

Abstract
Reports from Alzheimer’s disease (AD) biomarker work have shown a strong link between oxidative stress and AD neuropathology. The nonenzymatic antioxidant, glutathione (GSH), plays a crucial role in defense against reactive oxygen species and maintenance of GSH redox homeostasis. In particular, our previous studies on GSH redox imbalance have implicated oxidative stress induced by excessive reactive oxygen species as a major mediator of AD-like events, with the presence of S-glutathionylated proteins (Pr-SSG) appearing prior to overt AD neuropathology. Furthermore, evidence suggests that oxidative stress may be associated with dysfunction of the hypothalamic-pituitary-adrenal axis, leading to activation of inflammatory pathways and increased production of corticotropin-releasing factor (CRF). Therefore, to investigate whether oxidative insults can be attenuated by reduction of central CRF signaling, we administered the type-1 CRF receptor (CRFR1) selective antagonist, R121919, to AD-transgenic mice beginning in the preclinical/prepathologic period (30-day-old) for 150 days, a timepoint where behavioral impairments and pathologic progression should be measureable. Our results indicate that R121919 treatment can significantly reduce Pr-SSG levels and increase glutathione peroxide activity, suggesting that interference of CRFR1 signaling may be useful as a preventative therapy for combating oxidative stress in AD.
Keywords: Alzheimer’s disease, corticotropin-releasing factor-1 receptor (CRFR1), glutathione, glutathione peroxide, glutathione reductase, oxidative stress, oxidized glutathione, R121919, S-glutathionylated protein
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia and is characterized by progressive cognitive impairment and two neuropathological hallmarks, amyloid-β plaques (Aβ) and neurofibrillary tangles. Reactive oxygen species (ROS) are important metabolic products involved in the stimulation of cellular signaling pathways in mammalian cells, and are thought to accumulate under imbalanced redox conditions and can trigger oxidative stress [1]. Respiratory chain reactions are responsible for generating the majority of cellular ROS in mitochondria where electron transport chains are active. With progressive imbalance between ROS and antioxidant systems, oxidative stress and cell damage are thought to occur [2]. Evidence from AD biomarker studies has shown a strong link between oxidative stress and AD neuropathology, with greatly decreased antioxidant levels found in the AD brain [3]. In particular, elevated DNA lesions (8-hydroxy-2′-deoxyguanosine, 8-OHdG) [4, 5], increased oxidized proteins (S-glutathionylated proteins, Pr-SSG [6, 7], S-nitronsylated proteins [8], and carbonylated proteins [9]), and high levels of lipid peroxidation have been found to be positively correlated with oxidative stress in the AD brain [10]. Moreover, increased levels of metals may also be associated with stimulating free radicals in the AD brain [11].
Glutathione (GSH) consists of three amino acid residues (glutamic acid, cysteine, and glycine) and plays a crucial role in cellular defense against ROS and maintenance of GSH redox homeostasis, which is generally known as a non-enzymatic scavenger of oxidative stress in mammalian cells [12]. In the GSH redox cycle, GSH can be oxidized by oxidative stress in the presence of glutathione peroxidase (GPx) to form oxidized GSH (GSSG), which can be reduced back to GSH in the presence of glutathione reductase (GR) [13]. Our previous studies of GSH redox homeostasis have demonstrated that oxidative stress may have a significant contribution to AD neuropathology, because excessive ROS may induce oxidative stress as a disturbance of GSH redox balance in the AD brain [14]. Moreover, increased levels of hippocampal and cortical Pr-SSG have been reported in AD transgenic (Tg) mouse models compared to age-matched wild type (Wt) counterparts, suggesting the specific response of oxidative stress can be observed in AD-relevant brain regions [14].
Reports have suggested that hippocampal oxidative stress can induce dysfunction/dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, which may lead to an increase in circulating levels of stress steroids (cortisol in humans; glucocorticoid in rodents), resulting in oxidative neural damage [15]. In particular, corticotropin-releasing factor (CRF) and its receptors are involved in stress-signaling in the periphery, but are also implicated in central neuromodulation, as this signaling system is widely distributed throughout brain areas susceptible to AD neuropathology [16, 17]. Alerations in CRF peptides are evident early in disease progression [18], with reduction in cortical CRF immunoreactivity (in the face of increased hypothalamic levels) is a noticeable change in early AD progression [19]. Furthermore, epidemiologic studies demonstrate that AD patients are predisposed to psychological distress compared to age-matched controls [20].
To investigate whether oxidative insults in an Tg mouse model could be attenuated by CRFR1 blockade, a CRFR1 selective antagonist (R121919) [21–23] was tested on a well-characterized Tg mouse model of AD (PSAPP) in the present study. This Tg mouse model develops AD-like Aβ pathology and cognitive impairment in an age-dependent manner [24], while cognitive impairment and Aβ aggregates in the hippocampus and cortex can be detected in Tg mice as early as 4 month of age [25]. We treated 30-day-old Tg mice and age-matched Wt littermates with daily subcutaneous administration of R121919 for 5 months (150 days) continuously. Biochemical assays were conducted to evaluate GSH redox balance, Pr-SSG levels, and enzymatic activities.
