rationalization of current structure activity relationships (SAR) and provide a platform for further rounds of optimization to improve inhibitor potency, bioavailability and selectivity.
Interestingly, despite the fact that the HAA contact residues in IRE1α are highly conserved, yeast IRE1 is relatively resistant (~100 fold) to the fused dual ring core of MKC9989, whereas it is equally sensitive to the single ring core of OICR464 and OIRC573. This trend holds true also for the fused dual ring core of the naphthalene scaffold (Fig. 5, first panel). As only a conservative substitution of Phe889 to tyrosine in yeast IRE1 differentiates the respective HAA binding pockets, we speculate that this behavior may be due to differences in binding pocket plasticity, which may be less accommodating of chemical scaffolds employing planar dual-ring cores in yeast IRE1.
As IRE1α and β human isoforms are 100% identical in the composition of HAA binding pocket residues (Supplementary Fig. 4), we anticipate that both isoforms will be sensitive to inhibition by HAA inhibitors, although our findings with yeast IRE1 hold out the possibility for isoform selectivity despite similarities in binding pocket composition. The observation that HAA inhibitors centrally engage the RNase active site by exploiting three of four invariant and essential catalytic residues for binding, would explain their general effectiveness against both XBP1 splicing and RIDD cleavage targets 41,43. The exploitation of essential catalytic residues for binding may also impose a major impediment to the acquisition of resistance mutations if employed in a clinical setting, since mutations shown here that abrogate HAA binding also compromise IRE1α RNase catalytic activity. Lastly, RNase L, the sole paralogue of IRE1α in mammals, notably lacks an appropriately positioned lysine residue for Schiff base formation (substituted for leucine in human RNaseL) and shares only three of eight other contact residues (equivalent to the invariant catalytic residues Tyr892, Asn906, His910 in human IRE1α). These differences likely account for the observed resistance of RNase L to HAA inhibitors 41,43.
Although the core scaffold and ring substituents of MKC9989 make no overt clash with RNA, substitutions at position 3 with far bulkier groups have great potential to hinder RNA binding through clashes with the scissile phosphate group. Such substitutions would be expected to transition an HAA analogue from a substrate non-competitive to a substrate competitive mechanism of action. We speculate that elaboration of HAA inhibitor structure to exploit the RNA substrate-binding infrastructure (for example by covalently linking an HAA to a dinucleotide mimetic), to impart a competitive mechanism of action may also provide a means to improve inhibitor potency further. By capturing favorable or disfavourable interactions with specific RNA substrates, it may also be possible to engineer HAA analogues with inhibitory properties that discriminate between the numerous XBP1 and RIDD substrates.
Small molecule binders to the kinase domain of IRE1 that support a non-productive outwardly displaced conformation of helix αC, as promoted by ADP in the human and mouse IRE1α crystal structures, will function as protein kinase inhibitors by virtue of competitively inhibiting ATP binding, but might also function as RNase inhibitors by preventing attainment of the productive back-to-back dimer configuration required for RNase activity. As such, the crystal structures of mouse and human IRE1α would provide useful platforms for the design of small molecule kinase inhibitors that repress RNase function. Towards this end, we speculate that Compound 3 (1-(4-(8-amino-3-isopropylimidazo[1,5-a]pyrazin-1-yl)naphthalen-1-yl)-3-(3-(trifluoromethyl)phenyl)urea), a first in class ATP competitive protein kinase inhibitor that inhibits IRE1 RNase function 39, might act through such a mechanism.
Biomolecules containing aldehyde moieties are common in both food stuffs, essential nutrients and vitamins including, vanilla, sugars (glucose) and vitamins A and B6. Although some aldehyde containing compounds can form irreversible adducts, this is limited to unsaturated hydroxylated aliphatic chains such as lipid peroxidation products including 4-hydroxynonenal, which can induce ER stress 49, or hydroxylated saturated aliphatic chains where an Amadori rearrangement is required after the imine is formed. This is the case of HbA1c generation by glucose on hemoglobin 50, the major biomarker of long term glucose exposure in diabetics. Aromatic aldehydes including pyridoxal phosphate (vitamin B6) are unable to undergo such conversion. Furthermore, Schiff bases are not stable in the presence of water and studies indicate that dissociation from the mammalian IRE1α is rapid both on the enzyme in vitro and in cells after wash out 41,43. Therefore, stable adduct formation and creation of new epitopes by lysine conversion is not likely for IRE1 or other proteins limiting the possibility of immune recognition and sensitization in vivo; however, transient adduct formation of buried lysines in heterologous enzymes cannot be formally ruled out. HAA compounds designed to enhance the oxygen saturation of hemoglobin have long serum half-lives and excellent target binding properties in humans 51, contrary to conventional wisdom. Indeed, HAA inhibitors of IRE1α are active and well tolerated in vivo; firstly, the biphenyl HAA analog, Compound 2, synergized with an oncolytic virus 44,secondly, the naphthylene compound MKC3946 was readily tolerated with high exposure where it inhibited the splicing of XBP1 mRNA in tissues and tumors and reduced myeloma tumor growth 42, and lastly, the coumarin compound 4μ8C was effective in suppressing XBP1s, inflammatory cytokines and inflammation in a rheumatoid arthritis model 52. These studies suggest that HAA compounds may be highly selective for the RNase active site of IRE1α and can be administered in vivo to obtain desired effects.
Methods
XBP1 splicing and CD59 stability analysis
Human RPMI 8226 plasmacytoma cells were grown in monolayer culture using Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C and 5% CO2. MKC9989 was prepared as 10 mM stocks in DMSO, stored at −20°C, and diluted in medium as indicated. Thapsigargin (Tg) was resuspended in DMSO and diluted in medium. Cells were grown to 50% confluency, treated with 1 μM Tg and/or 10 μM MKC9989 at the indicated time points. Following incubation of cells for the indicated periods, cells were harvested. Procedures for preparation and measurement of XBP1s, XBP1u, and CD59 mRNAs were as described previously 41.
Protein Expression and Purification
Murine IRE1α residues 499 to 977 used for in vitro biochemical and biophysical analyses and IRE1α residues 549 to 977 used for x-ray crystallographic analyses, both harboring an Asn772Tyr substitution, were expressed in SF9 insect cells using the Bac-to-Bac baculovirus system (Invitrogen). IRE1α fragments were cloned into pFastBac HTA vector as N-terminal polyhistidine tag fusions and viruses were generated according to manufacturer’s instructions. Large-scale protein expression was performed from 8 liter cultures of SF9 cells grown in suspension. Following 3 days post infection, cells were harvested by centrifugation, lysed by sonication in lysis buffer containing: 0.1 M Tris pH 7.0, 1 M NaCl, 25 mM imidazole, 1 mM TCEP, 10% glycerol and purified by Ni Immobilized Metal Affinity Chromatography (IMAC). The polyhistidine tag was cleaved with Tobacco etch virus protease and subtracted by an additional IMAC step.
Following size exclusion chromatography (SEC) using a Superdex S-200 column equilibrated in 25 mM Hepes pH 7.0, 0.5 M NaCl, 2 mM TCEP, pure fractions were pooled and concentrated to 10 mg/ml and flash frozen in liquid nitrogen. Human IRE1α residues 462 to 977 and yeast IRE1 residues 658-1115 (with an internal deletion from Cys869 to Phe892), were expressed and purified as described previously 11,41. In brief, yeast IRE1 was expressed from a pProEX plasmid in E. coli BL21 cells as a poly histidine-tagged fusion. Human IRE1α was expressed using the Bac-N-Blue Baculovirus system and SF9 insect cells as a GST-tagged fusion. Proteins were purified using Ni-NTA and Glutathione resin chromatography respectively, followed by protease cleavage of the affinity tag and finally by size exclusion chromatography on a Superdex S200 column.
Crystallization and structure determination
Complexes were pre-formed on ice for 30 min prior to crystallization. The final solution contained 200 μM IRE1α, 1 mM inhibitor, 0.5% DMSO (v/v), 2 mM ADP, 2 mM MgCl2, 2 mM TCEP, 25 mM HEPES, pH 7.0. Crystals were grown at 20°C by mixing the pre-formed complex with precipitant solution containing 0.1 M HEPES, 12% PEG8000, 8% ethylene glycol at pH 7.5. Rod-shaped crystals ~0.15 mm in length grew after a week, and were flash-frozen in liquid-N2 prior to data collection.
All datasets were processed using XDS 53. Anisotropic truncation and scaling was applied using the UCLA MBI Diffraction Anisotropy Server (http://services.mbi.ucla.edu/anisoscale) 54. The structure of the IRE1α-MKC9989 complex was solved by molecular replacement using CCP4-PHASER 55 and the structure of human IRE1α (PDBID 3P23 46) as the search model. The other HAA complexes were solved using the IRE1α-MKC9989 structure as the search model. All structures were refined using PHENIX Refine 56. Real space inspection and refinement was performed using COOT 57. ADP, Mg2+, and the inhibitors were manually positioned in difference maps. Geometric restraints for the inhibitors were generated using eLBOW (PHENIX package) 58. All inhibitors were modeled covalently bound to the Lys907 through a Schiff base. For IRE1α-OICR573 and IRE1α-OICR464 structures a long tubular density at the surface of one protein protomer was modeled as a PEG molecule.