MATERIALS AND METHODS
Animals
Mice were housed (2 to 4 mice/cage) in a temperature controlled room (22°C) with a 12-h light-dark cycle. Tg mice reliably accumulate Aβ plaques in the cortex and hippocampus starting at 6 months of age. A total of 40 6-month-old mice were investigated in this study. All biological samples were collected between the hours of 8:00 to 10:00. The University of California at San Diego’s Institutional Animal Care and Use Committee approved all experimental protocols.
CRFR1 antagonist (R121919) treatment
For pharmacologic blockade of CRFR1, we used the well characterized, orally potent, small molecule CRFR1-selective antagonist, R121919 (gift of Dr. K. Rice, NIH) [21, 26]. Mice were given subcutaneous injections of vehicle or drug (R121919, 20 mg/kg/d) for 150 days based on daily body weight measurements. The dose of 20 mg/kg/d was adopted from our previous studies of R121919 treatment and stress-induced tau phosphorylation [23, 27]. R121919 was dissolved in 0.3% tartaric acid and 5% v/v polyethoxylated castor oil. Both R121919 and vehicle solution were vortexed and sonicated immediately before use. Wt and Tg animals were treated with vehicle or drug starting at 30 days of age. Five mice per condition (a total of 8 conditions) were used in this project, including 2 treatments (vehicle versus drug) × 2 genotypes (Wt versus Tg) × 2 genders (male versus female).
Sample preparation
After animals were transcardially perfused with cold PBS, brain and plasma samples were collected and stored at −80°C until use. Sample preparation for each assay was described below.
Measurement of GSH and GSSG levels in the brain and plasma samples
GSH, total GSH, protein thiol groups (Pr-SH), and total Pr-SH levels in the brain and plasma samples were assayed using GSH assay kits (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s instructions as described previously [14]. Plates were read using an iMark Microplate Reader (Bio-Rad Laboratories Inc., Hercules, CA) at 412 nm (spectrophotometry) with the kinetic method (see Cayman protocols). The concentration of GSSG was calculated by determining the difference between total GSH and native GSH. Data on GSH and GSSG contents are expressed as nanomole (nmol) per milligram (mg) of protein.
Measurement of total protein S-glutathionylation levels in the brain
To determine the relative levels of Pr-SSG, western blot analysis was employed. After adding the reducing agent, dithiothreitol (DTT), Pr-SSG reactive target bands disappear. Parallel experiments without DTT treatment were also studied. Hippocampal and cortical tissues were homogenized with 2 μl/mg in detergent-free RAB buffer, containing 100 mM 2-(N-morpholino) ethanesulphonic acid (MES; pH 7.0), 1 mM EGTA, 0.5 mM MgSO4, 750 mM NaCl, 20 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and protease and phosphatase inhibitor cocktail EDTA-free for measuring Pr-SSG levels. 10 μg of protein of each samples was loaded and then separated on a 10–20% gradient gel. S-glutathionylated proteins were detected with an anti-GSH monoclonal antibody (1:5000), and Enhanced Chemiluminescence (ECL) detection was performed using the Gel Doc XR+ Imager (Bio-Rad Laboratories Inc., Hercules, CA). A polyclonal anti-β-actin antibody (1:1000) was used as a loading control. All samples were normalized using β-actin levels.
Determination of enzymatic activity in the brain
GPx activity and GR activity (nmol/min/ml) were assayed using commercially available GPx and GR assay kits (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s protocols. Each absorbance reading was collected, 1 cycle/min at 340 nm for a total of 10 cycles using the kinetic method of calculation.
Protein concentration determination in the brain
Protein concentrations were determined by a BCA assay kit (Thermo Fisher Scientific, Waltham, MA) based on manufacturer’s manual. The absorbance of each protein sample at 595 nm was detected with an iMark plate reader (Bio-Rad Laboratories Inc., Hercules, CA).
Statistical analyses
Statistical analyses between groups were conducted with two-way ANOVA with Tukey’s Multiple Comparison post-hoc test using Graphpad Prism 6.02 software (La Jolla, CA). A value of p < 0.05 was accepted as statistically significant. All values are expressed as Mean ± SEM.