Data sets for MKC9989, OICR573 and OICR464 co-structures, were collected at 100K at 0.979 Å (NE-CAT beamline 24-ID-C), 1.540 Å (home source), and 0.979 Å wavelengths (NE-CAT-beamline 24-ID-C), respectively. Ramachandran statistics corresponding to outliers, allowed, and favoured regions for the MKC9989 co-structure were 0.79%, 6.9%, 93.02%, for the OICR573 co-structure were 0.33%, 4.09%, and 95.58%, and for the OICR464 co-structure were 0.47%, 4.93% and 94.61%, respectively.
In vitro kinase assays
Auto-phosphorylation kinase assay in Figure 6e was performed with 1.5μM murine IRE1α and 150 μM ATP spiked with 27.7 nM P32-γ-ATP. Reactions were pre-incubated with IRE1α and inhibitor for 1 hour at room temperature. After addition of ATP reaction mix, reactions were incubated at room temperature. Reactions were terminated by addition of SDS-PAGE 6X loading buffer and analyzed by SDS-PAGE using NuPage 4–12% Bis-Tris gels. P32-γ-ATP signal was detected by phosphor imaging using a GE Healthcare Typhoon Variable Mode Imager. Quantification was performed using ImageQuant5 software. Trans auto-phosphorylation assays in Figures 1c, 4c, and 6g were performed as above except with 150nM of IRE1α and 15μM of kinase dead (Asp797Ala) yeast IRE1, residues 658-1115, as a phospho-acceptor substrate.
Endoribonuclease assays
Fluorescence based RNA cleavage assays were performed with 10 nM IRE1α protein and 100 nM of a single hairpin RNA substrate (5′-CAU GUC CGC AGC GCA UG-3′), labeled on the 5′ and 3′ termini with Alexa Fluor 647 fluorophore and black hole quencher, respectively. Reaction buffer consisted of 50 mM Tris pH 7.0, 0.5 mM MgCl2, 10 mM KCl, 0.025% Tween-20, 0.063 mg/ml tRNA, and 2 mM DTT. Compounds were pre-incubated in reaction buffer with IRE1α proteins for 1 hour at room temperature. Upon addition of RNA substrate (diluted in 10 mM Tris pH 7.0, 0.1 mM EDTA), cleavage reactions were monitored in real time by fluorescence intensity (λexite= 651 nm, λemission= 672 nm) at two-minute intervals using a Molecular Dimensions Analyst HT microplate reader. Gel based RNA cleavage assays were performed in the same reaction buffer as above with the addition of 0.09mg/ml BSA. A single hairpin RNA substrate (5′-CAU GUC CGC AGC GCA UG-3′) labeled with fluorescein on the 5′ terminus was used at a concentration of 45nM. Reactions were pre-incubated with inhibitor compound for 1 hour at room temperature, and then upon addition of RNA substrate for 10–45 minutes at 37°C. Reactions were quenched by addition of 2X RNA loading buffer (90% (v/v) formamide, 25mM EDTA) and samples were analyzed using 20% Urea-TBE PAGE and imaged via a GE Healthcare Typhoon Variable Mode Imager. Images were quantified using ImageQuant5 software.
The gel based RNase cleavage assay shown in Figure 4d was performed in a reaction buffer containing 1 M Hepes pH 7.0, 100 mM ADP, 100 mM MgCl2 and100 mM TCEP, with 100 μM murine IRE1α and 60 μM of an unlabeled dual hairpin RNA substrate encompassing human XBP1 nucleotide positions 426 to 490 generated by in vitro transcription reactions. Cleavage reactions were pre-incubated with compound for 30 minutes on ice and after addition of RNA substrate further for 45 minutes at room temperature. Reactions were quenched by the addition of 2X RNA loading buffer (90% formamide, 25 mM EDTA, 0.02% xylene cyanol and 0.02% bromophenol blue) and samples were analyzed using 20% Urea-TBE PAGE, stained with RedSafe dye, and imaged via a GE Healthcare Typhoon Variable Mode Imager.
Direct HAA inhibitor binding analyses
Microscale thermophoresis (MST) was performed with 500 nM murine IRE1α and 0.05% Pluronic in non-coated standard capillaries. The reactions were performed in a buffer containing 25 mM Hepes pH 7.0, 0.5 M NaCl and 2 mM TCEP. MKC9989, OICR464 and OICR573 inhibitor binding reactions employed final DMSO concentrations of 1%, 5% and 2% respectively. All measurements were performed at 20% MST power and 20% LED power using the Monolith NT. LabelFree instrument (NanoTemper Technologies GmbH).
Chemical synthesis
MKC9989, OICR573, OICR464 and the napthol analogue series shown in Figure 5 were synthesized using methods described in WO 2011/127070 A2 (MKC9989) and WO 2008/154484 A1 (OICR573 and OICR464) with commercially available chemical reagents. (7-Hydroxy-6-methoxy-3-(2-(2-methoxyethoxy)ethyl)-4-methyl-2-oxo-2H-chromene-8-carbaldehyde) was synthesized as shown in Scheme 1.
Scheme 1.
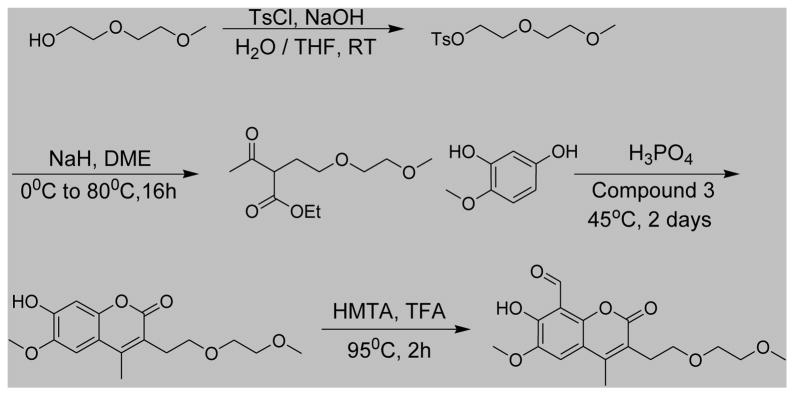
Toluene-4-sulfonic acid 2-(2-methoxy-ethoxy)-ethyl ester
A mixture of 125 mL aqueous NaOH (2.38 g, 0.0595 mol) solution and 200 mL THF 2-(2-methoxy-ethoxy)-ethanol (5 g, 0.042 mol) was cooled to 00C in a round bottom flask. A 200 mL THF solution of TsCl (7.38 g, 0.0388 mol) was added dropwise to the flask with stirring while keeping the inner temperature below 5°C. After the addition, the reaction mixture was stirred at 0°C ~ 5°C for additional 2 hours. The reaction mixture was poured into a flask with 500 mL ice-water. The resulting mixture was extracted with 150 mL DCM trice. The combined DCM solution was washed with water and brine, dried over Na2SO4 and concentrated to afford a light yellow needle-like solid as the desired product (9.50 g, yield: 89%). 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 8.4 Hz, 2H, ArH), 7.33(d, J = 8.4 Hz, 2H, ArH), 4.16 (t, J = 4.8 Hz, 2H, CH2), 3.68 (t, J = 4.8 Hz, 2H, CH2), 3.58-3.56 (m, 2H, CH2), 3.48-3.47 (m, 2H, CH2), 3.34 (s, 3H, OCH3), 2.44 (s, 3H, ArCH3).
2-[2-(2-Methoxy-ethoxy)-ethyl]-3-oxo-butyric acid ethyl ester
To a 25 mL DME suspension of NaH (0.992 g, 0.0248 mol) in a round bottom flask was added dropwise a 550 mL DME solution of acetyl ethylacetate (3.07 g, 0.0236 mol) at 0°C. After the addition, the resulting mixture was stirred at 20°C for 30 min. To the flask at 20°C was added successively a solution of toluene-4-sulfonic acid 2-(2-methoxy-ethoxy)-ethyl ester (6.79 g, 0.0247 mmol) in 100 mL DME and NaI (3.72 g, 0.0248 mol) powder. The resulting mixture was stirred at 20°C for 2 hours untill seeing some precipitate formed. The mixture was then heated to 80°C overnight under N2. Progress of the reaction was monitored by LCMS. The reaction was completed in 16 hours. After a filtration and removing the precipitate, the filtrate was collected and concentrated to a minimum volume. It was then diluted with H2O and acidified with 2N HCl to pH = 3, extracted with 100 mL DCM trice. The combined DCM solution was concentrated to a crude product which was purified by silica gel column (PE: EA = 6:1) chromatography affording a light yellow oil (1.8 g, 32% yield) as the product 2-[2-(2-methoxy-ethoxy)-ethyl]-3-oxo-butyric acid ethyl ester. 1H NMR (400 MHz, CDCl3): δ 4.19-4.16 (m, 2H, CH2), 4.12-4.10 (m, 1H, CH), 3.66 (m, 1H, CH), 3.52-3.48 (m, 6H, 3CH2), 3.36 (s, 3H, OCH3), 2.25 (s, 3H, COCH3), 2.17-2.11 (m, 2H, CH2), 1.49 (t, J = 7.2 Hz, 3H, CH3).