RESULTS
GSH and GSSG contents in the brain
To obtain a better understanding of the GSH redox cycle, GSH and GSSG levels as a function of R121919 treatment in AD mice, well characterized ELISA kits were used (see Methods). In terms of genotype effects on GSH contents, we observed a significant increase in GSH levels (+p < 0.05) in vehicle-treated male Tg mice compared to male Wt counterparts (Fig. 1A); whereas no statistical difference was found in female groups (Fig. 1B). Moreover, for gender effects on GSH levels, increased GSH levels was found in male Tg mice compared to female Tg counterparts treated with vehicle (†p < 0.05, Fig. 1D). However, no treatment effects were detected regardless of genotype and gender (Fig. 1A–D). As for genotype effects on GSSG levels, no significant changes were found in either male or female mice regardless of treatment condition (Fig. 1E, F). Additionally, based on gender effects on GSSG contents, male mice receiving vehicle had dramatically reduced GSSG levels (††p < 0.01, Fig. 1G) compared to female counterparts, while male mice treated with drug also had significantly decreased GSSG levels than their female littermates (†p < 0.05). Again, no alterations in GSSG levels were observed with the treatment effect (Fig. 1E–H). In summary, we found R121919 treatment may not have an impact on tissue GSH redox system.
Fig. 1.

Brain GSH and GSSG levels as a function of R121919 treatment. A) GSH levels in the brains of male mice; B) GSH levels in the brains of female mice; C) GSH levels in the brains of Wt mice; D) GSH levels in the brains of Tg mice; E) GSSG levels in the brains of male mice; F) GSSG levels in the brains of female mice; G) GSSG levels in the brains of Wt mice; H) GSSG levels in the brains of Tg mice. All values are expressed as nmol/mg protein, and presented as Mean ± SEM, n = 5 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. Genotype effect: Wt versus Tg. +p < 0.05. Gender effect: Male versus Female. †p < 0.05, ††p < 0.01, †††p < 0.005. Treatment effect: Vehicle versus Drug. *p < 0.05, ***p < 0.005.
Pr-SSG levels in the brain
To determine oxidative Pr-SSG levels in specific brain regions, RAB extracts from the hippocampus and cortex of Tg mice treated with vehicle or R121919 were analyzed using western blot. We found levels of Pr-SSG to be significantly reduced (49%) in the hippocampus of male Tg mice treated with R121919 (**p < 0.01) compared to vehicle counterparts (Fig. 2A). Similar effects were seen in female cohorts, with decreased Pr-SSG levels (38%) shown in the hippocampus of Tg mice treated with drug (**p < 0.01) compared to vehicle-treated mice. In terms of the cortex, a 24% decline of cortical Pr-SSG levels was found in male Tg mice treated with drug (*p < 0.05) compared to vehicle counterparts, while there was a reduction of 18% in cortical Pr-SSG contents of female Tg mice receiving drug (*p < 0.05) compared to their littermates receiving vehicle (Fig. 2B).
Fig. 2.

Quantification of brain Pr-SSG levels in male and female Tg animals as a function of R121919 treatment. Representative immunoblots showing reduced Pr-SSG levels in the hippocampus (A) and cortex (B) of both male and female mice treated with drug. All data are expressed as Mean ± SEM, n = 3 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. V, vehicle; D, drug. Treatment effect: vehicle versus drug. *p < 0.05, **p < 0.01.
In addition, a 32% reduction in hippocampal Pr-SSG levels in male Wt mice was observed, whereas no change in hippocampal Pr-SSG in female Wt mice was found (Supplementary Figure 1A). Also, cortical Pr-SSG contents were reduced by 34% in male Wt mice and 43% in female Wt mice, respectively (Supplementary Figure 1B). Thus, a significant decline in hippocampal and cortical Pr-SSG was found in both male and female Tg mice treated with R121919, suggesting that interfering with CRFR1 signaling can be beneficial for attenuating oxidative stress.
GPx activity in the brain
Because GPx activity is known to be a sensitive indicator of free radicals, brain GPx activity was analyzed using enzymatic activity kits (see Methods). In terms of genotype effects in male mice receiving vehicle, we observed a significant increase in GPx activity of Wt mice compared to Tg counterparts (+p < 0.05, Fig. 3A). Moreover, according to gender effects, we found elevated GPx activity in Wt female mice receiving R121919 mice compared to male counterparts (†p < 0.05, Fig. 3C), which was also seen in Tg mice receiving drug (†††p < 0.005, Fig. 3D). Additionally, significantly increased GPx activity was found in female Tg receiving R121919 compared to vehicle treated counterparts (*p < 0.05, Fig. 3B). Collectively, female Tg mice treated with R121919 had increased levels of GPx activity compared to vehicle or male littermates, indicating that female Tg mice may be more sensitive to CRFR1 antagonist treatment.
Fig. 3.

GPx activity in mouse brain with drug treatment. A) GPx activity in male mice under treatment and genotype conditions. B) GPx activity in female mice under treatment and genotype conditions. C) GPx activity in Wt mice under treatment and gender effects. D) GPx activity of Tg mice under treatment and gender effects. All data are expressed as Mean ± SEM, n = 5 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. Genotype effect: Wt versus Tg. +p < 0.05. Gender effect: Male versus Female. †p < 0.05, †††p < 0.005. Treatment effect: Vehicle versus Drug. *p < 0.05, ***p < 0.005.