7-Hydroxy-6-methoxy-3-[2-(2-methoxy-ethoxy)-ethyl]-4-methyl-chromen-2-one
2-[2-(2-Methoxy-ethoxy)-ethyl]-3-oxo-butyric acid ethyl ester (2.25 g, 0.0097 mol) and 2-hydroxyl-3-methoxyl-phenol (1.30 g, 0.0093 mol) were mixed with 200 mL H3PO4 in a round bottom flask. The reaction mixture was stirred at 45°C for 2 days. After the reaction was completed, the mixture was diluted with EA (100 mL) and poured into a flask with 1 liter ice-water. The resulting mixture was stirred for 15 minutes. After a filtration, the solid obtained was washed with cold saturated NaHCO3, brine and H2O successively. The solid was dried with toluene azeotropy distillation affording a light yellow solid as the product (1.25 g, 41% yield). 1H NMR ( 400 MHz, DMSO-d6): δ 7.14 (s, 1H, ArH), 6.74 (s, 1H, ArH), 3.85 (s, 3H, CH3), 3.49-3.47 (m, 4H, 2CH2), 3.41-3.33 (m, 2H, CH2), 3.20 (s, 3H, CH3), 2.78 (t, J = 7.2 Hz, 2H, CH2), 2.40 (s, 3H, CH3).
7-Hydroxy-6-methoxy-3-(2-(2-methoxyethoxy)ethyl)-4-methyl-2-oxo-2H-chromene-8-carbaldehyde
7-Hydroxy-6-methoxy-3-[2-(2-methoxy-ethoxy)-ethyl]-4-methyl-chromen-2-one (2.0 g, 0.0064 mol) and HMTA (3.65 g, 0.026 mol) were dissolved in 85 mL TFA in a round bottom flask. The reaction mixture was heated to 95°C for 2h. Progress After the reaction was completed, the reaction mixture was concentrated. The residue was diluted with H2O, extracted with 100 mL DCM trice. The combined DCM solution was washed with cold saturated NaHCO3, brine, dried over Na2SO4 and concentrated.
The remaining residue was purified with a silica gel column (DCM: MeOH = 100:1) chromatography affording 1.3 g of desired crude product. The product was further purified by recrystallization from EA affording 7-hydroxy-6-methoxy-3-(2-(2-methoxyethoxy)ethyl)-4-methyl-2-oxo-2H-chromene-8-carbaldehyde (0.8 g, 37% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 12.46 (s, 1H, OH), 10.66 (s, 1H, CHO), 7.30 (s, 1H, ArH), 4.01 (s, 3H, ArOCH3), 3.72 (t, J = 6.8 Hz, 2H, CH2), 3.65-3.64 (m, 2H, CH2), 3.56-3.55 (m, 2H, CH2), 3.40 (s, 3H, OCH3), 3.02 (t, J = 6.8 Hz, 2H, CH2), 2.49 (s, 3H, CH3); LCMS [M+H]+ = 337.2.
MKC3946 (2-Hydroxy-6-[5-(4-methyl-piperazine-1-carbonyl)-thiophen-2-yl]-naphthalene-1-carbaldehyde) was synthesized as shown in Scheme 2.
Scheme 2.

6-Bromo-2-hydroxy-naphthalene-1-carbaldehyde
A solution of titanium tetrachloride (16.5 mL, 28.5g, 150 mM) in methylene chloride (300 mL) was stirred under ice cooling in a 2 liter 3-necked flask, equipped with a thermometer, a dropping funnel and a gas outlet with a paraffin oil filled bubbler. To the flask, dichloromethyl methyl ether (24.4 mL, 31 g, 270 mM) was added dropwise during 10 minutes resulting in a yellow solution. Then a solution of 6-bromo-2-naphthol (20 g, 89.7 mM) in methylene chloride (300 mL) was added dropwise during 30 minutes whereupon the color of the mixture turned deep brownish-red and only a slight (3–4 °C) increase of the internal temperature was observed. Thereafter the cooling batch was removed and the mixture was stirred at ambient temperature overnight. The next day 1000 mL of 1 N hydrochloric acid were added under ice cooling whereupon, during the first 10–15 minutes of the addition, extensive gas evolution was observed. After the addition was complete and the color of the mixture turned yellow, a small amount (0.78 g after drying) of a brick-red insoluble substance was filtered off. The aqueous layer was separated and extracted with methylene chloride (2 × 100 mL). The combined organic solutions were washed with 1 N hydrochloric acid (100 mL), then with brine (5 × 100 mL), dried over anhydrous MgSO4 and evaporated to dryness. The brownish-yellow residue was triturated with diethyl ether; the yellow crystalline product was filtered off, washed with diethyl ether and dried over solid NaOH under reduced pressureas the desired product (16.95g, 75.3%), mp. 142-146 °C, 1H NMR (400 MHz, DMSO-d6) δ ppm 11.90 (s, 1H), 10.76 (s, 1H), 8.92 (d, J = 9.3 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 8.10 (d, J = 9.3 Hz, 1H), 7.72 (dd, J = 9.0, 2.3 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H). This product is used in the next step without any further purification.
2-Hydroxy-6-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-naphthalene-1-carbaldehyde
A mixture of 6-bromo-2-hydroxy-naphthalene-1-carbaldehyde (10.0 g, 39.8 mM), 1,4-dioxane (160 mL, distilled freshly from sodium metal, under nitrogen), bis(pinacolato)diborone (11.1 g, 43.8 mM), powdered potassium acetate (11.8 g, 120 mM) and palladium[1,1′-bis(diphenylphosphino)ferrocene]dichloride methylene chloride complex (3.9 g, 4.8 mM) was stirred at 80–85°C under a slow stream of nitrogen for 4 hours. After cooling the insolubles were filtered off and washed with 1,4-dioxane. The solvent was evaporated and the dark brown residue was purified by chromatography over silica (200 g) using methylene chloride as eluent to yield the product (10.4 g, 87.6%) a yellow powder in 86% HPLC purity, mp. 115–118°C.
The product can be further purified by recrystallization from 6-fold (v/w) diisopropyl ether with final yield as 54%. 1H NMR (400 MHz, CDCl3) δ ppm 13.23 (s, 1H), 10.82 (s, 1H), 8.33 (d, J = 8.8 Hz, 1H), 8.29 (s, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.98 (dd, J = 8.5, 1.3 Hz, 1H), 7.13 (d, J = 9.0 Hz, 1H), 1.39 (s, 12H).
5-Bromo-thiophene-2-carbonyl chloride
9.09 Grams 5-bromo-thiophene-2-carboxylic acid was dissolved in 100 mL thionyl chloride and the solution was refluxed for one hour. After completion of the reaction, the excess of thionyl chloride was removed under reduced pressure. 9.60 grams of a solid residue was obtained and used in the next step without further purification.
(5-Bromo-thiophen-2-yl)-(4-methyl-piperazin-1-yl)-methanone
4.27 grams of 1-Me-piperazine and 5.54 grams of DIPEA were dissolved in 80 mL of dry methylene chloride and the resulting solution was added dropwise at 0°C to a solution of 9.60 grams of 5-Bromo-thiophene-2-carbonyl chloride in dry methylene chloride. The stirred solution was allowed to warm to room temperature and was washed with water. The organic phase was separated, dried and evaporated. The obtained 12.08 grams of light brown oil were allowed to stand in the refrigerator overnight whereupon it solidified to a yellow-brownish solid (y = 98%). The product was used directly for next step without further purification.
2-Hydroxy-6-[5-(4-methyl-piperazine-1-carbonyl)-thiophen-2-yl]-naphthalene-1-carbaldehyde
2-Hydroxy-6-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)- naphthalene-1-carbaldehyde (5.50 g, 18.4 mmol), (5-Bromo-thiophen-2-yl)-(4-methyl-piperazin-1-yl)-methanone (5.60 g, 19.3 mmol) and sodium-carbonate (11.70 g) were dissolved in a mixture of 500 mL DMF and 500 mL water. To this mixture was added 644 mg tetrakis(triphenylphosphino)-palladium. The reaction mixture was heated to 100°C under nitrogen atmosphere. The progress was monitored by TLC. After one hour, the reaction mixture was evaporated and the solid residues were partitioned between 400 mL water and 300 mL chloroform. The aqueous phase was extracted with additional 300 mL chloroform. The combined organic layer was dried over sodium sulfate. Evaporation of the solvent resulted in 7.00 grams of a reddish powder which was purified by flash column chromatography with chloroform as eluent. The isolated 6.88 grams of crude material were suspended in ethanol and cooled to 0°C. 70 ml of saturated solution of HCl in ethyl acetate was added to the stirred suspension and was stirred overnight at room temperature. An additional portion of HCl/EtOAc was then added, the suspension was cooled again to 0°C. The product was filtered off, washed with EtOAc and diethyl ether to yield a yellow powder (5.67g, 74%) as the desired product. 1H NMR (400 MHz, CDCl3) δ ppm 13.13 (br. s, 1H), 10.82 (s, 1H), 8.37 (d, J = 9.0 Hz, 1H), 7.97 - 8.05 (m, 2H), 7.86 (dd, J = 8.8, 2.0 Hz, 1H), 7.30 - 7.35 (m, 2H), 7.19 (d, J = 9.0 Hz, 1H), 3.78 - 3.88 (m, 4H), 2.45 - 2.55 (m, 4H), 2.35 (s, 3H). LCMS [M+H]+ = 381.1.