GR activity in the brain
GR activity was measured to provide insight into R121919 effects on the GSH redox cycle in the brain. In terms of genotype effects, we found no significant differences in GR activity in either male or female mice (Fig. 4A, B). However, when genders were analyzed separately, R121919-treated female Wt mice had increased GR activity compared to R121919-treated male Wt counterparts. Additionally, based on treatment effects in female Wt mice, drug-treated mice had elevated GR activity than their vehicle littermates (*p < 0.05, Fig. 4B). Overall, increased GR activity was found in drug-treated female Wt mice compared to vehicle and male counterparts.
Fig. 4.
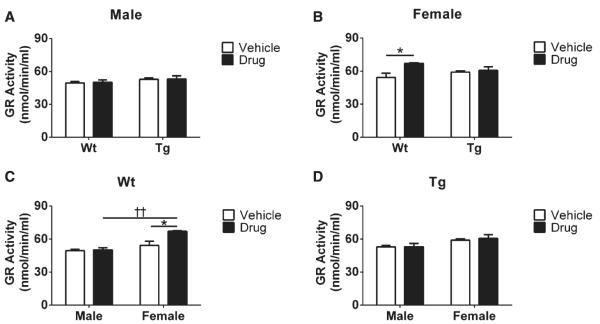
Comparison of GR activity in mouse brain as a function of drug treatment. A) GR activity of male mice under treatment and genotype conditions. B) GR activity of female mice under treatment and genotype conditions. C) GR activity of Wt mice under treatment and gender effects. D) GR activity of Tg mice under treatment and gender effects. All data are expressed as Mean SEM, n = 5 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. Gender effect: ± Male versus Female. ††p < 0.01. Treatment effect: Vehicle versus Drug. *p < 0.05, **p < 0.01.
GSH and GSSG contents in plasma as a function of R121919 treatment
Levels of GSH, GSSG, and total GSH in plasma samples from each group were detected using ELISA kits (see Methods). As for GSH levels, we found no significant genotype differences in either male or female mice treated with vehicle or drug, respectively, as shown in Fig. 5A and B. However, in terms of gender effects, male Wt mice receiving vehicle had higher GSH contents than their female Wt counterparts treated with vehicle (††p < 0.01, Fig. 5C), whereas no changes were observed in plasma GSH levels of Tg mice (Fig. 5D). Additionally, no effects were observed in plasma GSH levels in either gender or genotype as a function of R121919 treatment (Fig. 5A–D).
Fig. 5.

Plasma GSH and GSSG levels with drug treatment. A) GSH levels in plasma of male groups; B) GSH levels in plasma of female groups; C) GSH levels in plasma of Wt groups; D) GSH levels in plasma of Tg groups; E) GSSG levels in plasma of male groups; F) GSSG levels in plasma of female groups; G) GSSG levels in plasma of Wt groups; H) GSSG levels in plasma of Tg groups. All values are expressed as nmol/mg protein, and presented as Mean ± SEM, n = 5 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. The gender effect: Male versus Female. ††p < 0.01. The treatment effect: Vehicle versus Drug: **p < 0.01.
Moreover, for GSSG contents, no changes were seen as a function of genotype on GSSG levels in vehicle or drug-treated male and female mice (Fig. 5E, F), nor were significant differences seen in plasma GSSG levels regardless of treatment or gender (Fig. 5E-H). Notably, GSH and GSSG contents were much higher in plasma than in the brain, though no drug effects were observed.
Pr-SH and Pr-SSG contents in plasma
Pr-SH and Pr-SSG levels in plasma samples from each group were detected with ELISA kits (see Methods). As for genotype effects, in plasma Pr-SH levels, no differences were observed in either male or female mice at this stage (Fig. 6A, B). However, in terms of gender effects, female mice treated with vehicle had elevated Pr-SH levels compared to their male counterparts (†p < 0.05, Fig. 6D). As shown in Fig. 6A–D, we did not observe any treatment effect on plasma Pr-SH.
Fig. 6.

Plasma Pr-SH and Pr-SSG levels as a function of drug treatment. A) Pr-SH levels in plasma of male groups; B) Pr-SH levels in plasma of female groups; C) Pr-SH levels in plasma of Wt groups; D) Pr-SH levels in plasma of Tg groups; E) Pr-SSG levels in plasma of male groups; F) Pr-SSG levels in plasma of female groups; G) Pr-SSG levels in plasma of Wt groups; H) Pr-SSG levels in plasma of Tg groups. All values are expressed as nmol/mg protein, and presented as Mean ± SEM, n = 5 mice/group. Statistical analyses were conducted via 2-way ANOVA with Tukey’s Multiple Comparison post-hoc test. Gender effect: Male versus Female. †p < 0.05.