MKC3987 (7-Bromo-2-hydroxy-naphthalene-1-carbaldehyde): This compound was synthesized in a same manner as 6-Bromo-2-hydroxy-naphthalene-1-carbaldehyde as shown in Scheme 2 using 7-Bromo-naphthalen-2-ol as the starting material.
MKC4027 (2-Hydroxy-7-[4-(4-methyl-piperazine-1-carbonyl)-phenyl]-naphthalene-1-carbaldehyde): This compound was synthesized in a similar manner as shown in Scheme 2 using 7-bromo-2-hydroxy-naphthalene-1-carbaldehyde to make the Suzuki coupling boronic ester 2-hydroxy-7-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-naphthalene-1-carbaldehyde, which was then coupled with (4-bromo-phenyl)-(4-methyl-piperazin-1-yl)-methanone to form the target compound.
MKC3426 (2-Hydroxy-3-methoxy-naphthalene-1-carbaldehyde): Commercially sourced from Ambinter (Cat. No. Amb18653768) or Aurora Fine Chemicals LLC (Cat. No. A07.180.446).
MKC3437 (2-hydroxy-1-naphthylaldehyde): Commercially sourced from Sigma-Aldrich (Cat. No. H45353-25G).
MKC3820 (2-hydroxy-1-acetonaphthylaldehyde): Commercially sourced from Sigma-Aldrich (Cat. No. 303054-25G).
MKC3964 (1-naphthylaldehyde): Commercially sourced from Sigma-Aldrich (Cat. No. N109-2G-A).
Modeling of the IRE1α–hydroxy aryl aldehyde–RNA substrate complex
The tRNA splicing endonuclease in complex with RNA substrate (PDB: 2GJW)47 was used as a template to model RNA substrate concurrently bound to murine IRE1α and MKC9989 (Supplementary Figure 3e) as performed previously for yeast IRE111. In brief, the catalytic tetrad of the tRNA endonuclease (Tyr246, Ser258, His257 and Lys287) was superimposed manually onto the respective invariant catalytic tetrad (Tyr892, Asn906, His910 and Arg905) of mouse IRE1α. The two nucleotides flanking the scissile bond of the substrate RNA corresponding to chain E positions deoxy-uridine 14 and adenosine 15, were then excised and mutated to the IRE1α substrate consensus guanosine followed by cytosine. The full structure of IRE1α, dinucleotide and MKC9989 was then energy minimized using Refmac59,60.
Supplementary Material
Acknowledgments
This work was supported by a grant to F.S. from the Canadian Institutes of Health Research (MOP 84370) and a Multiple Myeloma Research Foundation Biotech Investment Award to MannKind Corporation. We thank Gary Flynn, David Lonergan, Peter Pallai, Warren Wade, Zoltan Zubovics, Yun Yang and Zhipeng Wu for chemistry support. We thank the Advanced Photon Source on the Northeastern Collaborative Access Team beam lines, supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health and by the U.S. DOE under contract No. DE-AC02-06CH11357. M.S. was supported through a Canadian Cancer Society Postdoctoral Fellowship.
Footnotes
Author contributions
M.S. and M.T. performed the co-structure determinations. N.D., M.T., L.L., and M.C. performed in vitro enzymatic assays and direct inhibitor binding measurements. D.C., N.T., and K.L. performed biophysical analyses and/or generated reagents. I.K. collected synchrotron diffraction data. D.U., R.A., G.P., M.P. and B.W chemically synthesized OICR573 and OICR464. V. T., C.S., A.T., J. L., D. V. and Q. Z performed in cell analyses, enzyme assays and directed the synthesis of MKC9989, MKC3946, MKC3987 and MKC4027. D.D. supervised M.C. F.S and J.P. designed experiments and interpreted results. F.S and J.P wrote the manuscript with contributions from all other authors.
Competing Financial Interests
The authors declare no competing financial interests. V. T., C. S., A.T., J. L., D. V.,Q. Z and J.P. are past or present employees of MannKind Corp. and F.S. was a paid consultant of MannKind Corp.
Accession codes. Final coordinates and structure factors for the IRE1α-MKC9989, IRE1α-OICR573, and IRE1α-OICR464 co-structures have been deposited to the PDB with the respective accession codes: 4PL3, 4PL4, and 4PL5.