In addition, similar Pr-SSG levels (no genotype effects) were identified in male and female mice shown in Fig. 6E and F. Also, we observed no changes in plasma Pr-SSG due to gender (Fig. 6G, H) or treatment effects (Fig. 6E–H). Overall, no significant differences were seen in plasma Pr-SH and Pr-SSG levels regardless of treatment, genotype, or gender, though a gender difference was found in Pr-SH levels. Our results likely indicate the lack of direct relationship of Pr-SSG and Pr-SH levels between plasma and brain.
DISCUSSION
The relationship between oxidative stress and the CRF system
The HPA axis is responsible for releasing CRF and stimulating the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. Stress steroids are then released from the adrenal cortex upon stimulation by ACTH [28]. Because oxidative stress can induce HPA axis dysfunction with aging, it may lead to over-excretion of glucocorticoids that can further inhibit the activity of the HPA axis and result in neuronal damage in vulnerable structures, such as the hippocampus [15]. Consequently, impairment in HPA axis function can further induce more oxidative stress in the hippocampus and adrenal cortex [29]. Increases in oxidative damage are also seen as a form of chronic stress, and studies also have shown that elevated hippocampal Aβ levels may be associated with corticosterone increases in rodent AD brains [30]. Moreover, lysosomal activation induced by oxidative stress has been reported in AD, which may be mechanistically involved in neurodegeneration, as results suggest that lysosomal integrity damage, lysosomal capacity increase, and induction of autophagy are common in AD [31]. Based on these observations and more recent data demonstrating a link between the CRF system and AD pathological processes (e.g., [23, 27, 32]), we hypothesize that CRFR1 antagonism may attenuate oxidative stress through regulating of central CRF signaling and the HPA axis, leading to a reduction in levels of oxidative biomarkers (S-glutathionylated proteins).
GSH redox status and protein S-glutathionylation in the brain
Tissue GSH and GSSG are sensitive and specific intermediates in the oxidative stress cascade that may be mechanistically involved in AD neurodegeneration. For example, Calabrese et al. have reported that decreased GSH contents and increased GSSG levels were observed in AD peripheral lymphocytes [33]. Furthermore, our previous work indicates that oxidative stress may play an important role in AD pathology, with age-dependent changes in GSH status and Pr-SSG levels seen in both brain and blood samples in Tg mice [14].
Stress has been suggested to be involved in AD development and progression [34–36]. Indeed, our results show that female Tg mice treated with vehicle have a decrease in tissue GSH content compared to male Tg counterparts (Fig. 1D), whereas no differences of GSSG levels are found in the brain among these mice (Fig. 1H), indicating that female Tg mice are more susceptible to oxidative stress at this age stage. Interestingly, female Wt mice show similar brain GSH levels (Fig. 1C) and elevated tissue GSSG contents than male counterparts (Fig. 1G), suggesting that female Wt mice might have a higher baseline level compared to male Wt mice. Also, similar GSSG levels (Fig. 1E) and increased amount of tissue GSH contents (Fig. 1A) are observed in male Tg treated with vehicle compared to their male Wt or female Tg counterparts, which may be a result of the fact that tissue GSH or GSSG contents in Tg mice may have reached an equilibrium of GSH redox cycle at this time point [14].
Additionally, as for genotype effects, tissue GSH or GSSG levels in female mice regardless of treatment indicate that no changes of the GSH redox cycle in female mice are present at this age (Fig. 1B, F). Our future studies aim to characterize optimal timepoints for chronic treatment of oxidative stress in Tg mice, because Tg brains are noticeably compromised by oxidative insults in an age-dependent manner. S-glutathionylation is known to modify intracellular Pr-SH to form protein disulfide (Pr-SSG) under oxidative conditions, which is considered as a reliable index of ROS [37] associated with changes in enzymatic activities, energy metabolism, and signaling pathways [38]. Recent studies have implicated S-glutathionylated proteins may be related to neurodegeneration, as increased levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and α-enolase have been reported in AD [39]. Both GAPDH and α-enolase enzymes are involved in the glycolytic pathway for energy production; compromised enzymatic activities induced by oxidative stress is correlated with a progressive decline in glucose metabolism during AD progression [39–41]. Moreover, p53 protein is known as a tumor suppressor that stabilizes and prevents genomic mutation [42] and the inactivation of p53 through S-glutathionylation protect cells from apoptosis [43–45]. p53 protein S-glutathionylation has been observed in human cancer cells [46] and increases of S-glutathionylated p53 monomer and dimer have been found in the inferior parietal lobule of AD patients [47]. In this study, although no treatment effects are shown in the GSH redox system at this timepoint, a significant reduction in oxidized proteins through S-glutathionylation is present (Fig. 2). The hippocampus and cortex are AD-vulnerable brain regions, which are exceedingly sensitive to ROS insults [27]. Our data demonstrate total Pr-SSG levels in both hippocampi and cortices are reduced in Tg mice treated with drug regardless of gender, indicating that CRFR1 antagonist may reduce oxidative stress and potentially rescue proteins from damage.
Enzymatic activities in the brain
GPx plays an essential role in reducing lipid and hydrogen peroxide in GSH redox cycling in mammalian cells. Its activity is recognized as a neuroprotective antioxidant intermediate involved in Huntington’s disease [48]. Reduced GPx activity may lead to elevated levels of hydrogen peroxide, resulting in inflammatory activation and tissue damage [49]. Furthermore, significantly reduced levels and activity of GPx have been reported in the brains of aged Tg and Wt mice [50]. Likewise, in human studies, aged cohorts have reduced GPx activity and increased levels of hydrogen peroxide compared to younger subjects [51]. Additionally, significant decreases of GPx activity have been reported in both mitochondria and synaptosomes of mild AD and AD subjects compared to their age-matched non-demented groups [52]. In the present study, GPx activity is increased in female Tg mice treated with R121919 compared to untreated female Tg cohorts or treated male Tg counterparts (Fig. 3B, D), which suggests that female Tg mice have increased antioxidant enzymatic activity with CRFR1 blockade. Additionally, vehicle treated male Wt mice had higher levels of GPx activity compared to male Tg-vehicle counterparts (Fig. 3A), suggesting that male Wt animals have high baseline levels of GPx activity. GPx is an antioxidant enzyme that counteracts hydroperoxide and ROS to maintain the antioxidative mechanisms, so cells utilize GPx to eliminate increased ROS. Because no change in GSH redox status and GR activity was observed in male mice, the decrease we identified in GPx activity may not be directly associated with the GSH redox system, whereas it may be regulated by changes in sex hormones. Alterations in stress, sex hormones, and other aspects of the neuroendocrine system are the subject of our current investigations and will likely be crucial for understanding sex-dependent differences in AD [53]. As for the GR activity, we found no differences in Tg mice regardless of treatment or gender (Fig. 4C, D), demonstrating that no abnormal GR activity can be detected at this age. Interestingly, because the female Wt-drug group has elevated levels in both GPx activity (Fig. 3C) and GR activity (Fig. 4A, B), female Wt mice treated with R121919 may be more sensitive to the changes in central CRF signaling and oxidative stress.
GSH redox status in plasma
Our data demonstrate that GSH and GSSG contents can be readily measured in blood samples, as reported previously [14]. However, no treatment or genotype differences are presented in plasma GSH, GSSG, and Pr-SSG levels (Figs. 5 and 6), which indicates that plasma samples may not accurately reflect the GSH redox status in the brain at this age stage.
Future studies
To better understand the relationship of oxidative damage and AD neuropathology, we will investigate the changes in oxidative DNA lesion [4], mitochondrial impairment [3], and cognitive deficit [54, 55] with AD progression.
CONCLUSIONS
In summary, our findings reveal that CRFR1 antagonism can reduce oxidized protein levels and improve the activity of antioxidant enzymes in the Tg mouse brain. These data suggest that modulation of the central CRF system may be an important approach to combat oxidative stress in neurodegenerative diseases.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health Grants AG032755, AG047484, DK026741, and AG010483; the Alzheimer’s Art Quilt Initiative; the Alzheimer’s Association; the Foundation for Medical Research. The work of the Drug Design and Synthesis Section, CBRB, NIDA, and NIAAA was supported by the NIH Intramural Research Programs of the National Institute on Drug Abuse (NIDA) and the National Institute of Alcohol Abuse and Alcoholism (NIAAA).
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/14-1722r1).
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-141722.