References
- 1.Cao SS, Kaufman RJ. Targeting endoplasmic reticulum stress in metabolic disease. Expert opinion on therapeutic targets. 2013;17:437–48. doi: 10.1517/14728222.2013.756471. [DOI] [PubMed] [Google Scholar]
- 2.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature reviews. Drug discovery. 2013;12:703–19. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 3.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 4.Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiological reviews. 2011;91:1219–43. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–7. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 7.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 8.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular biology of the cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida H, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Molecular and cellular biology. 2000;20:6755–67. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–9. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 11.Lee KP, et al. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132:89–100. doi: 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang XZ, et al. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. The EMBO journal. 1998;17:5708–17. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & development. 1998;12:1812–24. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 15.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 16.Dong B, Niwa M, Walter P, Silverman RH. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA. 2001;7:361–73. doi: 10.1017/s1355838201002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. Journal of virology. 2007;81:12720–9. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. The EMBO journal. 1996;15:3028–39. [PMC free article] [PubMed] [Google Scholar]
- 19.Welihinda AA, Kaufman RJ. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. The Journal of biological chemistry. 1996;271:18181–7. doi: 10.1074/jbc.271.30.18181. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez TN, Sidrauski C, Dorfler S, Walter P. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. The EMBO journal. 1999;18:3119–32. doi: 10.1093/emboj/18.11.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 22.Shen X, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 24.Harding HP, et al. An intact unfolded protein response in Trpt1 knockout mice reveals phylogenic divergence in pathways for RNA ligation. RNA. 2008;14:225–32. doi: 10.1261/rna.859908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and cellular biology. 2003;23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–7. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 27.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–7. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 28.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. The Journal of cell biology. 2009;186:323–31. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.So JS, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell metabolism. 2012;16:487–99. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature immunology. 2010;11:411–8. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao T, et al. PKA phosphorylation couples hepatic inositol-requiring enzyme 1alpha to glucagon signaling in glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15852–7. doi: 10.1073/pnas.1107394108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho JA, et al. The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell host & microbe. 2013;13:558–69. doi: 10.1016/j.chom.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Lin JH, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rutkowski DT, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS biology. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chawla A, Chakrabarti S, Ghosh G, Niwa M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. The Journal of cell biology. 2011;193:41–50. doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubio C, et al. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. The Journal of cell biology. 2011;193:171–84. doi: 10.1083/jcb.201007077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korennykh AV, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–93. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–7. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, et al. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nature chemical biology. 2012;8:982–9. doi: 10.1038/nchembio.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiseman RL, et al. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Molecular cell. 2010;38:291–304. doi: 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volkmann K, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. The Journal of biological chemistry. 2011;286:12743–55. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mimura N, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–81. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cross BC, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E869–78. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahoney DJ, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer cell. 2011;20:443–56. doi: 10.1016/j.ccr.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Oikawa D, Tokuda M, Iwawaki T. Site-specific cleavage of CD59 mRNA by endoplasmic reticulum-localized ribonuclease, IRE1. Biochemical and biophysical research communications. 2007;360:122–7. doi: 10.1016/j.bbrc.2007.06.020. [DOI] [PubMed] [Google Scholar]
- 46.Ali MM, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. The EMBO journal. 2011;30:894–905. doi: 10.1038/emboj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue S, Calvin K, Li H. RNA recognition and cleavage by a splicing endonuclease. Science. 2006;312:906–10. doi: 10.1126/science.1126629. [DOI] [PubMed] [Google Scholar]
- 48.Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17783–8. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vladykovskaya E, et al. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. The Journal of biological chemistry. 2012;287:11398–409. doi: 10.1074/jbc.M111.320416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lowrey CH, Lyness SJ, Soeldner JS. The effect of hemoglobin ligands on the kinetics of human hemoglobin A1c formation. The Journal of biological chemistry. 1985;260:11611–8. [PubMed] [Google Scholar]
- 51.Rolan PE, Mercer AJ, Wootton R, Posner J. Pharmacokinetics and pharmacodynamics of tucaresol, an antisickling agent, in healthy volunteers. British journal of clinical pharmacology. 1995;39:375–80. doi: 10.1111/j.1365-2125.1995.tb04465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu Q, et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. The EMBO journal. 2013;32:2477–90. doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabsch W. Xds. Acta crystallographica. Section D, Biological crystallography. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strong M, et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8060–5. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCoy AJ, et al. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta crystallographica. Section D, Biological crystallography. 2009;65:1074–80. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallographica. Section D, Biological crystallography. 2011;67:355–67. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.