REFERENCES
- [1].Bogaerts V, Theuns J, van Broeckhoven C. Genetic findings in Parkinson’s disease and translation into treatment: A leading role for mitochondria? Genes Brain Behav. 2008;7:129–151. doi: 10.1111/j.1601-183X.2007.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [3].Zhang C, Rissman RA, Feng J. Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J Alzheimers Dis. 2015;44:375–378. doi: 10.3233/JAD-141890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang C, Nestorova G, Rissman RA, Feng J. Detection and quantification of 8-hydroxy-2′-deoxyguanosine in Alzheimer’s transgenic mouse urine using capillary electrophoresis. Electrophoresis. 2013;34:2268–2274. doi: 10.1002/elps.201300036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nestorova G, Zhang C, Spaulding J, Feng J. Quantitative determination of 8-OHdG in Alzheimer transgenic mice urine using capillary electrophoresis with laser induced fluorescence detection. Free Radic Biol Med. 2010;49:S79. [Google Scholar]
- [6].Zhang C, Rodriguez C, Circu ML, Aw TY, Feng J. S-Glutathionyl quantification in the attomole range using glutaredoxin-3-catalyzed cysteine derivatization and capillary gel electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem. 2011;401:2165–2175. doi: 10.1007/s00216-011-5311-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang C, Kuo CC, Chiu AW, Feng J. Prediction of S-glutathionylated proteins progression in Alzheimer’s transgenic mouse model using principle component analysis. J Alzheimers Dis. 2012;30:919–934. doi: 10.3233/JAD-2012-120028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang S, Njoroge SK, Battle K, Zhang C, Hollins BC, Soper SA, Feng J. Two-dimensional nitrosylated protein fingerprinting by using poly (methyl methacrylate) microchips. Lab Chip. 2012;12:3362–3369. doi: 10.1039/c2lc40132k. [DOI] [PubMed] [Google Scholar]
- [9].Feng J, Navratil M, Thompson LV, Arriaga EA. Principal component analysis reveals age-related and muscle-type-related differences in protein carbonyl profiles of muscle mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:1277–1288. doi: 10.1093/gerona/63.12.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71:621S–629S. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- [11].Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- [12].Li W, Busu C, Circu ML, Aw TY. Glutathione in cerebral microvascular endothelial biology and pathobiology: Implications for brain homeostasis. Int J Cell Biol. 2012;2012:434971. doi: 10.1155/2012/434971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aw TY. Cellular redox: A modulator of intestinal epithelial cell proliferation. News Physiol Sci. 2003;18:201–204. doi: 10.1152/nips.01448.2003. [DOI] [PubMed] [Google Scholar]
- [14].Zhang C, Rodriguez C, Spaulding J, Aw TY, Feng J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;28:655–666. doi: 10.3233/JAD-2011-111244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kobayashi N, Machida T, Takahashi T, Takatsu H, Shinkai T, Abe K, Urano S. Elevation by oxidative stress and aging of hypothalamic-pituitary-adrenal activity in rats and its prevention by vitamin e. J Clin Biochem Nutr. 2009;45:207–213. doi: 10.3164/jcbn.09-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Van Pett K, Viau V, Bittencourt JC, Chan RKW, Li H-Y, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- [17].Rehman HU. Role of CRH in the pathogenesis of dementia of Alzheimer’s type and other dementias. Curr Opin Investig Drugs. 2002;3:1637–1642. [PubMed] [Google Scholar]
- [18].Davis KL, Mohs RC, Marin DB, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Neuropeptide abnormalities in patients with early Alzheimer disease. Arch Gen Psychiatry. 1999;56:981–987. doi: 10.1001/archpsyc.56.11.981. [DOI] [PubMed] [Google Scholar]
- [19].Whitehouse PJ, Vale WW, Zweig RM, Singer HS, Mayeux R, Kuhar MJ, Price DL, De Souza EB. Reductions in corticotropin releasing factor-like immunoreactivity in cerebral cortex in Alzheimer’s disease, Parkinson’s disease, and progressive supranuclear palsy. Neurology. 1987;37:905–909. doi: 10.1212/wnl.37.6.905. [DOI] [PubMed] [Google Scholar]
- [20].Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- [21].Chen C, Wilcoxen KM, Huang CQ, Xie Y-F, McCarthy JR, Webb TR, Zhu Y-F, Saunders J, Liu X-J. Design of 2, 5-dimethyl-3-(6-dimethyl-4-methylpyridin-3-yl)-7-dipropylaminopyrazolo [1, 5-a] pyrimidine (NBI 30775/R121919) and structure-activity relationships of a series of potent and orally active corticotropin-releasing factor receptor antagonists. J Med Chem. 2004;47:4787–4798. doi: 10.1021/jm040058e. [DOI] [PubMed] [Google Scholar]
- [22].Cottone P, Sabino V, Roberto M, Bajo M, Pockros L, Frihauf JB, Fekete EM, Steardo L, Rice KC, Grigoriadis DE, Conti B, Koob GF, Zorrilla EP. CRF system recruitment mediates dark side of compulsive eating. Proc Natl Acad Sci U S A. 2009;106:20016–20020. doi: 10.1073/pnas.0908789106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campbell SN, Zheng C, Monte L, Roe AD, Rice KC, Tache Y, Masliah E, Rissman RA. Increased tau phosphorylation and aggregation in mice overexpressing corticotropin-releasing factor. J Alzheimers Dis. 2015;43:967–976. doi: 10.3233/JAD-141281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ryan D, Koss D, Porcu E, Woodcock H, Robinson L, Platt B, Riedel G. Spatial learning impairments in PLB1 knock-in Alzheimer mice are task-specific and age-dependent. Cell Mol Life Sci. 2013;70:2603–2619. doi: 10.1007/s00018-013-1314-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gutman DA, Owens MJ, Skelton KH, Thrivikraman KV, Nemeroff CB. The corticotropin-releasing factor1 receptor antagonist R121919 attenuates the behavioral and endocrine responses to stress. J Pharmacol Exp Ther. 2003;304:874–880. doi: 10.1124/jpet.102.042788. [DOI] [PubMed] [Google Scholar]
- [27].Rissman RA, Staup MA, Lee AR, Justice NJ, Rice KC, Vale W, Sawchenko PE. Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc Natl Acad Sci U S A. 2012;109:6277–6282. doi: 10.1073/pnas.1203140109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: Implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- [29].Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol. 2013;9:228–240. doi: 10.1038/nrendo.2013.29. [DOI] [PubMed] [Google Scholar]
- [30].Rothman SM, Mattson MP. Adverse stress, hippocampal networks, and Alzheimer’s disease. Neuromolecular Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Butler D, Bahr BA. Oxidative stress and lysosomes: CNS-related consequences and implications for lysosomal enhancement strategies and induction of autophagy. Antioxid Redox Signal. 2006;8:185–196. doi: 10.1089/ars.2006.8.185. [DOI] [PubMed] [Google Scholar]
- [32].Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, Butterfield DA. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal. 2006;8:1975–1986. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- [34].Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: The glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- [35].Wilson R, Evans D, Bienias J, De Leon CM, Schneider J, Bennett D. Proneness to psychological distress is associated with risk of Alzheimer’s disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- [36].Wilson RS, Bennett DA, Mendes de Leon CF, Bienias JL, Morris MC, Evans DA. Distress proneness and cognitive decline in a population of older persons. Psychoneuroendocrinology. 2005;30:11–17. doi: 10.1016/j.psyneuen.2004.04.005. [DOI] [PubMed] [Google Scholar]
- [37].Carletti B, Passarelli C, Sparaco M, Tozzi G, Pastore A, Bertini E, Piemonte F. Effect of protein glutathionylation on neuronal cytoskeleton: A potential link to neurodegeneration. Neuroscience. 2011;192:285–294. doi: 10.1016/j.neuroscience.2011.05.060. [DOI] [PubMed] [Google Scholar]
- [38].Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid Redox Signal. 2011;15:233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer’s disease brain: Beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Butterfield DA, Hardas SS, Lange ML. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J Alzheimers Dis. 2010;20:369–393. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pocernich CB, Butterfield DA. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim Biophys Acta. 2012;1822:625–630. doi: 10.1016/j.bbadis.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Talos F, Moll UM. Role of the p53 family in stabilizing the genome and preventing polyploidization. Adv Exp Med Biol. 2010;676:73–91. doi: 10.1007/978-1-4419-6199-0_5. [DOI] [PubMed] [Google Scholar]
- [43].Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cenini G, Sultana R, Memo M, Butterfield DA. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45:81–85. doi: 10.1016/j.freeradbiomed.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cenini G, Sultana R, Memo M, Butterfield DA. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J Cell Mol Med. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. J Biol Chem. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Di Domenico F, Cenini G, Sultana R, Perluigi M, Uberti D, Memo M, Butterfield DA. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s disease brain: Implications for AD pathogenesis. Neurochem Res. 2009;34:727–733. doi: 10.1007/s11064-009-9924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mason RP, Casu M, Butler N, Breda C, Campesan S, Clapp J, Green EW, Dhulkhed D, Kyriacou CP, Giorgini F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat Genet. 2013;45:1249–1254. doi: 10.1038/ng.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Espinoza SE, Guo H, Fedarko N, DeZern A, Fried LP, Xue QL, Leng S, Beamer B, Walston JD. Glutathione peroxidase enzyme activity in aging. J Gerontol A Biol Sci Med Sci. 2008;63:505–509. doi: 10.1093/gerona/63.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang Q, Aluise CD, Joshi G, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: Toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010;88:2618–2629. doi: 10.1002/jnr.22422. [DOI] [PubMed] [Google Scholar]
- [51].Ito Y, Kajkenova O, Feuers RJ, Udupa KB, Desai VG, Epstein J, Hart RW, Lipschitz DA. Impaired glutathione peroxidase activity accounts for the age-related accumulation of hydrogen peroxide in activated human neutrophils. J Gerontol A Biol Sci Med Sci. 1998;53:M169–M175. doi: 10.1093/gerona/53a.3.m169. [DOI] [PubMed] [Google Scholar]
- [52].Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Friesen NT, Buchau AS, Schott-Ohly P, Lgssiar A, Gleichmann H. Generation of hydrogen peroxide and failure of antioxidative responses in pancreatic islets of male C57BL/6 mice are associated with diabetes induced by multiple low doses of streptozotocin. Diabetologia. 2004;47:676–685. doi: 10.1007/s00125-004-1367-x. [DOI] [PubMed] [Google Scholar]
- [54].Kuo CC, Zhang C, Rissman RA, Chiu AW. Long-term electrophysiological and behavioral analysis on the improvement of visual working memory load, training gains, and transfer benefits. J Behav Brain Sci. 2014;4:234–246. doi: 10.4236/jbbs.2014.45025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kuo CC, Luu P, Morgan KK, Dow M, Davey C, Song J, Malony AD, Tucker DM. Localizing movement-related primary sensorimotor cortices with multi-band EEG frequency changes and functional MRI. PLoS One. 2014;9:e112103. doi: 10.1371/journal.pone.0112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.